
Solid-phase synthesis and electrochemical pseudo-capacitance of nitrogen-atom interstitial compound Co3N
Jian-Fei
Gao
a,
Wei-Bin
Zhang
a,
Zhi-Yun
Zhao
a and
Ling-Bin
Kong
 *ab
*ab
aState Key Laboratory of Advanced Processing and Recycling of Non-Ferrous Metals, Lanzhou University of Technology, Lanzhou 730050, P. R. China. E-mail: konglb@lut.cn; Fax: +86 931 2976578; Tel: +86 931 2976579
bSchool of Materials Science and Engineering, Lanzhou University of Technology, Lanzhou 730050, P. R. China
First published on 27th March 2018
Abstract
Metal nitrides have great potential for electrochemical energy storage, but are relatively scarcely investigated. Herein, a novel metal nitride, Co3N, is prepared by nitridation using a Co3O4 precursor heated at 500 °C for 2 h in flowing NH3, and characterized by using X-ray diffraction (XRD), field emission-scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM), selected area electron diffraction (SAED) and energy dispersive spectroscopy (EDS) methods. Besides, the electrochemical properties of Co3N are investigated using cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS). The as-prepared Co3N demonstrates remarkable electrochemical performance with a high specific capacitance of 112.3 F g−1 at a current density of 0.5 A g−1, good rate capability (72.4%, from 0.5 to 5 A g−1) and superior cycling stability (109.7% retention over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at a current density of 2 A g−1). Finally, a Co3N//activated carbon (AC) asymmetric supercapacitor (ASC) is successfully assembled by using Co3N and AC as the positive electrode and negative electrode, respectively. The as-fabricated ASC device can achieve a maximum energy density of 12.1 W h kg−1 at a power density of 204.6 W kg−1, which shows that the Co3N material should be a potential electrode material for supercapacitors.
000 cycles at a current density of 2 A g−1). Finally, a Co3N//activated carbon (AC) asymmetric supercapacitor (ASC) is successfully assembled by using Co3N and AC as the positive electrode and negative electrode, respectively. The as-fabricated ASC device can achieve a maximum energy density of 12.1 W h kg−1 at a power density of 204.6 W kg−1, which shows that the Co3N material should be a potential electrode material for supercapacitors.
1. Introduction
The rapid depletion of non-renewable resources calls for the development of clean energy sources.1 This urgent demand has led to a global increase in the research of energy storage devices and systems.2,3 As one of the most important electrochemical energy storage devices, supercapacitors, including electrochemical double layer capacitors and pseudocapacitors, can provide means owing to their high power density, long cycle life and low maintenance cost.4–8 Thus, many researchers are engaged in investigating electrode materials for supercapacitors.In addition to the well-known carbon materials, some transition metal oxides, such as MnO2, Fe2O3, and NiO,9–12 are also common electrode materials for supercapacitors. Moreover, the interest in new materials based on bimetallic oxides or hydroxides is currently increasing.13–16 Although high capacitance (up to 1000 F g−1) has been obtained for transition metal oxides or hydroxides, as a result of their pseudocapacitances, the capacitance drastically decreased when stronger current regimes were applied because of their high electrical resistivity, which is undesirable for electrode materials in energy storage applications.17
To overcome this situation, highly conductive and ductile carbonaceous materials (i.e. AC, CNTs, graphene, and mesoporous carbon) have been introduced to prepare carbon material supported composites,18 such as N-doped carbon coated Fe3O4 composites,19 carbon-encapsulated tungsten oxide nanowires,20 and NiWO4/reduced graphene oxide nanocomposites.21 Although these composites can improve the electrical conductivity to some extent, the effect is unsatisfactory. So, it is desirable to exploit new electrode materials with intrinsic high electrical conductivity and outstanding electrochemical performance.
Recently, some transition metal nitrides (TMNs) have attracted great interest because of the unique nature of M(metal)–N(nitride) bonding which is described as simultaneous contributions from metallic, covalent and ionic bonding.22 In addition, the electronic structures of TMNs are similar to those of noble metals, which gives the expectation that TMNs should be able to provide similar performance to noble metal electrodes.23 Previous reports have proved that transition metal nitrides, such as niobium nitride, lanthanum nitride, vanadium nitride, and manganese nitride, can be used as electrode materials for supercapacitors owing to their outstanding electrochemical behavior.24–27 We studied Mo2N28 in an acid electrolyte, showing a specific capacitance of 171 F g−1 in the voltage range from −1.0 to −0.2 V (vs. SCE) that corresponds to a discharge current density of 0.5 A g−1. Choi et al.29 studied the capacitive behaviour of VN in an aqueous electrolyte, showing a high specific capacitance. Lu et al.30 synthesized VN nanowires showing an excellent capacitive behavior in the presence of LiCl/PVA gel electrolyte. Nevertheless, these previous reports on the application of transition metal nitrides in supercapacitors are limited to several nitrides formed using V, Nb, Mo, W, Mn and Ti, which inspires us to explore some new metal nitrides for application in supercapacitors.
Among metal nitrides, Co3N, as one of the binary nitride systems of 3d metals, has been regarded as a nitrogen-atom interstitial compound.31The nitrogen atoms incorporated into the interstices of the cobalt based-framework are bonded covalently to the cobalt atoms combined with the cobalt–cobalt interactions, giving metal-like properties. Currently, Co3N is recognized as the best choice as a highly efficient catalyst for the hydrogen evolution reaction due to its outstanding electrical conductivity, low cost, low resistance and environmental friendliness.32–35 In this regard, Co3N will be a promising electrode material for supercapacitors. Notably, a relative study has been ignored over the past years. Although there are some studies on the use of nickel nitride or iron nitride that belongs to the IIIVB group nitrides,36–38 there is no report about Co3N which also belongs to the IIIVB group nitrides as an electrode material for supercapacitors.
On the other hand, there have been several reports concerning the preparation and microstructure of Co3N in the past decades,39–41 in which magnetron sputtering was used to deposit these thin films. It is not easy to cause metal nitridation due to the inactivity of nitrogen gas. Therefore, it is necessary to exploit a simple preparation method for Co3N.
Thus, the objective of this research was to develop an innovative process for preparing a pure Co3N compound that can be employed in supercapacitors. Herein, as a proof-of-concept experiment, we developed a Co3N powder by a simple nitridation reaction for the first time and its electrochemical properties as an electrode material for supercapacitors were investigated. We also proposed a possible electrochemical energy storage mechanism to explain the results shown in the electrochemical test patterns. To the best of our knowledge, this is the first report of the Co3N powder as a positive electrode material for supercapacitors. Through this work, we extend earlier research on TMN compounds to prepare IIIVB group nitrides, which could provide some new opportunities for TMNs in high-performance energy storage applications.
2. Experimental
2.1. Chemicals and materials
All the reagents used in the experiment were of analytical grade and used without further purification. Cobalt(II) chloride hexahydrate (CoCl2·6H2O, >99%), ammonium hydroxide (NH3·H2O, 25–28%), AC, hydrochloric acid (HCl, 37%), potassium hydroxide (KOH, 90%), and ethanol (Et–OH, >99.7%) were purchased from Sinopharm Chemical Reagent Co. Ltd. Nickel foam (99.99%, 1.7 mm) was purchased from Lizhiyuan Co, Ltd (Shanxi, China). Prior to use, nickel foam was rinsed ultrasonically with 1 mol L−1 HCl solution for 15 min, and then washed three times with deionized water and absolute ethanol, respectively. Deionized water with a resistivity of >10.0 MΩ was used in experiments.2.2. Synthesis of cobalt nitride
2.3. Material characterization
The crystal structures of the products were characterized using X-ray diffraction (XRD, Rigaku, D/MAX2400) with Cu Kα radiation (wavelength 0.15418 nm) operating at 40 kV and 60 mA. Morphologies and structures were observed with a scanning electron microscope (SEM, JEOL JSM-6701F), transmission electron microscope (TEM, JEOL JEM-2010), high-resolution TEM (HR-TEM), and selected area electron diffractometer (SAED).2.4. Electrode preparation
The working electrode was prepared by mixing the electrode material (80 wt%), acetylene black (7.5 wt%), conducting graphite (7.5 wt%), PTFE (5 wt%), and then a few drops of ethanol and stirring for several minutes. Afterwards, the mixed and ground slurry was pressed at 10 MPa onto a fresh nickel foam (ca. 1 cm2) and kept in a vacuum oven at 60 °C for 12 h.2.5. Electrochemical evaluation
The electrochemical performance of the Co3N electrode was investigated with a three-electrode configuration and a two-electrode device. For a three-electrode configuration, the as-prepared samples directly acted as the working electrodes. A standard calomel electrode (SCE) and Pt foil (1 cm2) were used as the reference and counter electrodes, respectively. A solution of 6 mol L−1 aqueous KOH was chosen as the electrolyte for electrochemical measurements. Cyclic voltammetry (CV), galvanostatic charge–discharge measurements (GCD) and electrochemical impedance spectroscopy (EIS) were carried out on a CHI660E electrochemical workstation. The cycling performance was measured with a CT2001A tester (Wuhan, China). The specific capacitance (Cs) of a single electrode was calculated from the galvanostatic charge–discharge (GCD) profiles using eqn (1):42 | (1) |
An asymmetric supercapacitor (ASC) configuration was constructed by using Co3N and AC as positive and negative electrodes face to face in 6 mol L−1 aqueous KOH. The Q of each electrode depends on the specific capacitance (Cs), the potential range of the charge–discharge tests (V) and the mass of the active material (m) according to eqn (2):
| Q = Cs × m × V | (2) |
According to the charge-balance principle (Q+ = Q−), where Q+ and Q− represent the charges stored in the positive and negative electrodes, respectively, the specific mass ratio of AC to Co3N was thus designed as 1.43:1 in such an asymmetric device. And the total mass of electroactive materials in the device is 6.8 mg. The calculation of specific capacitance (Cdevice), energy density (E), and power density (P) is based on the total mass of the cathode and anode according to eqn (3)–(5).42
 | (3) |
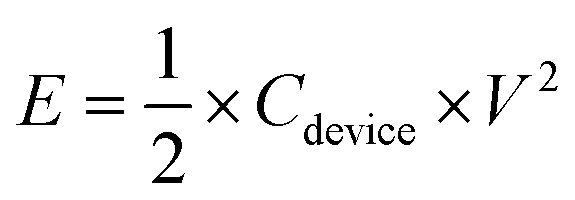 | (4) |
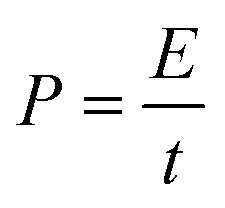 | (5) |
3. Results and discussion
3.1. Material structures
In order to confirm the complete transformation from Co(OH)2 to Co3O4 and from Co3O4 to Co3N, we conducted a series of experiments to study the temperature and time effect on the oxidation reaction of Co(OH)2 and the nitridation reaction of Co3O4. The XRD evolution of the Co–O precursor at different temperatures is shown in Fig. 1a. It is obvious that the diffraction peaks are invariable when the temperature is up to 100 °C, indicating that no phase change occurred at this temperature. When the temperature is up to 150 °C, diffraction peaks change and are consistent with the standard patterns of Co3O4 (JCPDF 43-1003), which demonstrated that the Co3O4 phase is formed at this temperature. However, some diffraction peaks of Co(OH)2 are also observed, indicating the existence of unreacted Co(OH)2. When the temperature is between 200 °C and 300 °C, the location of all diffraction peaks is consistent with the Co3O4 phase, which indicates that the Co(OH)2 phase has been removed and produced Co3O4 phase completely by heat-treatment at 200 °C. At the same time, we also studied the time effect on the oxidation reaction of Co(OH)2. In Fig. 1b, with the increase of annealing time, it is obvious that the diffraction peaks are invariable. Based on the above discussion, we found that the temperature has a great influence on the oxidation reaction of Co(OH)2, when the temperature rises to 200 °C, Co(OH)2 can completely translate into Co3O4. And the annealing time effect is negligible. | ||
| Fig. 1 XRD patterns. (a) Co–O precursor at different annealing temperatures; (b) Co–O precursor at different annealing times; (c) Co–N samples at different NH3 annealing temperatures; (d) Co–N samples at different NH3 annealing times. | ||
The XRD patterns of Co–N samples at different NH3 annealing temperatures are presented in Fig. 1c. When the temperature is up to 200 °C, the location of all diffraction peaks is consistent with the Co3O4 phase indicating that no phase change occurred at this temperature. When the temperature is up to 300 °C, the Co3O4 phase is successfully converted into the pure phase Co (JCPDS 15-0806). When the temperature is up to 500 °C, the location of all diffraction peaks is consistent with the pure phase Co3N (JCPDS 06-0691). When the temperature is up to 700 °C, only a Co diffraction peak is observed. At the same time, we also studied the time effect on the nitridation reaction of Co3O4. In Fig. 1d, when the time is 1 h, the XRD pattern reveals that some diffraction peaks are consistent with the CoN phase (JCPDS 83-0831). Besides, the XRD pattern also shows some diffraction peaks of the Co3O4 phase (JCPDF 43-1003) and Co phase (JCPDS 15-0806). When the time is 2 h, only a Co3N diffraction peak is observed. When the time is 3 h, the location of all diffraction peaks is consistent with the pure phase Co (JCPDS 15-0806). Based on the above discussion, we found that both the NH3 annealing time and temperature have a decisive influence on the nitriding reaction.
In order to demonstrate the reaction evolution process of products and their crystal structures in different steps, we also show the XRD patterns of Co(OH)2, Co3O4 (300 °C, 2 h), and Co3N (500 °C, 2 h). The corresponding XRD patterns of Co(OH)2, Co3O4, and Co3N are all recorded in Fig. 2a–c, respectively. In Fig. 2a, one can see that the XRD pattern of the first step product only shows the characteristic peaks of the Co(OH)2 phase. All of the diffraction peaks agree well with the standard diffraction data for bulk Co(OH)2 (JCPDS 46-0605). In Fig. 2b, the characteristic diffraction peaks of the second step product synthesized in an air atmosphere can be assigned to Co3O4, whereas the peaks at 2θ = 19.0°, 31.27°, 36.84°, 38.55°, 44.81°, 55.66°, 59.35°, 65.23°, 74.12°, 77.34° and 78.40° can be indexed to the (111), (220), (311), (222), (400), (422), (511), (440), (620), (533) and (622) crystal planes of Co3O4 as reported, which is in agreement with JCPDS (43-1003).43 In addition, all characteristic peaks appearing in Fig. 2c correspond to those in the standard pattern of pure phase Co3N (JCPDS 06-0691). This proves that Co(OH)2 was successfully converted into the Co3N based product (which is consistent with a typical hexagonal crystal system with a P63/mmc (194) space group obtained from the XRD standard card). In these three XRD patterns, no other characteristic peaks are observed showing the purity and single-phase nature of Co(OH)2, Co3O4, and Co3N. In particular, it should be noted that no additional peaks corresponding to any other phase are observed in the XRD patterns of Co3N. This indicates that the synthesized Co3N is stable and resistive to the formation of oxides. Briefly, the absolutely different phase of three samples demonstrated that our experimental design is successful. According to the XRD results, eqn (6)–(8) are responsible for the synthesis of Co3N at different reaction steps:
| CoCl2 + 2NH3·H2O → Co(OH)2 + 2NH4Cl | (6) |
| 6Co(OH)2 + O2 → 2Co3O4 + 6H2O | (7) |
| 6Co3O4 + 16NH3 → 6Co3N + 24H2O + 5N2 | (8) |
 | ||
| Fig. 2 XRD patterns and EDX spectrum and atomic ratio of Co3N. (a) XRD pattern of Co(OH)2; (b) XRD pattern of Co3O4; (c) XRD pattern of Co3N; (d) EDX spectrum of Co3N. | ||
The nitridation reaction of TiO2 with NH3 was reported in ref. 44 that can be expressed by eqn (9):
| 6TiO2 + 8NH3 → 6TiN + 12H2O + N2 | (9) |
Similar reactions can be written for the nitridation of Co3O4, as eqn (6)–(8), according to the XRD analysis. Moreover, the peak intensity of Co3N is very high, demonstrating improvement of the crystallization degree, which enhances the cycle stability due to the formation of a stable structure.45
In addition to the crystal structural analysis, the compositional analysis of the synthesized cobalt nitride was also carried out using the EDX spectroscopy analysis towards ascertaining the composition of the Co3N phase. The results of EDX analysis are provided in Fig. 2d. The presence of Co and N is confirmed in the synthesized Co3N compounds. However, there are some traces of non-indexed peaks in the samples, that correspond to carbon (C), oxygen (O) and copper (Cu). The Cu signal arises from the copper foil used as the substrate for the sample preparation towards the EDX analysis, the elemental C comes from the used conductive tape and the O signal can be attributed to the adsorbed oxygen on the surface of Co3N. No other elements are found in the samples, indicating that pure samples of Co3N are obtained. Further, the atomic ratio of the elements present in the composition is also obtained from the EDX spectrum that confirmed the Co to N atomic ratio as in the Co3N phase, which agrees with the results of XRD analysis.
The microstructure and morphology of the synthesized Co3N were analyzed using FESEM and TEM and the representative images obtained are shown in Fig. 3a–f. Fig. 3a and b show the SEM images in 10000× and 30000×, respectively. They reveal that the morphology of this material is found to be a spherical shape with a bumpy surface and the average particle size is found to be in the range of 100–200 nm in diameter. The bumpy surface morphology of Co3N renders a larger electrode/electrolyte contact area for electrolyte ion diffusion and access to Co3N, which is expected to enhance electrochemical properties. Moreover, these particles are linked and inlaid with each other to form well stable chains or agglomerations of a few grains, showing a macro-structure with empty spaces. This structure with a large amount of open space accelerated the diffusion of electrolyte ions, leading to a reduced internal resistance.
 | ||
| Fig. 3 SEM images, TEM images, HR-TEM image and SAED image of Co3N. (a and b) The SEM pattern in 10000× and 30000×, respectively; (c and d) TEM patterns; (e) the HR-TEM image over the crystallographic trait; (f) the SAED image. | ||
The TEM images presented in Fig. 3c and d also demonstrate that nitridation with ammonia leads to aggregated, spheroidal Co3N nanocrystals with a size of 150 ± 50 nm. This was in accordance with the result of the SEM image shown in Fig. 3a and b. The crystal plane spacing of Co3N is about 0.250 nm as shown in Fig. 3e, corresponding to the (100) crystal plane spacing (0.230 nm) of the hexagonal Co3N crystal, and this is consistent with the XRD and EDX analysis. The SAED image shown in Fig. 3f attests to the typical polycrystalline feature.
3.2. Electrochemical capacitance of the electrode
In order to perform the electrochemical characterization of the as-synthesized compound, the Co3N electrode was employed in a three-electrode configuration at room temperature for confirming its capacitive behavior using the CV, GCD, and EIS measurements. Also, all the electrochemical measurements were performed in 6 mol L−1 KOH aqueous solution. KOH was selected as the electrolyte owing to the smaller hydration sphere volume of K+ ions, promoting higher mobility for redox reactions and an effective interaction of ions with the electrode surface enabling rapid storage of charges.46Fig. 4a displays a set of typical CV curves of the Co3N sample recorded in the potential range of −0.2 to 0.5 V (vs. SCE) employing the scan rates of 5, 10, 20, 30, 40, 50, 60, 70, 80, 90 and 100 mV s−1. Notably, the shapes of the CV curves reveal that the charge-storage mechanism of cobalt nitride is distinct from the electric double-layer capacitance which is normally close to an ideal rectangular shape. The obvious typical redox couples corroborate the occurrence of surface redox reactions and the pseudocapacitive nature of the Co3N electrode.47 Two typical redox couples can be observed for all scan rates. This is clear evidence that the charge-storage mechanism in the Co3N material is significantly more faradaic in nature than the capacitive type. These peaks are assumed to be quasi reversible electron-transfer processes occurring on the surface of Co3N electrodes between different valence states of cobalt in alkaline electrolyte medium. Thus the electrochemical capacitance of Co3N is attributed to a quasi-reversible electron transfer process. Also, it can be seen from the curves that the anodic peaks shifted toward positive potential and the cathodic peaks shifted toward negative potential with the increase of scan rate indicating increment of the area of the CV curves and thus the improved charge storage capability of the electrodes. Furthermore, the anodic and cathodic peaks are similar even at a high scan rate of 100 mV s−1, revealing good rate capability, i.e. in other words fast charging and discharging processes at the interface between the Co3N matrix and the electrolyte solution. This is ascribed to the high electrical conductivity of as-prepared Co3N.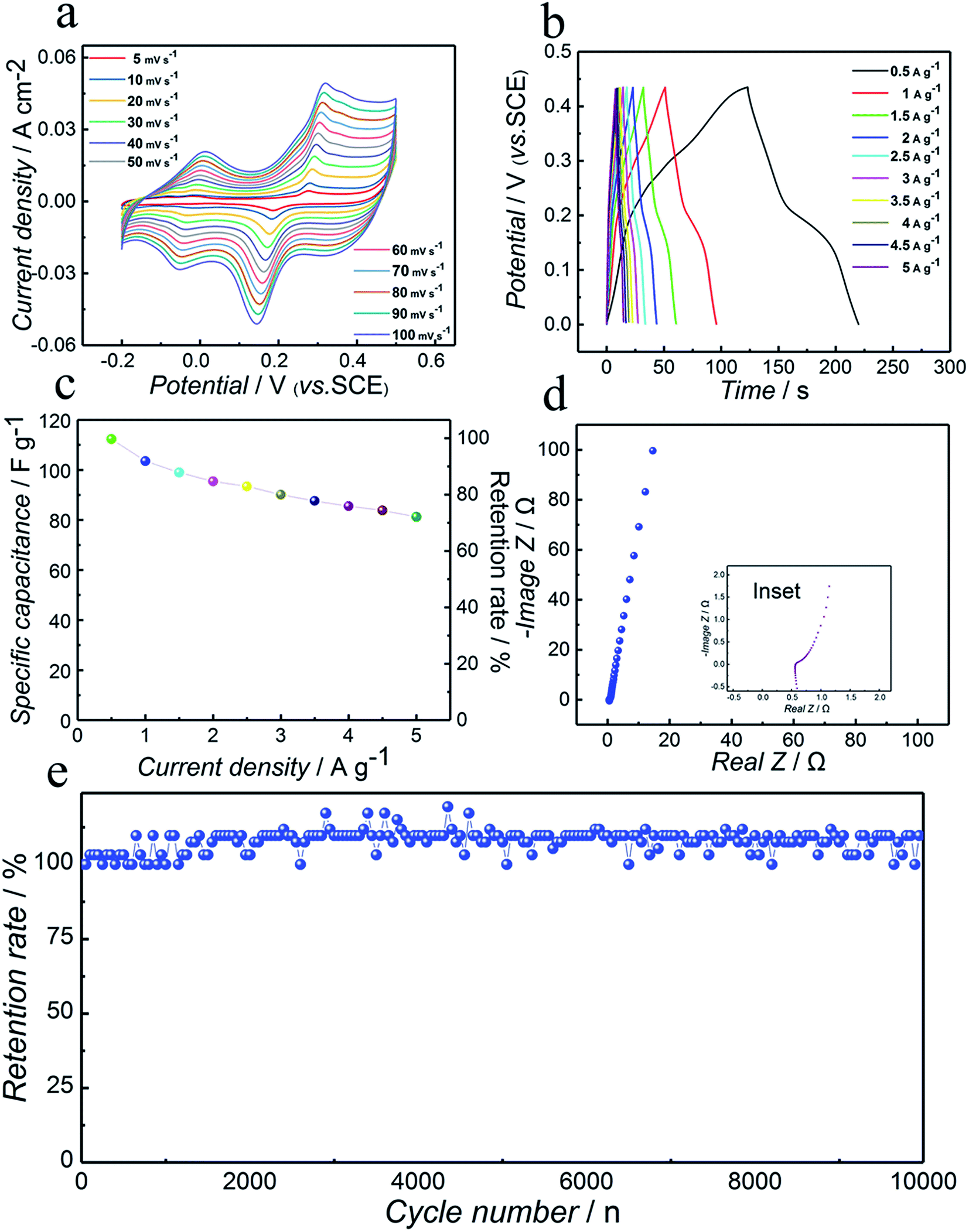 | ||
| Fig. 4 Three-electrode electrochemical measurements of the Co3N electrode. (a) Cyclic voltammograms at variable scan rates of 5, 10, 20, 30, 40, 50, 60, 70, 80, 90 and 100 mV s−1 within the potential window from −0.2 to 0.5 V vs. SCE; (b) galvanostatic charge–discharge curves at various current densities from 0.5 to 5 A g−1 within the potential window from 0 to 0.43 V vs. SCE; (c) specific capacitance as a function of the current densities and capacitance retention rate at various current densities; (d) Nyquist plots measured with an amplitude of 10 mV over the frequency range from 100000 to 0.1 Hz, inset: the magnified high frequency region; (e) cycling performance of synthesized electrode materials for 10000 cycles at a current density of 2 A g−1 within the potential window from 0 to 0.43 V vs. SCE. | ||
In order to evaluate the specific capacitance of the Co3N compound, GCD measurements have been conducted in a potential range of 0–0.43 V (vs. SCE) at various current densities from 0.5 to 5 A g−1. The typical GCD curves of the Co3N electrode at different current densities are presented in Fig. 4b. Apparently, in good agreement with the CV results in Fig. 4a, all of the curves present distinctly nonlinear features but not a triangular line, which confirmed the ideal pseudocapacitive nature of the electrode arising from the surface faradaic redox reactions.
Furthermore, according to eqn (1) mentioned above, the specific capacitance of Co3N can be calculated to be 112.3, 103.5, 98.9, 95.4, 93.4, 90.1, 87.6, 85.5, 83.8 and 81.3 F g−1 at a current density of 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5 and 5 A g−1 using GCD discharge curves, respectively. In order to better present the rate capability of the as-prepared electrodes, the specific capacitance as a function of current density is shown in Fig. 4c, and it is clear from the plot that about 72.4% specific capacitance retention with current density increasing 10 fold shows its stable electrochemical performance and high power output properties. As seen, the decrease of the specific capacitance of the electrodes with the increase of current density indicates that insufficient active materials are involved in the redox reaction and more obvious polarization phenomenon at higher current density.48 This excellent specific capacitance retention can be explained by the following reasons: low internal resistance and high conductivity are two important factors that are critical to achieve high rate supercapacitors.49 Furthermore, the very low IR drop observed even at high current densities could demonstrate the high conductivity and low internal resistance of the as-prepared electrode.50 As seen, the discharge curves with small IR drops at different current densities point to an improvement in the electrical and ionic conductivity of Co3N as shown in Fig. 4b. So, the Co3N electrode still maintains 72.4% of the initial capacitance after increasing the current density by a factor of 10, indicating its excellent rate capability.
To further understand the electrochemical performance characteristics, EIS was performed in the frequency range of 10 kHz to 0.01 Hz with an applied bias voltage of 10 mV. We can obtain the electrical conductivity and ion diffusion via EIS. Depending on the frequencies and characteristics of each part, the Nyquist plots can be divided into three segments, namely high, intermediate and low frequency regions. It is well known that the Nyquist plot for the electrode materials in redox supercapacitors should consist of a semicircle at high frequency (related to the faradaic reaction) and a linear section at low frequency (related to Warburg impedance).
As shown in the inset of Fig. 4d, the semicircle in the high frequency range corresponds to the faradaic charge-transfer resistance (Rct). Actually, Rct is representative of electrochemical reaction resistance, also known as faradaic resistance, which determines the response rate of the electrode material in the electrolyte. Also, the charge transfer resistance could be roughly estimated as the radius of the high frequency semicircle of the Nyquist plot. As displayed, the Nyquist plots for the Co3N electrode exhibit negligible semicircular shapes in the high frequency range, suggesting that the Co3N electrode possesses excellent interfacial charge transfer efficiency.
As shown in Fig. 4d, the linear part after the semicircle is related to the diffusive resistance (Warburg impendence, W) of the electrolyte into the interior of the electrode pores and ion diffusion into the host materials, while the high slope indicates the small diffusive resistance of electrodes owing to the fast ion diffusion during redox processes. This means that the ion and electrolyte can easily diffuse to the surface of Co3N electrode materials.
In the high frequency region, the intersection of the curve on the real axis represents Rs, where Rs is the series resistance of ionic resistance, intrinsic resistance and contact resistance of the electrochemical system. As observed from the right amplifying EIS data the Rs of Co3N was calculated to be 0.51 Ω, which implies that Co3N has a superior electrical conductivity. These EIS studies also verify that the better electrochemical performance of the Co3N electrode is ascribed to a lower solution resistance and charge-transfer resistance due to the high conductivity of Co3N. These results agree with the CV and GCD analysis.
Long cycling life is another important requirement for supercapacitors. In this context, cycling life tests for Co3N are carried out by repeating the GVD test between 0 V and 0.43 V (vs. SCE) at a current density of 2 A g−1 for 10000 cycles. As shown in Fig. 4e, it is evidenced that Co3N can retain 109.7% of its original capacity after 10000 charge–discharge cycles. Good cycling stability performance may be attributed to the unique structure and high conductivity of Co3N. It is because that solid-phase synthesis can form a more stable structure compared with liquid-phase synthesis. In constant charging–discharging tests, the structures of the products from liquid-phase synthesis methods tend to collapse, resulting in the decrease of cycling life. In contrast, products from solid-phase synthesis methods can still maintain a stable structure in constant charging–discharging tests.
It can be seen that, according to the CV, GCD and EIS analysis, the redox reaction in the present Co3N specimens is a surface or near-surface reaction, which can be attributed to the excellent conductivity of the compound. Besides this, the high rate capability and improved cycling stability of the composite could come from its low resistance revealed by the EIS spectrum.
3.3. Electrochemical performance of the cell
To demonstrate the practical application of the as-synthesized Co3N electrode, an aqueous ASC has been assembled. AC has been intensively investigated as a negative electrode for supercapacitor applications due to its high surface area and good electrical conductivity.The CV curves of ASC Co3N//AC are recorded between 0 and 1.63 V at various scan rates of 5, 10, 20, 30, 40 and 50 mV s−1 and shown in Fig. 5a, and these curves show a nearly rectangular mirror-image current response on voltage reversal combining the characteristics of faradaic pseudocapacitance and double layer capacitance. Furthermore, the shape of the CV curves remained unchanged with increasing the scan rate, even at a high scan rate of 50 mV s−1, suggesting better diffusion of electrons and ions to the inner regions of the electrode. Therefore, ideal rate capability and excellent reversibility can be expected, which are desirable for high-power supercapacitors.
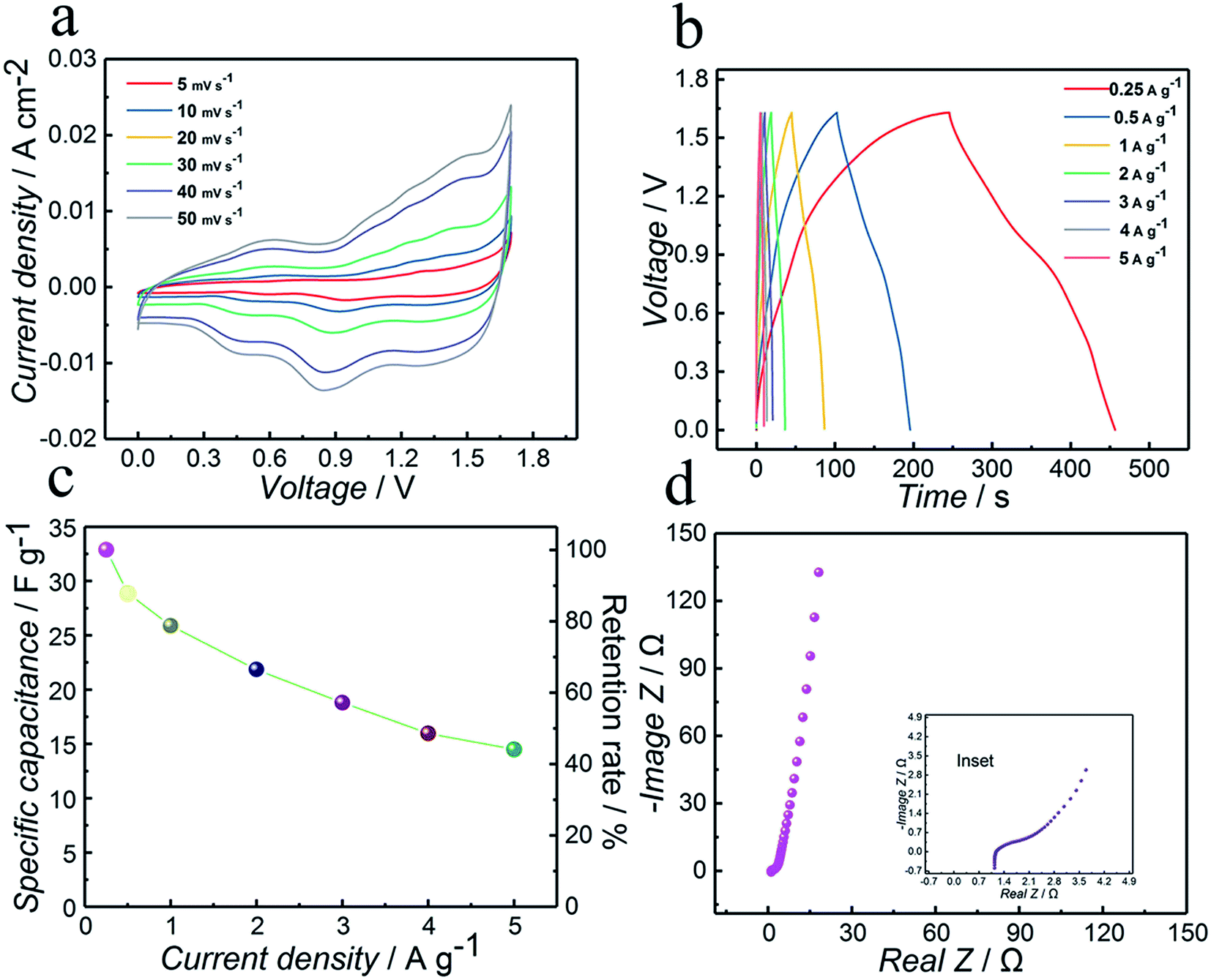 | ||
| Fig. 5 Co3N//AC asymmetric supercapacitor measurements within the voltage window from 0 to 1.63 V. (a) Cyclic voltammograms at variable scan rates of 5, 10, 20, 30, 40 and 50 mV s−1; (b) GCD curves at various current densities from 0.25 to 5 A g−1; (c) specific capacitance as a function of the current densities and capacitance retention rate at various current densities; (d) Nyquist plots measured with an amplitude of 10 mV over the frequency range from 100000 to 0.1 Hz, inset: the magnified high frequency region. | ||
In order to further evaluate the performance of the ASC and support the CV studies, GCD was conducted at various current densities within the potential window of 1.63 V. As illustrated in Fig. 5b, the GCD curves of the ASC at various current densities show a nearly linear and symmetrical shape and the initial voltage loss (i.e. IR drop) observed in the discharge curve is rather small, demonstrating its excellent electron conductivity and good coulombic efficiency again. As a result, the specific capacitance values are 32.9, 28.9, 25.9, 21.9, 18.8, 15.0 and 14.5 F g−1, respectively, calculated based on the GCD curves at different current densities of 0.25, 0.5, 1, 2, 3, 4 and 5 A g−1. The specific capacitance of the asymmetric capacitor Co3N//AC is summarized in Fig. 5c, where one can see that its specific capacitance still reaches up to 14.5 F g−1 even at a very high current density of 5 A g−1, which corresponds to the retention of 44.1%, indicating a high rate capability.
Fig. 5d shows the Nyquist plot of the ASC by EIS. From the extended spectrum in the inset, the value of Rs is about 1.1 Ω, which is one of the desirable factors for achieving high power capability. This manifests the favorable conductivity and very low internal resistance of the ASC.
Fig. 6a shows the Ragone plots of the ASC based on GCD at different current densities. It is worth noting that the maximum energy density obtained for the ASC is 12.1 W h kg−1 at a power density of 204.6 W kg−1, what is more, it can still retain 5.4 W h kg−1 at a higher power density of 4101.2 W kg−1. This superior energy density property of the ASC can be attributed to both its high specific capacitance and its much elevated cell voltage of 1.63 V in the aqueous electrolyte.
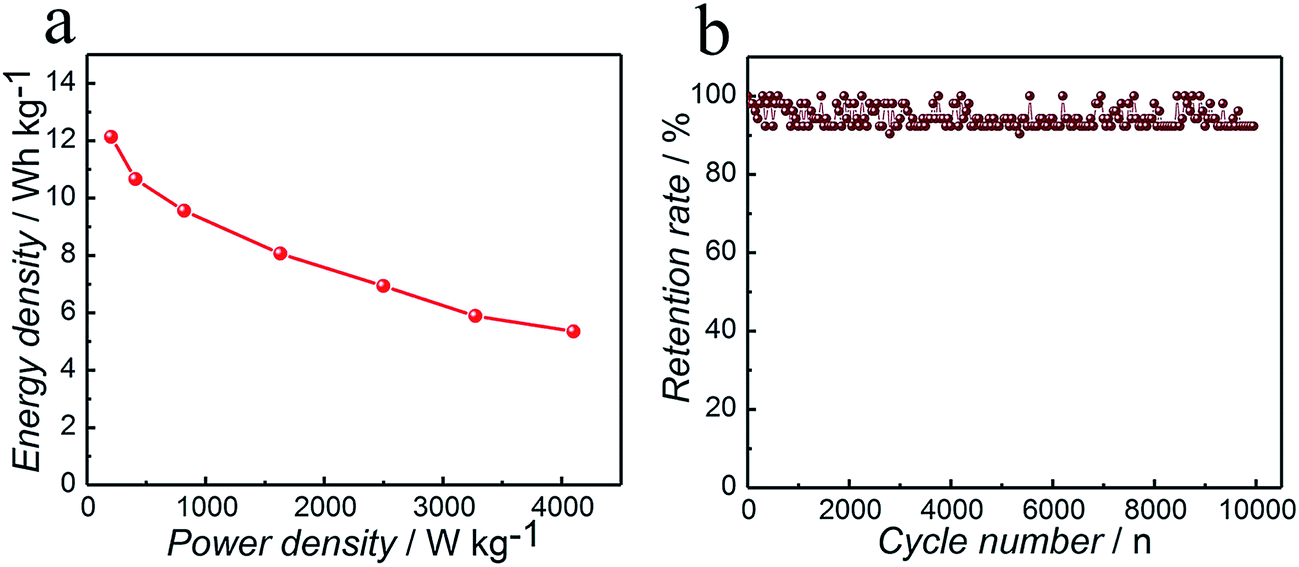 | ||
| Fig. 6 Co3N//AC asymmetric supercapacitor with a voltage of 1.63 V. (a) Ragone plot related to energy and power densities of the asymmetric device; (b) cycling performance of the device with a voltage of 1.63 V at a current density of 1 A g−1. | ||
The long-term cycling performance of the ASC is a critical parameter for supercapacitor practical applications. In order to assess the cycling stability of the asymmetric supercapacitor, the Co3N//AC asymmetric supercapacitor was operated between 0 and 1.63 V at a current density of 1 A g−1, as shown in Fig. 6b. From the cycle number vs. capacitance retention curve, the specific capacitance was retained at 92.3% after 10000 cycles. The gradual fading of capacitance is due to the poor interfacial contact between the active materials and the current collecting substrate. In addition, the active materials might peel off the substrate over cycling, which would induce a decrease of the capacitance further.
The excellent pseudocapacitive behavior of Co3N//AC can be attributed to the following: (i) the structure of as-prepared Co3N with a large amount of open space accelerated the diffusion of electrolyte ions, leading to a reduced internal resistance. (ii) The as-prepared Co3N compound demonstrates high electrical conductivity along with electrochemical activity favoring the electrochemical stability and a fast redox reaction.
Comparison of the electrochemical properties of Co3N in three-electrode electrochemical measurements vs. some previous reports of transition metal compounds is given in Table 1.
| Sample | Gravimetric capacitance or areal specific capacitance based on the electrode geometric area | Rate capability | Ref. |
|---|---|---|---|
| ZnMn2O4 | 60 F g−1 at 0.5 A g−1 | — | 51 |
| Co3O4-TH | 67.9 F g−1 at 0.2 A g−1 | 73.5% from 0.2 to 1 A g−1 | 52 |
| ZnWO4 | 23.72 F g−1 at 0.25 A g−1 | — | 53 |
| NiO | 29 F g−1 at 0.5 A g−1 | — | 54 |
| CoFe2O4 | 164 F g−1 at 0.2 A g−1 | 19.5% from 0.2 to 0.4 A g−1 | 55 |
| CoS | 41.1 F g−1 at 1.5 A g−1 | 60.8% from 1.5 to 9 A g−1 | 56 |
| GaN | 24 F g−1 at 0.5 A g−1 | — | 57 |
| Mo2N/rGo | 142 mF cm−2 at 1 mA cm−2 | 72.4% from 1 mA cm−2 to 150 mA cm−2 | 58 |
| CrN | 12.8 mF cm−2 at 1 mA cm−2 | 61% from 0.5 mA cm−2 to 10 mA cm−2 | 59 |
| Nb4N5 | 225.8 mF cm−2 at 0.5 mA cm−2 | 60.8% from 0.5 mA cm−2 to 10 mA cm−2 | 60 |
| Nb4N5 NBAs | 30.2 mF cm−2 at 1 mA cm−2 | — | 61 |
| VN/CNT | 178 mF cm−2 at 1.1 mA cm−2 | — | 62 |
| Co3N | 112.3 F g−1 at 0.5 A g−1 (corresponding to 449.2 mF cm−2 at 2 mA cm−2) | 72.4% from 0.5 to 5 A g−1 (corresponding to 72.4% from 2 mA cm−2 to 20 mA cm−2) | This work |
4. Conclusions
This work evaluates the potential of cobalt nitride, a rather unexplored simple binary nitride material, for energy conversion applications. Studying cobalt nitride in the context of energy storage applications provides a conceptual advance in the field of nitrides used in supercapacitors, a field that has so far been limited to group IIIB, IVB and VB nitrides. Briefly, we have obtained the Co3N powder, by a simple chemical precipitation method followed by annealing under ammonia gas. Moreover, this study constitutes the first demonstration of employing Co3N as an electrode material for supercapacitors. Benefiting from better conductivity that favours fast charge transport, this Co3N electrode exhibits excellent capacitive performances, including a high capacitance, a great rate capability and good stability during prolonged cycle measurements. When constructing an asymmetric supercapacitor with activated carbon, excellent performance was also achieved. Consequently, this work creates more opportunities for the utilization of metal nitrides as electrode materials. However, further insight is required to better understand the energy storage mechanism of this newly emerged electrode material used in supercapacitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51762031) and the Foundation for Innovation Groups of Basic Research in Gansu Province (No. 1606RJIA322).References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287 CAS.
- S. Gao, Y. Chen, H. Fan, X. Wei, C. Hu, H. Luo and L. Qu, J. Mater. Chem. A, 2014, 2, 3317 CAS.
- M. He, K. Fic, E. Fr
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) ckowiak, P. Novák and E. J. Berg, Energy Environ. Sci., 2016, 9, 623–633 Search PubMed.
ckowiak, P. Novák and E. J. Berg, Energy Environ. Sci., 2016, 9, 623–633 Search PubMed. - H. Hu, B. Y. Guan and X. W. Lou, Chem, 2016, 1, 102–113 CAS.
- J. Huang, J. Wang, C. Wang, H. Zhang, C. Lu and J. Wang, Chem. Mater., 2015, 27, 2107–2113 CrossRef CAS.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41–53 CAS.
- T. Cottineau, M. Toupin, T. Delahaye, T. Brousse and D. Bélanger, Appl. Phys. A: Mater. Sci. Process., 2005, 82, 599–606 CrossRef.
- T. Nathan, A. Aziz, A. F. Noor and S. R. S. Prabaharan, J. Solid State Electrochem., 2007, 12, 1003–1009 CrossRef.
- M. Huang, F. Li, F. Dong, Y. X. Zhang and L. L. Zhang, J. Mater. Chem. A, 2015, 3, 21380–21423 CAS.
- M. Liu, X. Wang, D. Zhu, L. Li, H. Duan, Z. Xu, Z. Wang and L. Gan, Chem. Eng. J., 2017, 308, 240–247 CrossRef CAS.
- F. Shi, L. Li, X.-l. Wang, C.-d. Gu and J.-p. Tu, RSC Adv., 2014, 4, 41910–41921 RSC.
- H. Yang, J. Xie and C. M. Li, RSC Adv., 2014, 4, 48666–48670 RSC.
- Z. Zhang, Z. Huang, L. Ren, Y. Shen, X. Qi and J. Zhong, Electrochim. Acta, 2014, 149, 316–323 CrossRef CAS.
- X. Li, Q. Li, Y. Wu, M. Rui and H. Zeng, ACS Appl. Mater. Interfaces, 2015, 7, 19316–19323 CAS.
- D. Yang, J. Power Sources, 2011, 196, 8843–8849 CrossRef CAS.
- L. L. Zhang, R. Zhou and X. S. Zhao, J. Mater. Chem., 2010, 20, 5983 RSC.
- C. Lei, F. Han, D. Li, W. C. Li, Q. Sun, X. Q. Zhang and A. H. Lu, Nanoscale, 2013, 5, 1168–1175 RSC.
- C. Yao, B. Wei, H. Li, G. Wang, Q. Han, H. Ma and Q. Gong, J. Mater. Chem. A, 2017, 5, 56–61 CAS.
- X. Xu, L. Pei, Y. Yang, J. Shen and M. Ye, J. Alloys Compd., 2016, 654, 23–31 CrossRef CAS.
- S. Dong, X. Chen, X. Zhang and G. Cui, Coord. Chem. Rev., 2013, 257, 1946–1956 CrossRef CAS.
- G. R. Li, J. Song, G. L. Pan and X. P. Gao, Energy Environ. Sci., 2011, 4, 1680 CAS.
- H. Cui, G. Zhu, X. Liu, F. Liu, Y. Xie, C. Yang, T. Lin, H. Gu and F. Huang, Adv. Sci., 2015, 2, 1500126 CrossRef PubMed.
- W.-B. Zhang, X.-J. Ma, A. Loh, X. Li, F. C. Walsh and L.-B. Kong, ACS Energy Lett., 2017, 2, 336–341 CrossRef CAS.
- A. Djire, J. Y. Ishimwe, S. Choi and L. T. Thompson, Electrochem. Commun., 2017, 77, 19–23 CrossRef CAS.
- W.-B. Zhang, X.-J. Ma, L.-B. Kong, Y.-C. Luo and L. Kang, J. Electrochem. Soc., 2016, 163, A2830–A2834 CrossRef CAS.
- W.-B. Zhang, X.-J. Ma, L.-B. Kong, M.-C. Liu, Y.-C. Luo and L. Kang, J. Electrochem. Soc., 2016, 163, A1300–A1305 CrossRef CAS.
- D. Choi, G. E. Blomgren and P. N. Kumta, Adv. Mater., 2006, 18, 1178–1182 CrossRef CAS.
- X. Lu, M. Yu, T. Zhai, G. Wang, S. Xie, T. Liu, C. Liang, Y. Tong and Y. Li, Nano Lett., 2013, 13, 2628–2633 CrossRef CAS PubMed.
- R. Juza and W. Sachsze, Z. Anorg. Chem., 1945, 253, 95–108 CrossRef CAS.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186–19192 CrossRef CAS PubMed.
- P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 14710–14714 CrossRef CAS PubMed.
- Y. Yu, W. Gao, Z. Shen, Q. Zheng, H. Wu, X. Wang, W. Song and K. Ding, J. Mater. Chem. A, 2015, 3, 16633–16641 CAS.
- M.-S. Balogun, Y. Zeng, W. Qiu, Y. Luo, A. Onasanya, T. K. Olaniyi and Y. Tong, J. Mater. Chem. A, 2016, 4, 9844–9849 CAS.
- A. Śliwak, A. Moyseowicz and G. Gryglewicz, J. Mater. Chem. A, 2017, 5, 5680–5684 Search PubMed.
- K. Oda, T. Yoshio and K. Oda, J. Mater. Sci., 1987, 22, 2729–2733 CrossRef CAS.
- K. K. Shih and J. Karasinski, J. Appl. Phys., 1993, 73, 8377–8380 CrossRef CAS.
- H. Asahara, T. Migita, T. Tanaka and K. Kawabata, Vacuum, 2001, 62, 293–296 CrossRef CAS.
- S. Liu, K. San Hui, K. N. Hui, J. M. Yun and K. H. Kim, J. Mater. Chem. A, 2016, 4, 8061–8071 CAS.
- Z. Xiao, L. Fan, B. Xu, S. Zhang, W. Kang, Z. Kang, H. Lin, X. Liu, S. Zhang and D. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 41827–41836 CAS.
- K. Kamiya, T. Yoko and M. Bessho, J. Mater. Sci., 1987, 22, 937–941 CrossRef CAS.
- W.-B. Zhang, X.-J. Ma, L.-B. Kong, M.-C. Liu, Y.-C. Luo and L. Kang, J. Electrochem. Soc., 2016, 163, A2441–A2446 CrossRef CAS.
- K. Bindu, K. Sridharan, K. M. Ajith, H. N. Lim and H. S. Nagaraja, Electrochim. Acta, 2016, 217, 139–149 CrossRef CAS.
- I. Y. Y. Bu and R. Huang, Mater. Sci. Semicond. Process., 2015, 31, 131–138 CrossRef CAS.
- K. P. Annamalai, L. Liu and Y. Tao, J. Mater. Chem. A, 2017, 5, 9991–9997 CAS.
- Y. Wang, B. Chen, Z. Chang, X. Wang, F. Wang, L. Zhang, Y. Zhu, L. Fu and Y. Wu, J. Mater. Chem. A, 2017, 5, 8981–8988 CAS.
- S. K. Kaverlavani, S. E. Moosavifard and A. Bakouei, J. Mater. Chem. A, 2017, 5, 14301–14309 CAS.
- A. Sahoo and Y. Sharma, Mater. Chem. Phys., 2015, 149–150, 721–727 CrossRef CAS.
- J. Jiang, F. Wei, G. Yu and Y. Sui, J. Nanomater., 2015, 16, 80 Search PubMed.
- R. D. Kumar, Y. Andou and S. Karuppuchamy, J. Phys. Chem. Solids, 2016, 92, 94–99 CrossRef CAS.
- Y.-g. Wang and Y.-y. Xia, Electrochim. Acta, 2006, 51, 3223–3227 CrossRef CAS.
- D. H. Deng, H. Pang, J. M. Du, J. W. Deng, S. J. Li, J. Chen and J. S. Zhang, Cryst. Res. Technol., 2012, 47, 1032–1038 CrossRef CAS.
- Y. Anil Kumar, S. Srinivasa Rao, D. Punnoose, C. Venkata Tulasivarma, C. V. V. M. Gopi, K. Prabakar and H.-J. Kim, R. Soc. Open Sci., 2017, 4, 170427 CrossRef PubMed.
- S. Wang, L. Zhang, C. Sun, Y. Shao, Y. Wu, J. Lv and X. Hao, Adv. Mater., 2016, 28, 3768–3776 CrossRef CAS PubMed.
- G. Ma, Z. Wang, B. Gao, T. Ding, Q. Zhong, X. Peng, J. Su, B. Hu, L. Yuan, P. K. Chu, J. Zhou and K. Huo, J. Mater. Chem. A, 2015, 3, 14617–14624 CAS.
- B. Wei, H. Liang, D. Zhang, Z. Wu, Z. Qi and Z. Wang, J. Mater. Chem. A, 2017, 5, 2844–2851 CAS.
- H. Cui, G. Zhu, X. Liu, F. Liu, Y. Xie, C. Yang, T. Lin, H. Gu and F. Huang, Adv. Sci., 2015, 2, 1500126 CrossRef PubMed.
- B. Gao, X. Xiao, J. Su, X. Zhang, X. Peng, J. Fu and P. K. Chu, Appl. Surf. Sci., 2016, 383, 57–63 CrossRef CAS.
- X. Xiao, X. Peng, H. Jin, T. Li, C. Zhang, B. Gao, B. Hu, K. Huo and J. Zhou, Adv. Mater., 2013, 25, 5091–5097 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |