
Understanding homogeneous hydrogen evolution reactivity and deactivation pathways of molecular molybdenum sulfide catalysts†
M.
Dave‡
a,
A.
Rajagopal‡
a,
M.
Damm-Ruttensperger
a,
B.
Schwarz
a,
F.
Nägele
b,
L.
Daccache
b,
D.
Fantauzzi
*cd,
T.
Jacob
*bcd and
C.
Streb
 *acd
*acd
aInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: carsten.streb@uni-ulm.de
bInstitute of Electrochemistry, Ulm University, Albert-Einstein-Allee 47, 89081 Ulm, Germany. E-mail: timo.jacob@uni-ulm.de
cHelmholtz Institute Ulm for Electrochemical Energy Storage, 89081 Ulm, Germany
dKarlsruhe Institute of Technology, P.O. Box 3640, 76021 Karlsruhe, Germany. E-mail: donato.fantauzzi@kit.edu
First published on 1st March 2018
Abstract
Molybdenum sulfides are highly active hydrogen evolution reaction (HER) catalysts based on earth abundant elements. Here, the molybdenum sulfide anion [Mo3S13]2− is used as a molecular model to rationalize HER reactivity of Mo–S-catalysts. For the first time, homogeneous, visible light-driven HER activity of [Mo3S13]2− is reported and high reactivity is observed (turnover number TON ∼23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, maximum turnover frequency TOFmax ∼156 min−1). Experimental and theoretical studies shed light on the catalytic role of terminal disulfide ligands (S22−) and show that these ligands modulate catalyst redox-activity and electron transfer in solution. Partial substitution of the terminal disulfides with water ligands leads to the most active catalytic species, e.g. [Mo3S11(H2O)2]. In contrast, complete substitution of the terminal disulfides results in a significant loss of reactivity. These results could lay the foundations for the knowledge-based development of homogeneous and heterogeneous molybdenum sulfide catalysts.
000, maximum turnover frequency TOFmax ∼156 min−1). Experimental and theoretical studies shed light on the catalytic role of terminal disulfide ligands (S22−) and show that these ligands modulate catalyst redox-activity and electron transfer in solution. Partial substitution of the terminal disulfides with water ligands leads to the most active catalytic species, e.g. [Mo3S11(H2O)2]. In contrast, complete substitution of the terminal disulfides results in a significant loss of reactivity. These results could lay the foundations for the knowledge-based development of homogeneous and heterogeneous molybdenum sulfide catalysts.
Introduction
Over the last decade, amorphous solid-state molybdenum sulfides (MoS2+x) have emerged as technologically important, noble-metal-free catalysts, which enable the (photo-) electrochemical hydrogen evolution reaction (HER).1–4 However, due to their amorphous structure, molecular-level mechanistic insights are difficult to obtain and the hydrogen evolution mechanism might depend on catalyst preparation and the exact catalytic conditions.5 As a consequence, several HER catalytic pathways for amorphous Mo–S HER catalysts are discussed, involving protonated disulfide ligands,6–9 Mo-oxo10–12 or MoV-hydride species.12,13 An unique option for gaining mechanistic understanding of amorphous Mo–S HER catalysts was recently reported by Artero, Tran et al.: the group proposed that amorphous molybdenum sulfides are based on polymeric chains of molecular molybdenum sulfides (thiomolybdates),14 [Mo3S13]2−, as illustrated in Fig. 1.12 Thus, studies on the catalytic HER activity of molecular molybdenum sulfide anions could have direct relevance for the most active solid-state molybdenum sulfides.6,9,10,15 Pioneering heterogeneous HER studies on molecular molybdenum sulfides such as [Mo3S13]2− and related species have been focused on catalyst deposition on (photo-)electrodes, leading to efficient solid-state catalysts for electrocatalytic6,9,16,17 or photoelectrocatalytic18–20 HER. To the best of our knowledge, only one study of homogeneous thiomolybdate HER activity has been reported so far.17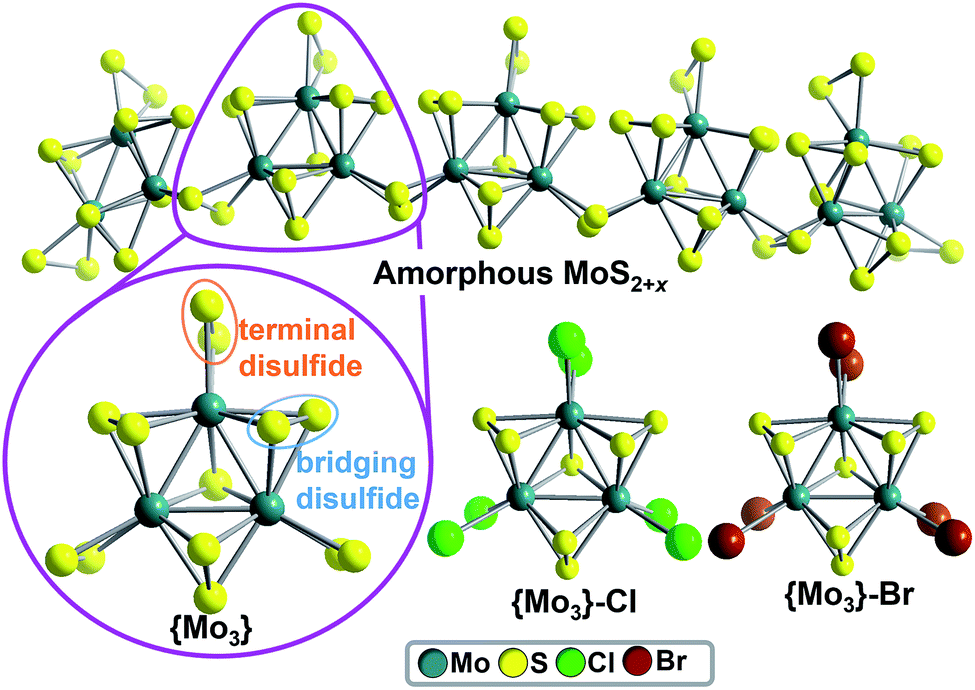 | ||
| Fig. 1 Molecular molybdenum sulfides as models for amorphous MoS2+x catalysts. Top: simplified structure of amorphous MoS2+x based on disulfide-bridged [Mo3S13]2− units, as proposed by Artero, Tran et al.12 Bottom: Ball-and-stick representation of the molecular molybdenum sulfide prototypes [Mo3S13]2− (={Mo3}), [Mo3S7Cl6]2− (={Mo3}–Cl) and [Mo3S7Br6]2− (={Mo3}–Br). Color scheme: MoIV: teal; S: yellow, Cl: green, Br: brown. | ||
In this study, we build on these ground-breaking results and report the high visible light-driven HER activity of a molecular molybdenum sulfide together with experimental and theoretical studies to rationalize the high reactivity observed. Terminal disulfide ligands are identified as key species, which tune energetics of the hydrogen formation, control the redox-properties of the catalyst and modulate catalyst–photosensitizer interactions in solution. Exchange of the terminal disulfides with water or halide ligands results in significant changes in HER reactivity. These changes are rationalized using experimental and theoretical methods, providing unprecedented molecular-level insights into catalyst reactivity and deactivation.
Results and discussion
Here, we introduce the Müller-type molecular molybdenum sulfide (NH4)2[Mo3S13] (=(NH4)2{Mo3})14,21,22 as a prototype homogeneous light-driven HER catalyst. Our approach builds on the established heterogeneous (photo-)electrochemical HER activity of {Mo3} and explores the relevance of {Mo3} as a molecular model for homogeneous and heterogeneous Mo–S catalysts. To-date, virtually no mechanistic insights into {Mo3} stability and reactivity under homogeneous, light-driven HER conditions is available and as a result, knowledge-driven development of this catalyst class is challenging.Structural features of {Mo3}
{Mo3} is based on three MoIV centers arranged in a trigonal fashion and linked by one central μ3-S2− ligand. Neighboring Mo ions are connected by bridging disulfide (μ,η2-S22−) ligands. Each Mo center features one terminal disulfide (η2-S22−), giving the formula [Mo3S13]2− =[Mo3(S2,bridging)3(S2,terminal)3S]2− = {Mo3}, see Fig. 1. In the current literature on Mo–S catalysts, bridging6,7,9 as well as terminal8,9,23 disulfide ligands are discussed as reactive sites in HER catalysis. To investigate the roles of bridging and terminal disulfide ligands in {Mo3}, we independently synthesized the literature-known {Mo3} analogues [Mo3(S2,briding)3SX6]2− (X = Cl− (={Mo3}–Cl) and Br− (={Mo3}–Br)),24 where all terminal disulfides are replaced by chloride or bromide ligands, respectively (Fig. 1). In all other respects, {Mo3}–Cl and {Mo3}–Br are isostructural to {Mo3}. For all compounds, purity was confirmed using spectroscopic, elemental-analytic, mass-spectrometric and crystallographic methods, see ESI.†HER-catalytic reactivity of {Mo3} in water
Initial studies explored the homogeneous, visible light-driven HER activity of {Mo3} by combining the catalyst (0.3 μM) with the molecular photosensitizer [Ru(bpy)3]2+ (20 μM, bpy = 2,2′-bipyridine) and the sacrificial electron donor ascorbic acid (0.1 M) in de-gassed water (pH 6.0). An established colloid detection protocol combining dynamic light scattering (DLS), UV-vis-spectroscopy and microfiltration was used for all catalytic studies and no colloid formation was detected for the catalytic processes reported. Note that at higher catalyst concentrations (>10−6 M), colloid formation by electrostatic aggregation of the catalyst anions with the photosensitizer cations is observed. The colloids are HER-catalytically inactive, see ESI† for details.25 Hydrogen evolution was initiated by irradiating the reaction solution at room temperature with monochromatic visible light (LED light source, λmax = 470 nm, P ∼ 40 mW cm−2). H2 formation was quantitatively monitored over a period of 6 h using head-space gas chromatography, all measurements were carried out in triplicate. Control experiments showed no hydrogen evolution when the samples were not irradiated, or when the catalyst, [Ru(bpy)3]2+ or ascorbic acid were absent. Under aqueous homogeneous conditions and irradiation for 6 h, {Mo3} gives turnover numbers (TONs) of ∼2850 (based on {Mo3}).Notably, the initial turnover frequency (TOF) in water (∼37 min−1, determined after tirrad = 5 min) decreases sharply to ∼12 min−1 (tirrad = 30 min), after which TOF values remain constant (Fig. 2b). This behaviour was interpreted as a structural change of the catalyst, leading to the formation of a less HER-active species. Structural considerations led us to suggest that the most likely deactivation mode is an exchange of the terminal disulfide ligands with solvent (i.e. water) ligands. This mode of ligand exchange is documented in the literature22,26,27 and leads to species such as [Mo3(S2,bridging)3S(H2Oterminal)6]4+, see Fig. 2d.26 In contrast, exchange of the bridging disulfide ligands on {Mo3} (or related thiomolybdates) is not known and would most likely lead to cluster decomposition and formation of colloidal molybdenum sulfide particles.14 This was not observed.
 | ||
| Fig. 2 Mechanistic studies on the light-driven HER reactivity of {Mo3}. Top left (a): increased HER activity by {Mo3} when going from water to MeOH/H2O mixed solvents. Top centre (b): change of turnover frequencies (TOFs) during HER catalysis, indicating faster catalyst deactivation under aqueous conditions. Note that an initial increase of TOFs is observed in MeOH/H2O mixtures (tirrad = 0–20 min), suggesting the in situ formation of a catalytically more active species (see below for details). Conditions: [{Mo3}] = 0.3 μM, [Ru(bpy)3]2+] = 20 μM, [ascorbic acid] = 0.1 M. Top right (c): in situ Raman spectroscopy showing the exchange of terminal disulfides under catalytic conditions. Conditions: solvent: MeOH:H2O (1:1, v/v), [{Mo3}] = 0.3 mM. Bottom (d): proposed ligand exchange, leading to the introduction of terminal aquo ligands on the molybdenum sulfide [Mo3S7]4+ core. | ||
To support that exchange of the terminal disulfides occurs under reaction conditions, we performed in situ Raman spectroscopy on {Mo3} in the water-containing reaction solution. Raman spectroscopy is an ideal tool to examine structural changes in {Mo3} as the bridging and terminal disulfide ligands give two well-separated signals at 552 cm−1 (bridging) and 521 cm−1 (terminal), respectively (Fig. 2c).7 A mixture of MeOH and H2O (1:1, v/v) was used as solvent to reduce the water content of the solution and slow down ligand exchange so that spectroscopic measurements became possible. Further, higher catalyst concentrations (0.3 mM) were used to obtain detectable Raman signals. HER catalytic experiments verified that the system is still active under these conditions and indeed shows increased activity (Table 1, entry 2) compared with the purely aqueous system. As shown in Fig. 2c, with increasing reaction time, a decrease of the signal intensity for terminal disulfides is noted, indicating exchange of these ligands. In contrast, the signal associated with the bridging disulfides remains constant. The ligand exchange processes occur over a period of ∼1 h, which qualitatively corresponds to the period of time during which TOF decrease is observed (Fig. 2b). Note that quantitative correlation between both measurements is not directly possible due to different experimental conditions required for catalysis and Raman spectroscopic analyses. Further evidence of the proposed ligand exchange was provided by high resolution ESI mass spectrometry on the {Mo3} reaction solution, where deprotonated aquo species such as [Mo3S7O3]2− (exp: 280.76 m/z; calcd: 280.76 m/z) were detected, see ESI.† In sum, these experimental findings suggest that exchange of the terminal disulfide ligands by aquo ligands occurs under the given homogeneous HER conditions in the presence of water, leading to significant reactivity changes (see Fig. 2d for proposed exchange mechanism). Note that we also examined whether exchange of the terminal disulfides with MeOH ligands could occur, however, UV-vis spectroscopy showed no ligand exchange when {Mo3} is dissolved in pure MeOH (see ESI†).
| No | Catalyst | Solvent | TONb/— | TOFmax/min−1 |
|---|---|---|---|---|
| a catalytic conditions: [catalyst] = 0.3 μM, [Ru(bpy)3]2+ = 20 μM, [ascorbic acid] = 0.1 M, light source, LED, λmax = 470 nm, P ∼ 40 mW cm−2. b Determined after tirrad = 6 h, standard deviation ± 3–4%. | ||||
| 1 | {Mo3} | H2O | 2850 | 37 |
| 2 | {Mo3} | MeOH:H2O (1:1, v/v) |
3950 | 54 |
| 3 | {Mo3} | MeOH:H2O (10:1, v/v) |
23000 |
156 |
| 4 | {Mo3}, tirrad = 24 h | MeOH:H2O (10:1, v/v) |
41000 |
156 |
| 5 | {Mo3} | MeOH | 8850 | 47 |
| 6 | {Mo3}–Cl | MeOH:H2O (10:1, v/v) |
500 | 1.0 |
| 7 | {Mo3}–Br | MeOH:H2O (10:1, v/v) |
60 | 0.7 |
Optimized catalytic HER reactivity of {Mo3}
The results described above suggest that the rate of ligand exchange is correlated with the concentration of water. Thus, the next set of HER experiments was performed under standard catalytic conditions using a mixture of MeOH and H2O (10:1, v/v) as solvent instead of water to slow down ligand exchange by reducing the amount of water in solution. Under these conditions, significantly higher TONs (∼23000) were observed after tirrad = 6 h. In addition, longer irradiation up to 24 h lead to TONs of ∼41000, making this one of the most active visible light-driven homogeneous HER catalysts to-date (see Table 2 for comparison). Notably, the TOF changes in the MeOH:H2O mixture show striking differences compared with the purely aqueous system (Fig. 2b): for the MeOH:H2O system, a sharp initial TOF increase from ∼91 min−1 (tirrad = 5 min), to ∼156 min−1 (tirrad = 20 min) was noted. This was followed by a continuous drop of TOFs to ∼61 min−1 at tirrad = 6 h, see Fig. 2b. These changes are in line with the in situ-formation of aquo-substituted catalyst species [Mo3S13−x(H2O)x](2−x)−, where HER activity depends on the number of aquo ligands present (x = 0, 2, 4, 6, see Fig. 2d). It can therefore be suggested, that partial exchange of the disulfide ligands with aquo ligands leads to more active species, while complete exchange of the terminal disulfides results in decreased catalytic activity. Theoretical calculations support this proposed mechanism and indicate that hydrogen evolution is indeed energetically more favoured for species featuring mixed disulfide and aquo ligands, see below.
| No | Catalysta | Photosensitizer | Electron donor | TON/—([catalyst]) | TOFmax/min−1 | Reference |
|---|---|---|---|---|---|---|
| a Ligand abbreviations: bdt = 1,2-benzene dithiolate; PentPy = mono-hydroxylated pentapyridyl ligand; DPA-py = N,N-bis(2-pyridinylmethyl)-2,2′-bipyridine-6-methanamine; pyS = pyridine-2-thiolate, tacn = 1-[(4-CO2Et-3,5-H-2-pyridyl)methyl]-4,7-dimethyl-1,4,7-triazacyclononane. | ||||||
| 1 | [Co(bdt)2]− | [Ru(bpy)3]2+ | Ascorbic acid | 2700 (5 μM) | ∼15 | 28 |
| 2 | [CoBr(PentPy)]Br | [Ru(bpy)3]2+ | Ascorbic acid/P(C2H4COOH)3 × HCl | 33300 (1 μM) |
∼100 | 29 |
| 3 | [Fe2S2]-dendrimer | [Ir(ppy)2(bpy)]+ | Triethyl amine | 22200 (1 μM) |
∼120 | 30 |
| 4 | [Co(DPA-py) (OH2)]3+ | [Ru(bpy)3]2+ | Ascorbic acid | 4400 (0.5 μM) | ∼25 | 31 |
| 5 | [Ni(pyS)3]− | Fluorescein | Triethyl amine | 5500 (4 μM) | ∼5 | 32 |
| 6 | [CoII(OTf)2(tacn)] | [Ir(ppy)2(bpy)]+ | Triethyl amine | 9000 (0.25 μM) | ∼870 | 33 |
| 7 | [Mo3S13]2− | [Ru(bpy)3]2+ | Ascorbic acid | 41000 (0.3 μM) |
156 | This work |
Based on these insights, we hypothesized that lower HER reactivity can be expected for {Mo3} in the absence of aquo ligands. Thus, we performed HER catalysis under our standard conditions using pure MeOH as a solvent (Table 1, entry 5). This led to a significantly lower performance (TON ∼ 8,850, TOFmax ∼ 47 min−1) compared with the MeOH:H2O mixtures described above, thereby substantiating our hypothesis that exchange of terminal disulfides with aquo ligands can be used to optimize HER activity of {Mo3}.
To investigate the reactivity of the [Mo3(S2,bridging)3S]4+-core in the absence of any terminal disulfide ligands, we assessed the catalytic performance of the halide-terminated species {Mo3}–Cl and {Mo3}–Br (see Fig. 1) under our standard HER-catalytic conditions in MeOH:H2O (10:1, v/v). Both halide-substituted species show significantly lower reactivity based on TON and TOF values compared with {Mo3}, see Fig. 3a and Table 1, entries 6/7. This demonstrates that simple replacement of the terminal disulfides with other common anionic ligands does not lead to a stabilization of the HER-catalytic activity and emphasizes that the terminal disulfide ligands introduce specific structural and electronic effects, which contribute to the increased HER activity of {Mo3}, see below.
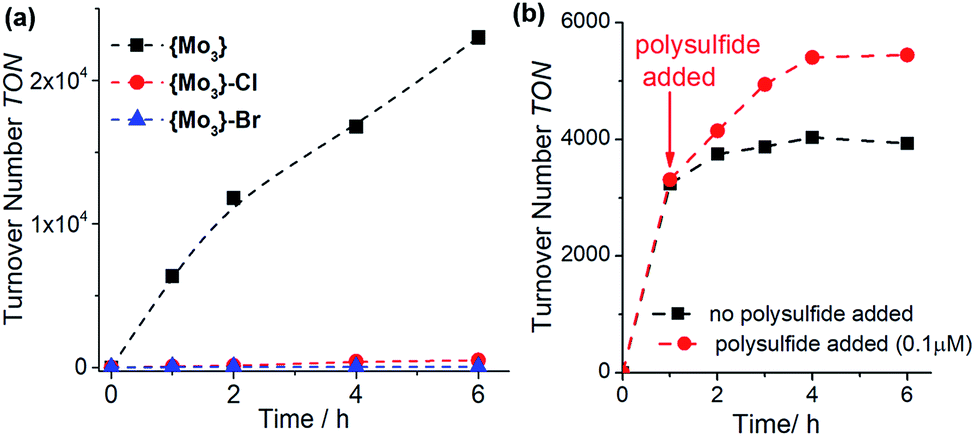 | ||
| Fig. 3 Influence of terminal ligands on the visible-light driven HER activity of {Mo3}. (a): replacement of the terminal disulfides with halide ligands ({Mo3}–Cl = [Mo3S7Cl6]2−, {Mo3}–Br = [Mo3S7Br6]2−) leads to a significant decrease of catalytic activity. Conditions: solvent: MeOH:H2O (10:1, v/v), [catalyst] = 0.3 μM, [[Ru(bpy)3]2+] = 20 μM, [ascorbic acid] = 0.1 M. (b): increased catalytic activity of {Mo3} under standard HER conditions in the presence of the disulfide source ammonium polysulfide ((NH4)2Sx). Solvent: MeOH:H2O (1:1, v/v), [{Mo3}] = 0.3 μM, [[Ru(bpy)3]2+] = 20 μM, [ascorbic acid] = 0.1 M, [(NH4)2Sx] = 0.1 μM. | ||
Since the disulfide-aquo ligand exchange is dependent on the underlying chemical equilibria shown in Fig. 2d, we suggested that using Le Chatelier's principle, it should be possible to slow down the full exchange of terminal disulfides. As an initial proof of principle, we performed the standard {Mo3} HER catalysis in MeOH:H2O (1:1, v/v) in the presence of ammonium polysulfide (NH4)2Sx, which is used as disulfide source in the synthesis of {Mo3}.14,21,22 Under these conditions, we note that catalyst deactivation proceeds at a slower rate compared to the polysulfide-free reference reaction (Fig. 3b, TON increase ∼30%), suggesting that polysulfides could improve the HER activity of {Mo3}.
Theoretical analysis of the {Mo3} catalytic reactivity
Density functional theory (DFT) calculations were performed to assess the hydrogen evolution energetics of {Mo3}-based catalysts featuring terminal disulfide, chloride or aquo ligands.To this end, free energies (ΔGs) of formation of intermediate states involved in the catalytic process were calculated, which provide information on the thermodynamic contributions to activation energies.34 Of course, a more rigorous analysis of the overall reaction kinetics would additionally require detailed investigations on the energetics of transition states.
In our studies special care was taken to define the reference energies of the solvated proton and electron to be in agreement with the experimental conditions.15,35,36 Careful referencing is important, as even small changes of the reference energies might perturb the free energy values. Details of the theoretical approach used and a full list of the pathways studied are given in the ESI.†
Our calculated reaction path analysis is in line with previous experimental reports on heterogenized {Mo3} electrocatalysts15 and suggests a Volmer–Heyrovsky-type mechanism:6,15 in step one, a proton-coupled electron transfer (Volmer step, Fig. 4a) is energetically favored and leads to a hydrogen atom bound at a bridging disulfide. Note that this step leads to a cleavage of the disulfide bond, resulting in the formation of a hydrogen sulfide (HS−) ligand, see Fig. 4e.37 In step 2 (Heyrovsky step, Fig. 4a), a second proton-coupled electron transfer to the catalyst is energetically favored, resulting in the formation and release of a H2 molecule and re-generation of the native catalyst. In addition to proton-coupled electron transfers, we also studied individual protonation or electron-transfer steps (Fig. 4b). In comparison to the proton-coupled electron transfers, these individual steps were always energetically non-favoured, see details in the ESI.†15,34
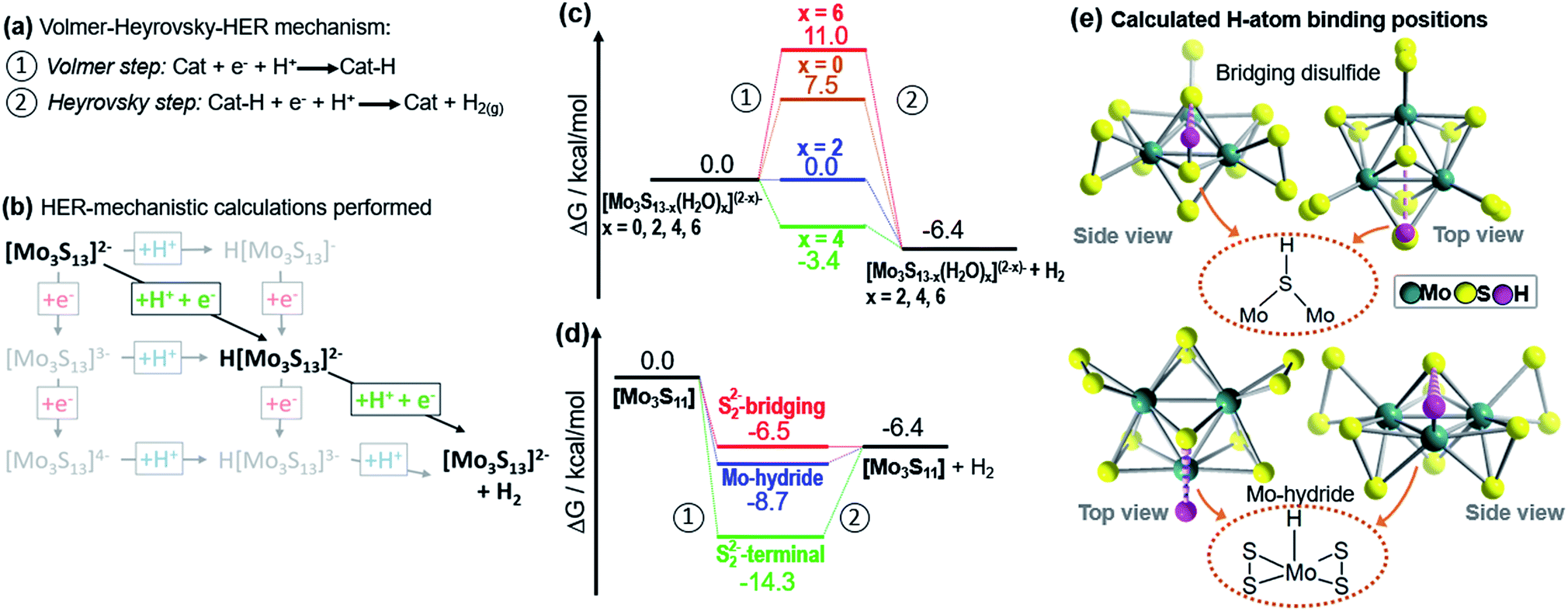 | ||
| Fig. 4 Theoretical investigation of the HER catalytic reactivity of proposed catalytic intermediates. For each species, free energy changes during a Volmer–Heyrovsky-type H2 evolution was calculated. (a) Illustration of the Volmer–Heyrovsky mechanism: step 1 corresponds to the Volmer step, i.e. a proton-coupled electron transfer to the catalyst. Step 2 corresponds to the Heyrovsky step, i.e. a proton-coupled reductive hydrogen evolution. (b) Illustration of the H2 formation processes calculated, exemplified for [Mo3S13]2−. Note that proton-coupled electron transfers (diagonal lines) were the energetically favoured processes. Other pathways such as stepwise electron-proton transfer were energetically non-favoured and therefore unlikely (see ESI†). (c) Proton reduction free-energies for {Mo3} and {Mo3}-derivatives where one, two or three terminal disulfides have been replaced by two, four or six water ligands. H atom binding occurs at the bridging disulfide ligands. (d) Free energies for hydrogen binding by [Mo3S11], where a vacant coordination site is generated by loss of one terminal disulfide ligand. Here, the energies for H atom binding on a bridging disulfide, on the Mo (i.e. formation of a MoV hydride, MoV–H) and on the terminal disulfide were calculated. (e) Calculated binding modes for H-atom binding at bridging disulfide ligands in [Mo3S13]2− and at a Mo center (forming a MoV-hydride) in [Mo3S11]. Note that weakly stabilizing S⋯H hydrogen bonds37 (d(S⋯H) ∼2.6–2.8 Å) to the apical sulfide ligand are observed in both binding modes (dashed magenta lines). | ||
We then examined, how the exchange of terminal disulfides by aquo ligands on {Mo3} affects the hydrogen evolution energetics. To this end, we studied the species [Mo3S13−x(H2O)x](2−x)− with x = 0, 2, 4, and 6. As shown in Fig. 4c, the free energy of the Volmer step for native {Mo3} (7.5 kcal mol−1) is relatively high. Significantly lower free energies are observed when one or two disulfides are exchanged with water ligands, i.e. x = 2 (0.0 kcal mol−1) and x = 4 (−3.4 kcal mol−1). Exchange of all three terminal disulfides (x = 6) leads to the highest free energies of the Volmer step (11 kcal mol−1).
These findings are in line with the experimental results discussed above: in MeOH:H2O mixtures (Fig. 2b), we note a reproducible initial TOF increase (tirrad = 0–20 min), which indicates the conversion of {Mo3} into a catalytically more active species. This would be in line with the substitution of aquo ligands on {Mo3}, leading to the species [Mo3S11(H2O)2] and/or [Mo3S9(H2O)4]2+. The TOF decrease after tirrad = 20 min indicates that catalyst deactivation becomes the dominant process. This would be in line with the formation of [Mo3S7(H2O)6]4+ as main species, where the highest formation energies for the Volmer step (Fig. 4c) suggest low HER reactivity.
The calculations also shed light on the significantly faster deactivation of {Mo3} under aqueous conditions: the high water concentration results in faster exchange of terminal disulfides with aquo ligands and thus, faster formation of the less reactive species [Mo3S7(H2O)6]4+.
In the next step, we compared how complete substitution of terminal disulfides with chloride ligands affects the HER energetics: for {Mo3}–Cl we calculated a free energy of the Volmer step (12.1 kcal mol−1, see ESI†). This is in line with the experimentally observed low HER activity for {Mo3}–Cl (Table 1, entry 6). As a final step, we assessed whether molybdenum hydride (MoV–H) species could be relevant for hydrogen evolution by {Mo3}. This is a relevant question as recent studies on solid-state Mo–S HER electrocatalysts suggest that MoV–H species might be involved in the catalytic cycle.12,13 Here, we investigated the proton reduction by [Mo3S11], i.e. a molecular model for a Mo–S catalyst featuring vacant terminal molybdenum coordination sites (Fig. 4e). The species can be formed from {Mo3} by loss of one terminal disulfide (Fig. 4d). For this species, we also obtained a Volmer–Heyrovsky type mechanism. In the Volmer step, H atom binding at a bridging disulfide (−6.5 kcal mol−1) and at the vacant Mo center (−8.7 kcal mol−1, leading to a MoV–H) are energetically comparable (Fig. 4d). In contrast, H atom binding at the terminal disulfide leads to an energetically stabilized species where the subsequent Heyrovsky step would be significantly uphill (Fig. 4d). This finding suggests that for {Mo3} and related Mo–S catalysts, two HER pathways could be energetically possible, and the exact route might depend on the chosen reaction conditions. This could indeed help to consolidate the divergent reports on Mo–S HER catalysts where either hydrogen binding by bridging disulfides6–9 or MoV hydride formation12,13 are described as the key mechanistic steps.
In summary, theoretical analysis supports our experimental findings: partial exchange of terminal disulfides with aquo ligands leads to species such as [Mo3S7(H2O)x](2−x)- (x = 2, 4) where hydrogen evolution should be energetically favoured. In contrast, complete exchange of the terminal disulfides with aquo or chloride ligands (giving [Mo3S7(H2O)6]4+ or [Mo3S7Cl6]2−, respectively) results in species where HER is expected to be energetically more demanding. These findings are in line with recent studies on structurally related homogeneous10,38 and heterogeneous12 Mo–S electrocatalysts, where partial replacement of terminal disulfides with oxo ligands is suggested to give catalytically more active species.
Electrochemical and photophysical studies
To gain further insights into the effects of terminal disulfide ligands on the HER-activity of {Mo3}, we investigated the catalyst redox properties as well as the interactions between photosensitizer and catalyst in solution.Electrochemical studies were performed to assess the redox-activity of {Mo3}, {Mo3}–Cl and {Mo3}–Br under reductive conditions. To this end, linear sweep voltammetry was performed between E = 0 and −1.5 V (referenced against Fc+/Fc in de-gassed DMF containing 0.5 vol% H2O and 0.1 M nBu4NPF6 as electrolyte, [catalyst] = 1 mM). Under identical experimental conditions, the onset of reduction for {Mo3} is observed at less negative potentials (Eonset ∼ −1.2 V) compared to the halide-substituted species {Mo3}–Cl and {Mo3}–Br (Eonset ∼ −1.4 V), see Fig. 5a. This suggests that electron transfer to {Mo3} features favorable redox-potentials for the reductive electron transfer required to initiate HER catalysis. The observed overpotentials are a result of the given experimental conditions (low proton donor concentration, use of glassy carbon electrode).
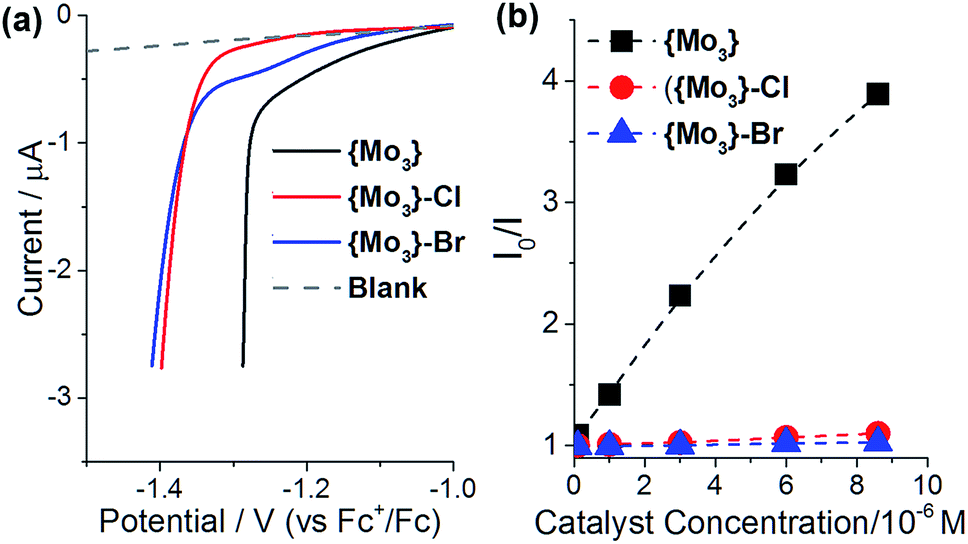 | ||
| Fig. 5 Redox-and photophysical properties affecting the homogeneous light-driven HER reactivity of {Mo3}. (a) Linear sweep voltammetry showing the onset of electrochemical reduction for {Mo3}, {Mo3}–Cl and {Mo3}–Br. The reduction onset for {Mo3} is observed at less negative potentials (shift ∼ 200 mV) indicating that a lower overpotential is required for electron transfer to {Mo3} compared with {Mo3}–Cl and {Mo3}–Br. Conditions: [catalyst] = 1 mM, working electrode: glassy carbon, solvent: de-gassed DMF containing 0.5% (v/v) H2O and 0.1 M nBu4PF6, scan rate: 50 mV s−1. (b) Stern–Volmer plot showing the quenching of the [Ru(bpy)3]2+ emission by the catalysts. Conditions: solvent: MeOH:H2O (10:1, v/v), [[Ru(bpy)3]2+] = 20 μM, excitation wavelength λ = 459 nm. | ||
The solution interactions between [Ru(bpy)3]2+ and {Mo3}, {Mo3}–Cl or {Mo3}–Br were explored in MeOH:H2O (10:1, v/v) using emission spectroscopy to analyze the quenching of the [Ru(bpy)3]2+-3MLCT emission depending on catalyst concentration. The resulting Stern–Volmer plots (Fig. 5b) show significantly higher quenching efficiency for {Mo3} compared to {Mo3}–Cl and {Mo3}–Br in the catalytic concentration range. Quenching is assigned to energy transfer between photosensitizer and catalyst, so that these initial data suggest higher solution interactions between {Mo3} and [Ru(bpy)3]2+ compared with the halide-substituted species.
Conclusions
The first example of homogeneous, visible light-driven HER catalysis by molecular molybdenum sulfide clusters is presented together with molecular-level rationalization of the observed catalytic reactivity. In water, [Mo3S13]2− shows promising HER activity but fast catalyst deactivation (TON ∼ 2850, TOFmax ∼ 37 min−1). In mixed methanol–water solvents, significantly higher reactivity is achieved by stabilizing highly active catalytic species (TON ∼ 41000, TOFmax ∼ 156 min−1). Mechanistic analyses show that substitution of one or two terminal disulfides with water ligands leads to the most active catalytic species (i.e. [Mo3S11(H2O)2] and [Mo3S9(H2O)4]2+) while complete exchange of the terminal disulfides results in catalytically less reactive species ([Mo3S7(H2O)6]4+). Reactivity studies show that the terminal disulfides modulate the electronic structure and redox-properties of the catalyst and facilitate catalyst–photosensitizer interactions in solution. Further, theoretical studies suggest that the first steps of the hydrogen evolution could involve H atom binding at bridging disulfides as well as the formation of molybdenum(V) hydrides. The study could lead to knowledge-based design routes for Mo–S catalysts and could enable catalyst stabilization schemes based on reversible ligand exchange.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Ulm University, the Schlumberger Foundation Faculty for the Future program (M.D.), the Deutsche Forschungsgemeinschaft DFG (Graduate School GRK1626) and EU COST Actions CM1202 (PerspectH2O) and CM1203 (PoCheMoN) are gratefully acknowledged. Maximilian Dürr and Prof. Ivana Ivanovic-Burmazovic (FAU Erlangen-Nürnberg) are gratefully acknowledged for ESI-MS measurements.Notes and references
- C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671–2681 CrossRef CAS PubMed
.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878 CAS
- T. Wang, H. Xie, M. Chen, A. D'Aloia, J. Cho, G. Wu and Q. Li, Nano Energy, 2017, 42, 69–89 CrossRef CAS
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS
- B. Seo and S. H. Joo, Nano Converg., 2017, 4, 19 CrossRef PubMed
- Z. Huang, W. Luo, L. Ma, M. Yu, X. Ren, M. He, S. Polen, K. Click, B. Garrett, J. Lu, K. Amine, C. Hadad, W. Chen, A. Asthagiri and Y. Wu, Angew. Chem., Int. Ed., 2015, 54, 15181–15185 CrossRef CAS PubMed
- L. R. L. Ting, Y. Deng, L. Ma, Y.-J. Zhang, A. A. Peterson and B. S. Yeo, ACS Catal., 2016, 6, 861–867 CrossRef CAS
- B. Lassalle-Kaiser, D. Merki, H. Vrubel, S. Gul, V. K. Yachandra, X. Hu and J. Yano, J. Am. Chem. Soc., 2015, 137, 314–321 CrossRef CAS PubMed
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS PubMed
- B. R. Garrett, K. A. Click, C. B. Durr, C. M. Hadad and Y. Wu, J. Am. Chem. Soc., 2016, 138, 13726–13731 CrossRef CAS PubMed
- B. R. Garrett, S. M. Polen, K. A. Click, M. He, Z. Huang, C. M. Hadad and Y. Wu, Inorg. Chem., 2016, 55, 3960–3966 CrossRef CAS PubMed
- P. D. Tran, T. V. Tran, M. Orio, S. Torelli, Q. D. Truong, K. Nayuki, Y. Sasaki, S. Y. Chiam, R. Yi, I. Honma, J. Barber and V. Artero, Nat. Mater., 2016, 15, 640–646 CrossRef CAS PubMed
- Y. Huang, R. J. Nielsen, W. A. Goddard and M. P. Soriaga, J. Am. Chem. Soc., 2015, 137, 6692–6698 CrossRef CAS PubMed
- A. Müller, E. Diemann, R. Jostes and H. Bögge, Angew. Chem., Int. Ed. Engl., 1981, 20, 934–955 CrossRef
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed
- J. Kibsgaard and T. F. Jaramillo, Angew. Chem., Int. Ed., 2014, 53, 14433–14437 CrossRef CAS PubMed
- Y. Shang, X. Xu, B. Gao and Z. Ren, ACS Sustain. Chem. Eng., 2017, 5, 8908–8917 CrossRef CAS
- D. Recatalá, R. Llusar, A. L. Gushchin, E. A. Kozlova, Y. A. Laricheva, P. A. Abramov, M. N. Sokolov, R. Gómez and T. Lana-Villarreal, ChemSusChem, 2015, 8, 148–157 CrossRef PubMed
- M. Kan, J. Jia and Y. Zhao, RSC Adv., 2016, 6, 15610–15614 RSC
- K. Du, L. Zheng, T. Wang, J. Zhuo, Z. Zhu, Y. Shao and M. Li, ACS Appl. Mater. Interfaces, 2017, 9, 18675–18681 CAS
- A. Müller, R. G. Bhattacharyya and B. Pfefferkorn, Chem. Ber., 1979, 112, 778–780 CrossRef
-
A. Müller and E. Krickemeyer, in Inorganic Syntheses, ed. A. P. Ginsberg, New York, 1990, pp. 47–51 Search PubMed
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed
- V. P. Fedin, M. N. Sokolov, Y. V. Mironov, B. A. Kolesov, S. V. Tkachev and V. Y. Fedorov, Inorganica Chim. Acta, 1990, 167, 39–45 CrossRef CAS
- B. Kirchhoff, S. Rau and C. Streb, Eur. J. Inorg. Chem., 2016, 2016, 1425–1429 CrossRef CAS
- V. P. Fedin, G. J. Lamprecht and A. G. Sykes, J. Chem. Soc., Chem. Commun., 1994, 2685–2686 RSC
- D. Recatala, R. Llusar, A. Barlow, G. Wang, M. Samoc, M. G. Humphrey and A. L. Guschin, Dalton Trans., 2015, 44, 13163–13172 RSC
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS PubMed
- C. Bachmann, B. Probst, M. Guttentag and R. Alberto, Chem. Commun., 2014, 50, 6737–6739 RSC
- T. Yu, Y. Zeng, J. Chen, Y.-Y. Li, G. Yang and Y.-Y. Li, Angew. Chem., Int. Ed., 2013, 52, 5631–5635 CrossRef CAS PubMed
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou, N. J. Deyonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef CAS PubMed
- Z. Han, W. R. McNamara, M.-S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667–1670 CrossRef CAS PubMed
- J. Lloret Fillol, A. Call, F. Franco, N. Kandoth, S. Fernández, M. González-Béjar, J. Perez-Prieto and J. M. Luis, Chem. Sci., 2018, 9, 2609–2619 RSC
- K. S. Exner, J. Anton, T. Jacob and H. Over, Angew. Chem., Int. Ed., 2016, 55, 7501–7504 CrossRef CAS PubMed
- S. Losse, J. G. Vos and S. Rau, Coord. Chem. Rev., 2010, 254, 2492–2504 CrossRef CAS
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS PubMed
-
H. S. Biswal, ed. S. Scheiner, Hydrogen Bonds Involving Sulfur: New Insights from ab Initio Calculations and Gas Phase Laser Spectroscopy, Noncovalent Forces, Springer International Publishing, Cham, 2015, pp. 15–45 Search PubMed
- B. R. Garrett, S. M. Polen, M. Pimplikar, C. M. Hadad and Y. Wu, J. Am. Chem. Soc., 2017, 139, 4342–4345 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1533536 and 1533537. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7se00599g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |