
Combining battery-like and pseudocapacitive charge storage in 3D MnOx@carbon electrode architectures for zinc-ion cells†
Jesse S.
Ko
 a,
Megan B.
Sassin
b,
Joseph F.
Parker
b,
Debra R.
Rolison
b and
Jeffrey W.
Long
*b
a,
Megan B.
Sassin
b,
Joseph F.
Parker
b,
Debra R.
Rolison
b and
Jeffrey W.
Long
*b
aNaval Research Laboratory – National Research Council Postdoctoral Associate, Surface Chemistry Branch (Code 6170), Washington, DC 20375, USA
bU.S. Naval Research Laboratory, Surface Chemistry Branch (Code 6170), Washington, DC 20375, USA. E-mail: jeffrey.long@nrl.navy.mil
First published on 19th December 2017
Abstract
We demonstrate that electrodes comprising nanoscale, birnessite-type manganese oxide affixed to carbon nanofoam paper (MnOx@CNF) exhibit two distinct charge-storage mechanisms—battery-like Zn2+ insertion/de-insertion and pseudocapacitance—when electrochemically cycled in aqueous electrolytes that include both Na+ and Zn2+ salts. When the mixed-electrolyte composition is 0.75 M Na2SO4 + 0.25 M ZnSO4 (i.e., “6[Na+]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1[Zn2+]”), the MnOx@CNF electrode delivers high specific capacity at low rates, approaching theoretical capacity for Zn2+ insertion/de-insertion at MnOx. At high rates (>10C) the Na+-supported pseudocapacitance mechanism maintains charge-storage capacity well above that observed with electrolytes that contain only ZnSO4. Impedance analysis was performed to discriminate between these distinct charge-storage mechanisms by visualizing the frequency- and potential-dependent capacitance as 3D Bode plots. In the 6[Na+]:1[Zn2+] electrolyte, the potential-independent pseudocapacitance is augmented by reversible Zn2+-based redox processes between 1.4 and 1.8 V vs. Zn/Zn2+. Galvanostatic testing with two-electrode zinc-ion cells that pair MnOx@CNF with a zinc foil negative electrode proves the practical performance advantages of combining pseudocapacitance and Zn2+-insertion mechanisms: higher energy efficiency and greater specific power in the 6[Na+]:1[Zn2+] electrolyte compared to 1 M ZnSO4.
:1[Zn2+]”), the MnOx@CNF electrode delivers high specific capacity at low rates, approaching theoretical capacity for Zn2+ insertion/de-insertion at MnOx. At high rates (>10C) the Na+-supported pseudocapacitance mechanism maintains charge-storage capacity well above that observed with electrolytes that contain only ZnSO4. Impedance analysis was performed to discriminate between these distinct charge-storage mechanisms by visualizing the frequency- and potential-dependent capacitance as 3D Bode plots. In the 6[Na+]:1[Zn2+] electrolyte, the potential-independent pseudocapacitance is augmented by reversible Zn2+-based redox processes between 1.4 and 1.8 V vs. Zn/Zn2+. Galvanostatic testing with two-electrode zinc-ion cells that pair MnOx@CNF with a zinc foil negative electrode proves the practical performance advantages of combining pseudocapacitance and Zn2+-insertion mechanisms: higher energy efficiency and greater specific power in the 6[Na+]:1[Zn2+] electrolyte compared to 1 M ZnSO4.
1. Introduction
Manganese oxides (MnOx) have had a long and impactful history as active materials for electrochemical energy storage, the most prominent example being the now–ubiquitous primary alkaline Zn/MnO2 battery, in which charge storage is facilitated by proton/electron insertion into the oxide.1 The desire for high-performance secondary (rechargeable) batteries drove the subsequent development of manganese-based oxides, such as LiMn2O4, that undergo reversible Li+ insertion to serve as positive-electrode materials for lithium-ion batteries.2,3 The 1999 report by Lee and Goodenough showing capacitor-like charge storage (i.e., “pseudocapacitance”) for disordered MnOx when cycled in mild-pH aqueous electrolytes4 launched a new direction for the use of manganese oxides in electrochemical capacitors (ECs).5,6 The most recent concept for charge storage in MnOx involves the reversible insertion of Zn2+ into nanocrystalline forms of the oxide in mild-pH aqueous electrolytes (e.g., 1 M ZnSO4) as the basis for “rechargeable zinc-ion batteries”.7–19The ability of manganese oxides to exhibit the charge-storage characteristics of either batteries or ECs offers an intriguing opportunity to design electrode materials that can be tailored to deliver optimized combinations of pulse power and high capacity. Charge-storage behavior at nanoscale MnOx, in terms of both charge/discharge time scale and current–voltage profiles, depends on many factors including specific crystal structure (or lack thereof), particle/crystal size, and the type of charge-compensating ions supplied from the contacting electrolyte, such as H+, Li+, or Na+. Electrode architecture also plays a critical role in electrochemical performance, as we have shown for nanoscale MnOx conformally deposited on fiber paper-supported carbon nanofoams.20–22 In such electrode designs, interpenetrating nanoscale networks of void and conductive carbon facilitate the transport of ions and electrons, respectively, to the charge-storing MnOx coating present at high weight loadings.
Lamellar birnessite-type MnOx expressed in such 3D architectures amplifies charge storage in neutral-pH electrolytes via pseudocapacitance mechanisms. Yet, when these same electrode materials are cycled in alkaline electrolytes (LiOH + KOH), the electrode exhibits either capacitive or battery-like voltammetric features, depending on the potential region examined.23 If the carbon-anchored MnOx layers are crystal engineered in situ to the crystalline spinel LiMn2O4via ion-exchange and thermal processing,24,25 the resulting material exhibits well-defined, battery-like peaks for lithium-ion insertion, yet can be charged and discharged in tens of seconds, time scales that approach those of relevance for ECs.
Herein, we use established electrode architectures of birnessite MnOx-affixed carbon nanofoam paper (MnOx@CNF) to extend “battcap” performance beyond Li+-containing alkaline electrolytes. By using simple aqueous electrolyte mixtures of ZnSO4 and Na2SO4, we demonstrate high-capacity, battery-like Zn2+ insertion and pseudocapacitive charge storage in a single electrode composition.
2. Experimental
2.1. Materials
Resorcinol (“R”; Aldrich), formaldehyde (“F”; 37%, Aldrich), sodium carbonate (“C”; Aldrich), sodium sulphate (Aldrich), and sodium permanganate monohydrate (Aldrich) were used as received. All solutions were prepared with ultrapure water from an in-house water purification system (>18 MΩ cm).2.2. Preparation of MnOx@CNF
Carbon fiber paper (Lydall) was cut into 4 × 4 cm2 squares and exposed to an air–ice plasma for 45 min (Harrick radio-frequency glow-discharge plasma cleaner; 30 W setting).26 Glass slides (VWR, 5 cm2) were cleaned by soaking in a KOH/ethanol base bath for 16 h, rinsed thoroughly with ultrapure water, and then heated in a muffle furnace (Thermolyne 47900) under static air at 500 °C for 2 h.Carbon nanofoams were fabricated as previously described for the RF 40 R/C 500 recipe,22,26i.e., 40 wt% R + F catalysed with a molar R/C ratio of 500. Briefly, carbon fiber papers were placed in a glass Petri dish containing the oligomerized RF sol and soaked under vacuum for 1 min. The RF-infiltrated carbon fiber paper was placed between two glass slides and the assembly was secured on each side with a mini-binder clip and then wrapped in duct tape. The duct tape-wrapped assemblies were placed in a single Al foil pouch containing ∼2 mL of water and cured for 12 h at room temperature. The Al pouch containing the nanofoams was placed in a pressure cooker (Nesco 3-in-1, target, steam setting) on “slow” cook (∼88–94 °C) for 9.5 h, followed by “warm” cook (∼80 °C) for 14.5 h and then removing the Al pouch from the pressure cooker. The polymer nanofoam-filled papers were released from the glass slides, soaked in ultrapure water for 1 h, soaked in acetone for 1 h, and then dried under ambient conditions for 1 h. The RF nanofoams were pyrolyzed in a tube furnace (Thermo Scientific Lindberg Blue M) by ramping to 1000 °C at 1 °C min−1 and holding at 1000 °C for 2 h under flowing argon.
Manganese oxide-decorated carbon nanofoam papers (MnOx@CNF) of the “1-ply 40/500” type were prepared via electroless deposition using a previously published protocol.22 Briefly, carbon nanofoams were infiltrated with 0.1 M Na2SO4 under vacuum and soaked for 8 h. The 0.1 M Na2SO4 solution was decanted and replaced with 0.1 M NaMnO4·H2O + 0.1 M Na2SO4 and soaked for 20 h under ambient laboratory conditions. The MnOx@CNF papers were removed from the MnO4− solution, rinsed well with ultrapure water, and then vacuum infiltrated with ultrapure water and allowed to soak for 1 h. The rinse/vacuum infiltrate protocol was repeated until the rinse water was clear and colorless (approximately twice more). The MnOx@CNF papers were dried under flowing N2 at 50 °C for 8 h.22
2.3. Electrochemical characterization
Voltammetric and impedance measurements were performed using a Gamry Reference 600 potentiostat. For three-electrode measurements, the working electrode was prepared by adhering a 0.5 × 0.5 cm2 piece of MnOx@CNF (∼4–5 mg cm−2) onto a 1 × 2 cm2 Ti foil (Alfa-Aesar) using conductive carbon glue (Pelco). These electrodes were baked at 150 °C for 10 min to cure the carbon adhesive and then dried in an oven at 100 °C for several hours. The counter and reference electrodes consisted of zinc foil and zinc wire, respectively. A series of electrolytes were prepared comprising: (i) 1 M Na2SO4; (ii) 0.75 M Na2SO4 + 0.25 M ZnSO4; (iii) 0.5 M Na2SO4 + 0.5 M ZnSO4; and (iv) 1 M ZnSO4. The electrolyte series is respectively designated as 2[Na+]:0[Zn2+], 6[Na+]:1[Zn2+], 2[Na+]:1[Zn2+], and 0[Na+]:1[Zn2+] (Table 1).
:ZnSO4 electrolyte in which sulphate concentration is maintained at 1 M. Electrolyte designations: Na+:Zn2+ = 2[Na+]:0[Zn2+], 6[Na+]:1[Zn2+], 2[Na+]:1[Zn2+], 0[Na+]:1[Zn2+]
| Molar concentration of electrolyte | Electrolyte notation (atom:atom ratio) |
Ionic strength of electrolyte (M) | Ionic conductivity (mS cm−1) | Specific capacity at 1 mV s−1 (mA h gMnOx−1) | Specific capacity at 10 mV s−1 (mA h gMnOx−1) | Integrated capacitive charge from Bode plots (mA h gMnOx−1) at 0.01 Hz |
|---|---|---|---|---|---|---|
| 1 M Na2SO4 | 2[Na+]:0[Zn2+] |
3 | 71 | 88 | 89 | 43 |
| 0.75 M Na2SO4 + 0.25 M ZnSO4 | 6[Na+]:1[Zn2+] |
3.25 | 62 | 178 | 117 | 51 |
| 0.5 M Na2SO4 + 0.5 M ZnSO4 | 2[Na+]:1[Zn2+] |
3.5 | 53 | 201 | 118 | 24 |
| 1 M ZnSO4 | 0[Na+]:1[Zn2+] |
4 | 41 | 233 | 83 | 18 |
The cyclic voltammetric measurements were made within a voltage window of 0.9–1.9 V vs. Zn/Zn2+ cycling at 1–100 mV s−1 while purging under N2. The specific capacity was calculated as the average of the integrated current of both charge–discharge curves. The AC impedance measurements were performed from 10 mHz to 100 kHz with an AC amplitude of 10 mV. Impedance cycling was carried out at 50 mV intervals within a 1.0–1.9 V vs. Zn/Zn2+ window collecting over 10 mHz to 100 kHz at each step.
Two-electrode measurements were configured using a PFA Swagelok® assembly with Ti rods serving as current collectors. The working electrodes were prepared in a similar fashion as described above by gluing MnOx@CNF onto a 3/8′′ diameter Ti foil (Alfa-Aesar). The counter electrode was 1/2′′ diameter zinc foil separated from the working electrode using a glass-fiber separator. The AC impedance measurements were performed before the cell was cycled using the same conditions described above. Cycling measurements were carried out using an Arbin BT2000 battery cycler from 0.9–1.9 Vcell with all C-rates normalized to the theoretical capacity of MnOx (308 mA h g−1 for a one-electron reaction).
2.4. Materials characterization
Samples of MnOx@CNF papers were analysed by scanning electron microscopy (SEM) and energy-dispersive spectroscopy (EDS) using a Leo Supra 55 equipped with an Oxford Instruments Aztec EDS system at an accelerating voltage of 20 keV. The samples were prepared by cutting small sections of the MnOx@CNF papers with a fresh razor blade and mounting on flat Al stubs (Ted Pella) using conductive carbon tape.3. Results and discussion
3.1. Cyclic voltammetry
The “one-ply 40/500” type manganese oxide-affixed carbon nanofoam papers (MnOx@CNF; Fig. 1) were chosen for their balance of hierarchical pore sizes (10–80 nm range) and high specific surface area (262 m2 g−1).22 This version of CNF paper retains sufficient nanofoam surface area at which high weight loadings (50 wt%) of 10 nm-thick, X-ray amorphous (Fig. S1†),25 birnessite–MnOx can be electrolessly deposited, while the co-continuous mesoporous network supports the ions/mass transport necessary for high-rate cycling of the MnOx pseudocapacitance reaction in mild-pH electrolytes.22 The ∼80 μm thickness of the one-ply nanofoam paper structure as well as the active material loading (5.4 mg MnOx per cm2)22 matches the metrics of typical powder-composite electrodes in commercial ECs, making MnOx@CNF a drop-in, binder-free electrode for technologically relevant devices.22 | ||
| Fig. 1 Scanning electron micrograph of MnOx@CNF electrodes (inset shows an energy-dispersive spectroscopic mapping of the Mn (purple)). | ||
We examined the electrochemical properties of one-ply 40/500 MnOx@CNF electrodes in a series of Zn2+-containing aqueous electrolytes with varying Zn2+-to-Na+ composition. Initial tests performed in a three-electrode, half-cell configuration isolated the performance of MnOx@CNF from any limitations that might be imposed by the zinc negative electrode in a two-electrode device. However, we do use a zinc-wire quasi-reference electrode in the half-cell tests in order to convey the redox characteristics of these electrodes on a voltage scale relevant to two-terminal aqueous zinc-ion energy-storage devices.
When cycling MnOx@CNF at modest scan rate (1 mV s−1) in aqueous 1 M ZnSO4, a common electrolyte used for zinc-ion studies, reversible redox peaks appear centered at ca. +1.4 V vs. Zn (Fig. 2a). Such features are often attributed to Zn2+-insertion/de-insertion reactions at MnOx of various crystalline habits,7–19 although recently Lee et al. proposed that the voltammetric peaks commonly observed in ZnSO4 electrolytes arise from precipitation of zinc complexes mediated by redox reactions at the MnOx surface.27 At ∼300 mV, the oxidation/reduction peak splitting for the Zn2+-supported redox process at MnOx@CNF is competitive with or superior to that reported in the literature for conventional powder-composite electrode structures containing MnOx.7–17 Integrating the charge under the voltammogram yields a preliminary estimate of the electrode capacity as 94 mA h gT−1 with respect to total electrode mass (T) and 235 mA h gMnOx−1 normalized to the ∼36 wt% loading of MnOx in the nanofoam paper electrode.
 | ||
| Fig. 2 Cyclic voltammograms from 0.9–1.9 V vs. Zn/Zn2+ for MnOx@CNF cycled in mixed Na2SO4:ZnSO4 electrolyte in which sulphate concentration is maintained at 1 M but with varied Na+:Zn2+ content (2[Na+]:0[Zn2+]; 6[Na+]:1[Zn2+]; 2[Na+]:1[Zn2+]; and 0[Na+]:1[Zn2+]) at (a) 1 mV s−1; (b) 10 mV s−1; and (c) 100 mV s−1. (d) Corresponding rate capability (specific capacity vs. scan rate). | ||
While the zinc-ion insertion properties of MnOx@CNF are promising, we also have the opportunity to expand the functionality of this electrode architecture beyond that based only on a battery-like charge-storage mechanism. Our prior work showed that the nanoscopic birnessite–MnOx that coats the walls of the CNF exhibits pseudocapacitive charge storage in aqueous alkali-metal salt neutral electrolytes (e.g., Na2SO4, Li2SO4),20–22 supported by redox reactions that involve charge-compensating cations (or in some cases, anions28). In order to add pseudocapacitance functionality to Zn–MnOx faradaic reactions, we explored the voltammetric response of MnOx@CNF in mixed-salt aqueous electrolytes in which we systematically substituted Na2SO4 for ZnSO4 while maintaining an invariant sulphate concentration of 1 M.
Replacing half of the ZnSO4 content (0.5 M Na2SO4 + 0.5 M ZnSO4, designated “2[Na+]:1[Zn2+]”) i.e., 2 Na+ are present for every Zn2+, only modestly lowers peak current by ∼20% (Fig. 2a). However, with a cationic charge equivalent of monovalent (2 Na+) and divalent (1 Zn2+) in the electrolyte, the zinc-ion insertion peaks now ride on a capacitor-like envelope. Pseudocapacitance behavior for birnessite–type MnOx is well established for both CNF-based20–22 and powder-composite electrodes29,30 cycled in mild aqueous electrolytes. Suib and coworkers recently reported capacitive cyclic voltammograms for powder-composite birnessite–MnOx electrodes in Na2SO4 electrolytes containing less than 0.2 g L−1 ZnSO4 as a means to extract Zn2+ from aqueous media.31 Other reports describe the use of mixed Na2SO4:ZnSO4 electrolytes in conjunction with powder-composite electrodes containing Na0.44MnO2 and Na4Mn9O18 for the purpose of charge storage.15,16,32 However, the composite electrodes in these cases did not exhibit an unambiguous pseudocapacitive background; only redox peaks are seen in the published voltammograms.
Further increasing the Na2SO4 concentration from 0.5 M to 0.75 M (i.e., 6[Na+]:1[Zn2+]) minimally changes the voltammogram. Total capacity of MnOx@CNF when cycling at 1 mV s−1 is 180 mA h gMnOx−1; so with four-fold less Zn2+ present, the specific capacity drops only 23%. In 1 M Na2SO4, MnOx@CNF exhibits a nearly rectangular pseudocapacitance-dominated background (Fig. 2a), with an integrated capacity of 35 mA h gT−1, corresponding to 88 mA h gMnOx−1. Thus the pseudocapacitance mechanism, involving Na+ or SO42−,20,28 delivers less than half of the capacity delivered by the Zn2+-based battery-like reaction, at least at this slow scan rate (1 mV s−1), but still greatly amplifies charge-storage capacity vs. double-layer contributions, which can be estimated by cycling native CNFs in similar aqueous electrolytes (Fig. S2†).
Having established that MnOx@CNF electrodes exhibit both battery-like (zinc-ion insertion) and capacitor-like (pseudocapacitive) character in mixed Na2SO4:ZnSO4 electrolytes under slow-sweep conditions, we extended our scan rates to 100 mV s−1 to explore the relative contributions of these two mechanisms as a function of their time/power response. At 10 mV s−1, the response is bimodal in this series of electrolytes: <90 mA h gMnOx−1 in the Na+-only and Zn2+-only electrolytes and ∼120 mA h gMnOx−1 in the two Na+:Zn2+ mixed electrolytes (Table 1). Note that at this faster scan rate, the charge-storage capacity for the Zn2+-only electrolyte is 64% lower with broadening and separation of the redox peaks (Fig. 2b). Yet the baseline pseudocapacitance that is characteristic of Na+-only electrolyte persists.
These effects are further amplified at 100 mV s−1 (Fig. 2c). Although all voltammograms exhibit significant distortion at this scan rate (due to iR drop and RC time-constant effects), the Na2SO4-containing electrolytes yield an improved voltammetric response compared to Na+-free 1 M ZnSO4, where no significant charge storage, either redox or pseudocapacitance, is observed. Note that all Na+-containing electrolytes support comparable specific capacity ∼40 mA h gMnOx−1 at this high scan rate (Fig. 2d). The higher ionic conductivity of Na+-based electrolytes vs. Zn2+-based electrolytes (Table 1) may also play a secondary role in the observed high-rate capability. Individual cyclic voltammograms of MnOx@CNF electrodes cycled in their respective electrolytes are shown from 1–10 mV s−1 (Fig. S3†) and 10–100 mV s−1 (Fig. S4†). Specific capacity expressed as a function of charging time is also presented in Fig. S5.†
The dynamics of charge storage were further examined by applying a “b-value” analysis to a series of scan rates and electrolyte compositions.33 In this analysis, the measured current, i, at a selected potential, V, obeys a power-law relation with the sweep rate, v:
| i(V) = avb | (1) |
i vs. logv offers insight into the mass-transport character of the underlying charge-storage mechanism. A b-value of 1 corresponds to a diffusion-independent process, such as that expected for faradaic electron transfer to a surface-adsorbed electroactive species or a capacitive process, whereas a value of 0.5 indicates a process controlled by semi-infinite diffusion, such as Zn2+ insertion/de-insertion.
The b-values of the cathodic reaction at 1.2 V (Fig. 3 legend) are comparable to those for the anodic process at 1.7 V (Fig. S6†): both branches provide linear regressions of R2 > 0.99. Analysis of MnOx@CNF cycled in a Zn2+-free electrolyte yields a b-value of 1, indicating that charge storage is dominated by diffusion-independent redox processes, likely involving Na+or H+-compensation of electrogenerated Mn(III). In a purely Zn2+-containing electrolyte, a b-value of 0.5 signifies a semi-infinite diffusion-controlled process, as would be expected for Zn2+ insertion/de-insertion redox processes into a solid-state material on the time-scale of the experiment. At 1 mV s−1, the insertion process has 1000 s to occur into the 10 nm-thick MnOx, thus the solid-state diffusion coefficient for Zn2+ in MnOx must be less than 10−16 cm2 s−1 for this process to follow semi-infinite diffusive transport.
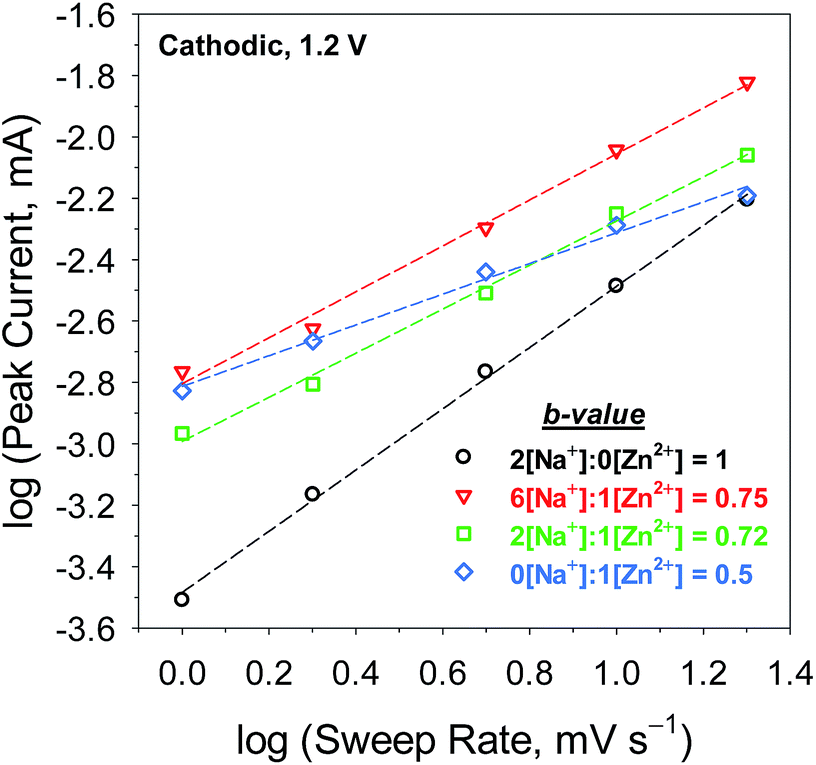 | ||
| Fig. 3 The b-value analyses (log cathodic peak current vs. log sweep rate) for MnOx@CNF cycled in mixed Na2SO4:ZnSO4 electrolytes (Na+:Zn2+ = 2[Na+]:0[Zn2+], 6[Na+]:1[Zn2+], 2[Na+]:1[Zn2+], 0[Na+]:1[Zn2+]). | ||
The mixed Na+:Zn2+ electrolytes (6[Na+]:1[Zn2+] and 2[Na+]:1[Zn2+]) show b-values of ∼0.73, intermediate of the previous two cases. An intermediate b-value is characteristic of finite diffusion,34,35 which indicates that on the time scale of the electrolysis, a diffusing reactant samples a boundary or the flux lines intersect. It is unlikely that with a surface-independent process and a semi-infinite Zn2+-insertion process, the mass transport now becomes finite. We posit that we see the intermediate b-values because both processes are operative (Scheme 1) on this time scale (50 to 1000 s). The only way one can physically access finite diffusion is if Na+ association affects solid-state diffusion of Zn2+.
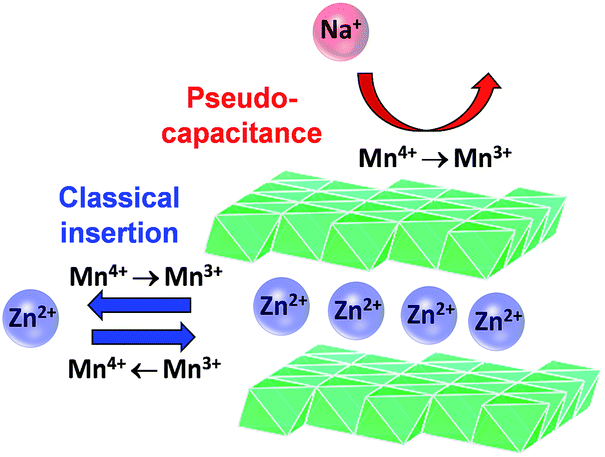 | ||
| Scheme 1 Proposed electrochemical charge-storage mechanisms for MnOx@CNF from Na+ pseudocapacitance and Zn2+ insertion/de-insertion. | ||
3.2. Impedance analysis
Qualitatively similar Nyquist plots are found for the MnOx@CNF electrodes across the series of electrolytes (Fig. S7†), where the high-frequency intercepts approximately track electrolyte ionic conductivity (which ranges from 41 mS cm−1 for 1 M ZnSO4 to 71 mS cm−1 for 1 M Na2SO4; Table 1). Charge-transfer arcs corresponding to area-normalized resistances of 2.5–5 Ω cm2 are observed at intermediate frequencies, consistent with values we previously reported for related MnOx@CNFs in aqueous electrolytes.22,24 We note, however, that the lowest charge-transfer resistance in this series occurs with the 6[Na+]:1[Zn2+] electrolyte, although the ionic conductivity of this solution is lower than 1 M Na2SO4. At low frequency, the Nyquist plots exhibit the expected capacitive branches.
For a more detailed analysis, we turned to Bode plot representations of the impedance data. Analyses using Bode plots provide a much less ambiguous deconvolution of the time-based response of an electrode, and mapping those data onto the third axis (potential), provides a clear map of the electrode response. The real component of capacitance (C′) derived from impedance measurements by the following relation:
| Z′′ = 1/2πfC′ | (2) |
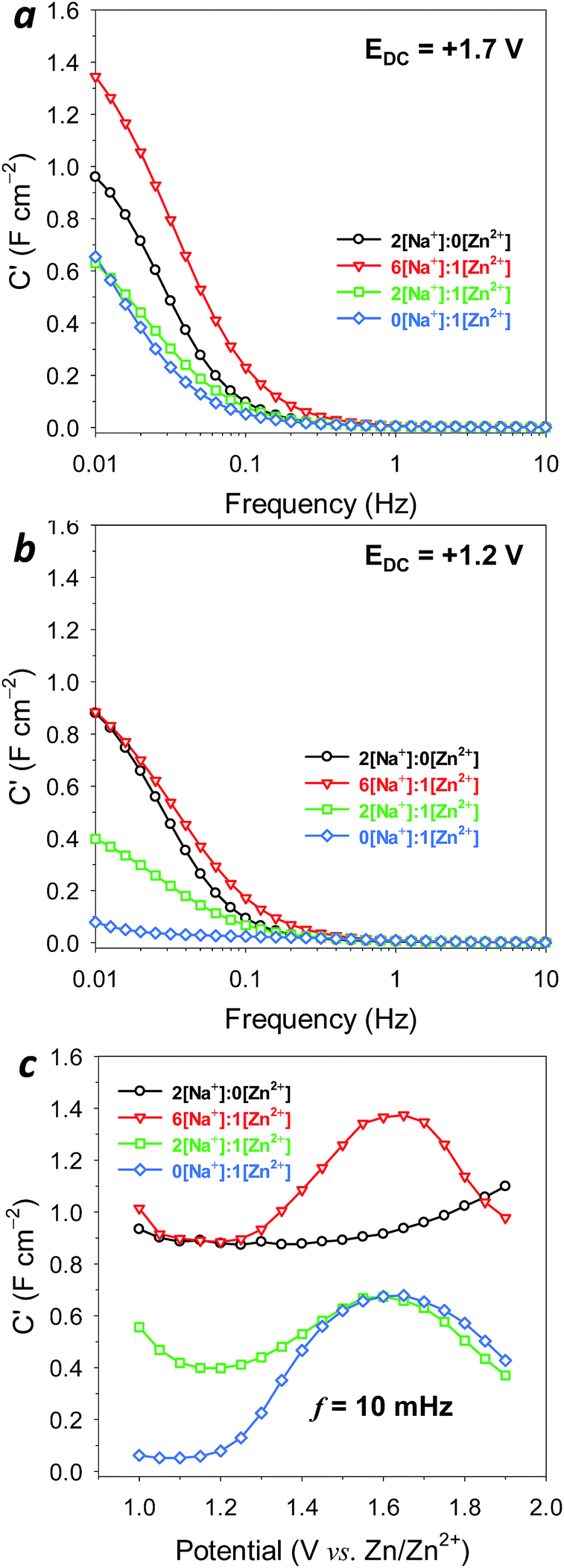 | ||
| Fig. 4 The Bode plots for MnOx@CNF cycled in mixed Na2SO4:ZnSO4 electrolyte (Na+:Zn2+ = 2[Na+]:0[Zn2+], 6[Na+]:1[Zn2+], 2[Na+]:1[Zn2+], 0[Na+]:1[Zn2+]) obtained at an applied DC bias of (a) +1.2 V and (b) +1.7 V vs. Zn/Zn2+. (c) Impedance cycling from 1.0–1.9 V vs. Zn/Zn2+; the area-normalized capacitance is determined at 10 mHz. | ||
In the more oxidized, Mn(IV)-dominant state, the capacitance for MnOx@CNF begins to rise rapidly at ∼0.3 Hz for both 6[Na+]:1[Zn2+] and Zn2+-free electrolytes, maximizing at ∼1.4 and ∼1 F cm−2, respectively (Fig. 4a). These values are consistent with pseudocapacitive charge storage in the birnessite MnOx coating.21,22 The capacitance response is notably lower in 2[Na+]:1[Zn2+] and 1 M ZnSO4 electrolyte, reaching only 0.6 F cm−2 at low frequency. Yet the persistence of such levels of capacitance, even in the absence of Na+, suggests that either Zn2+ or SO42− enables surface-sited pseudocapacitance reactions at MnOx.
When conditioned at a potential (E = +1.2 V) where the oxide is Mn(III)-rich, presumably compensated by associated Zn2+, Na+, or H+, the capacitance response in Zn2+-free, 6[Na+]:1[Zn2+], and 2[Na+]:1[Zn2+] electrolytes is not markedly changed (Fig. 4b). However, capacitance is almost entirely suppressed at this potential in the 1 M ZnSO4 electrolyte. We posit that in such a reduced state, Mn sites are strongly associated with inserted Zn2+ cations, and therefore are unavailable for the surface-sited redox reactions needed for pseudocapacitance. This response at 0.06 F cm−2 represents an upper bound on the double-layer capacitance of this oxide-coated nanofoam architecture. This finding is further confirmed from impedance measurements at native CNF paper electrodes (MnOx-free), which express approximately the same area-normalized capacitance at low frequency (Fig. S8†).
To further explore the potential dependence of the capacitive response, we measured impedance spectra at 50 mV intervals for DC potentials between 1.0–1.9 V, and generated corresponding Bode plots at 10 mHz (Fig. 4c) and 31.6 and 100 mHz (Fig. S9†). In 1 M ZnSO4, electrode capacitance remains low at <0.05 F cm−2 for potentials from 1.0–1.2 V, but significantly increases to ∼0.75 F cm−2 between 1.2 and 1.55 V. This rise in capacitance can be explained by overlaying the information contained in the voltammogram (Fig. S10a†) because it occurs concomitantly with de-insertion of Zn2+ from the MnOx, likely reflecting that Mn sites are becoming available for surface-sited pseudocapacitance reactions. A similar rise in capacitance through this potential region is also seen with the 6[Na+]:1[Zn2+] electrolyte (Fig. 4c), but it is superimposed on a large baseline capacitance of ∼0.9 F cm−2, a value similar to the relatively potential-independent pseudocapacitance exhibited by MnOx in 1 M Na2SO4 (Fig. S10b†). Integrating the area beneath these capacitance-vs.-potential curves provides another measure of charge-storage capacity for fast, reversible reactions (Table 1). For example, the 6[Na+]:1[Zn2+] electrolyte provides 51 mA h gMnOx−1 of such charge-storage capacity compared to 44 mA h gMnOx−1 in 1 M Na2SO4, indicating an additional fast charge-storage contribution from Zn2+-based mechanisms. Capacitance is notably depressed for the 2[Na+]:1[Zn2+] electrolyte, likely due to competition from Zn2+ for redox sites at the MnOx surface, thereby limiting Na+-supported pseudocapacitance.
Inspired by prior computational data treatments proposed by Bai and Conway,36,37 we turned to three-dimensional representations of the Bode plot data to provide a more complete experimental understanding of the complex interplay of capacitance values as a function of frequency, potential, and electrolyte composition (Fig. 5). With only Na+ present in the electrolyte, the 3D Bode plot appears as a simple “waterfall”, with relatively potential-independent capacitance gently falling off as frequency increases (Fig. 5a). In 1 M ZnSO4, capacitance maximizes near 1.5 V at low frequency, from which point it quickly falls as either frequency or potential is changed (Fig. 5b).
 | ||
| Fig. 5 The 3D Bode plot representations of the real area-normalized capacitance vs. frequency vs. potential for MnOx@CNF in mixed Na2SO4:ZnSO4 electrolyte: (a) 2[Na+]:0[Zn2+], (b) 0[Na+]:1[Zn2+], (c) 2[Na+]:1[Zn2+], (d) 6[Na+]:1[Zn2+]. | ||
The superposition of multiple charge-storage processes becomes more evident for mixed electrolytes in this 3D representation (Fig. 5c and d). The capacitance magnitude and shape in 2[Na+]:1[Zn2+] electrolyte is similar to that in 1 M ZnSO4, but with additional capacitance captured between 1.0–1.2 V at ∼10–30 mHz. The best combination of capacitive contributions occurs with the 6[Na+]:1[Zn2+] electrolyte, where the full pseudocapacitance response of the MnOx is restored, while continuing to express capacitance characteristic of potential-dependent Zn2+-supported reactions at >1.4 V (contrast Fig. 5a with Fig. 5d). Individual 3D Bode plots at different viewing angles are illustrated for the series of electrolytes in Fig. S11.† The native CNF exhibits a near-constant, but significantly lower, capacitance over this same potential range in 3D Bode plots that extends to higher frequencies as expected for double-layer capacitance mechanisms (Fig. S8†).
3.3. Assessing two-electrode zinc-ion cells
Following the half-cell electroanalytical characterization of MnOx@CNF electrodes, we evaluated oxide-affixed architecture as a positive electrode vs. zinc foil in a two-terminal Swagelok cell, a configuration that should reflect practical device performance. For cell-level testing, we down-selected two electrolyte compositions: 1 M ZnSO4 and the mixed 6[Na+]:1[Zn2+] electrolyte. Cells were subjected to a galvanostatic cycling sequence comprising increasing rates from 1C–20C (ten discharge–charge cycles at each rate), followed by the same sequence in reverse. At the initial imposed 1C rate (normalized to the MnOx mass in the CNF and the one-electron theoretical capacity of MnO2, 308 mA h g−1), galvanostatic cycling in 1 M ZnSO4 shows a plateau-like feature (1.25–1.50 V; Fig. 6a) superimposed on a sloping discharge profile, ultimately reaching a near-theoretical discharge capacity of ∼300 mA h gMnOx−1. Upon charging at 1C, full capacity is recovered, while charge–discharge voltage hysteresis averages a competitive 300 mV. As the C-rate increases to 2C in this cycling sequence, capacity falls by nearly two-thirds.
 | ||
| Fig. 6 Galvanostatic charge–discharge curves (10th cycle) at 1C–20C depicted for MnOx@CNF-zinc cells. Electrolytes were (a) 0[Na+]:1[Zn2+] (1C charge–discharge curve taken for the 2nd cycle) and (b) 2[Na+]:1[Zn2+] electrolyte with corresponding rate capability trends presented as specific capacity vs. cycle number in (c) 0[Na+]:1[Zn2+] and (d) 2[Na+]:1[Zn2+]. | ||
With the mixed 6[Na+]:1[Zn2+] electrolyte, we observe sloping voltage profiles on both charge and discharge (Fig. 6b), a trend that is reminiscent of pseudocapacitive processes.38 At a 1C rate, charge and discharge capacity is 305 mA h gMnOx−1, comparable to that observed in 1 M ZnSO4, but with the additional advantages of higher average discharge voltage (∼1.6 V) (Fig. S12†) and lower voltage hysteresis (∼200 mV). Even though the 6[Na+]:1[Zn2+] electrolyte is four-fold lower in Zn2+-concentration, this mixed electrolyte supports higher capacities in the face of increasing C-rate. For example, specific capacity at 2C is 260 mA h gMnOx−1; at 10C, capacity is 140 mA h gMnOx−1, which is three-fold higher than that observed in 1 M ZnSO4. Thus, at increasing rates, where insertion becomes limiting, we posit that both Zn2+-insertion and Na2SO4-based pseudocapacitance are operative in maintaining high capacity.
Capacity vs. rate trends (Fig. 6c and d) for the entire sequence demonstrate that the mixed electrolyte sustains enhanced electrochemical performance at all C-rates. At 1C in 1 M ZnSO4, MnOx capacity begins to fall in successive cycles even at this moderately slow rate whereas capacity is relatively constant over multiple cycles at 1C in the 6[Na+]:1[Zn2+] electrolyte. In the reverse sequence, from 20C back to 1C, capacity recovers only to ∼125 mA h gMnOx−1 in 1 M ZnSO4. In the mixed electrolyte, specific capacity shows good recovery back to 250 mA h gMnOx−1 at the end of the C-rate sequence.
In addition to trends in electrode capacity with increasing rate, we also accounted for differences in discharge voltage and charge–discharge voltage hysteresis by calculating specific energy, normalized only to the MnOx mass in the CNF (Wh kgMnOx−1), as well as the round-trip energy efficiency. Note that these MnOx-specific energy values are reported only for contrasting the behavior of the MnOx@CNF electrodes in the two electrolytes, and are not intended to reflect device-level performance energy content, which would be a factor of 5–10 lower than values in Fig. S13a and b.† With 1 M ZnSO4, MnOx-normalized specific energy rapidly falls off as the rate is increased to 20C, primarily due to the decline in accessible capacity, with lower average discharge voltage as a secondary effect. Upon a sequential return from 20C to 1C, specific energy is significantly lower than in the initial cycles. Greater voltage hysteresis with increasing C-rates also reduces the round-trip energy efficiency in 1 M ZnSO4, from 75% down to 64% (Fig. S13a and b†).
The MnOx@CNF electrodes cycled in the mixed 6[Na+]:1[Zn2+] electrolyte show superior performance in terms of MnOx-normalized specific energy and energy efficiency. At 5C, MnOx@CNF achieves a specific energy of ∼250 W h kgMnOx−1 (discharge) and an energy efficiency of ∼83%, whereas equivalent tests yield 75 W h kgMnOx−1 (discharge) and an energy efficiency of ∼75% with 1 M ZnSO4. This nearly 3× improvement in MnOx-normalized specific energy is maintained even at high rates (10–20C); energy efficiency is also sustained at >80% at all rates with the mixed electrolyte (Fig. S13a and b†).
In separate experiments, pristine MnOx@CNF electrodes were cycled at the challenging 10C rate to assess long-term stability. Although the MnOx@CNF electrode exhibits modestly higher initial capacity in 1 M ZnSO4 at this rate, capacity continuously declines thereafter, falling from 190 to 60 mA h gMnOx−1 after 1000 cycles (Fig. 7 and S14†). When the MnOx@CNF electrode is cycled in the 6[Na+]:1[Zn2+] electrolyte, the loss of capacity is much less severe, with 90 mA h gMnOx−1 still available after 1000 cycles. Future studies will focus on improving the cyclability of the MnOx@CNF electrode, for example by the use of electrolyte additives that suppress Mn dissolution,13,14 in conjunction with the improved performance obtainable in the 6[Na+]:1[Zn2+] electrolyte.
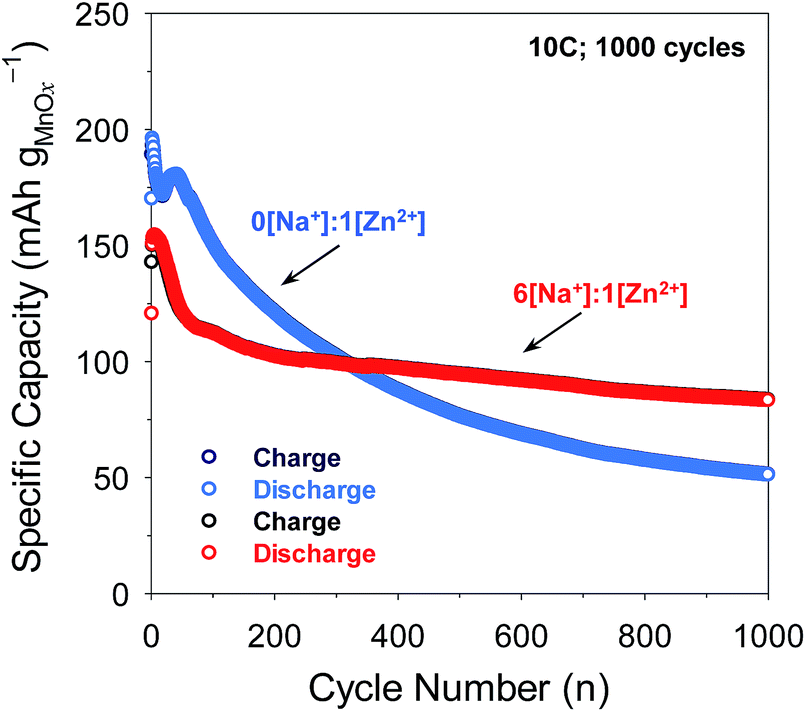 | ||
| Fig. 7 Comparison of long-term cycling at 10C for 1000 cycles of MnOx@CNF electrode compared in 0[Na+]:1[Zn2+] vs. 6[Na+]:1[Zn2+]. | ||
Conclusions
The results reported herein highlight the ability to combine characteristics of high power in the form of Na2SO4-supported pseudocapacitance and high energy from battery-like Zn2+-insertion/de-insertion processes by employing a MnOx@CNF electrode architecture and a mixed Na+:Zn2+ aqueous electrolyte. The relative contributions of these physically distinct charge-storage processes were successfully assessed via voltammetric scan-rate dependence and impedance analysis, while practical performance improvements were demonstrated in two-electrode cell configurations with zinc foil anodes. Using an electrode material that can alternately express capacitor- and battery-like behaviour creates new opportunities to design energy-storage devices that deliver optimal combinations of energy and power performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the financial support from the U.S. Office of Naval Research. J. S. K. is an NRL-NRC Postdoctoral Associate (2016–2018).References
- Y. Chabre and J. Pannetier, Structural and electrochemical properties of the proton/γ-MnO2 system, Prog. Solid State Chem., 1995, 23, 1–130 CrossRef CAS.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Lithium insertion into manganese spinels, Mater. Res. Bull., 1983, 18, 461–472 CrossRef CAS.
- M. M. Thackeray, M. F. Mansuetto and J. B. Bates, Structural stability of LiMn2O4 electrodes for lithium batteries, J. Power Sources, 1997, 25, 153–158 CrossRef.
- H. Y. Lee and J. B. Goodenough, Supercapacitor behavior with KCl electrolyte, J. Solid State Chem., 1999, 144, 220–223 CrossRef CAS.
- D. Bélanger, T. Brousse and J. W. Long, Manganese oxides: Battery materials make the leap to electrochemical capacitors, ECS Interface, 2008, 17, 49–52 Search PubMed.
- C. Xu, F. Kang, B. Li and H. Du, Recent progress on manganese dioxide based supercapacitors, J. Mater. Res., 2010, 25, 1421–1432 CrossRef CAS.
- C. Yuan, Y. Zhang, Y. Pan, X. Liu, G. Wang and D. Cao, Investigation of the intercalation of polyvalent cations (Mg2+, Zn2+) into λ-MnO2 for rechargeable aqueous battery, Electrochim. Acta, 2013, 116, 404–412 CrossRef.
- C. Xu, B. Li, H. Du and F. Kang, Energetic zinc ion chemistry: the rechargeable zinc ion battery, Angew. Chem., Int. Ed., 2012, 41, 933–935 CrossRef PubMed.
- J. Lee, J. B. Ju, W. I. Cho, B. W. Cho and S. H. Oh, Todorokite-type MnO2 as a zinc-ion intercalating material, Electrochim. Acta, 2013, 112, 138–143 CrossRef CAS.
- B. Lee, C. S. Yoon, H. R. Lee, K. Y. Chung, B. W. Cho and S. H. Oh, Electrochemically-induced reversible transition from the tunneled to layered polymorphs of manganese dioxide, Sci. Rep., 2014, 4, 6066 CrossRef CAS PubMed.
- B. Lee, H. R. Lee, H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, Elucidating the intercalation mechanism of zinc ions into α-MnO2 for rechargeable zinc batteries, Chem. Commun., 2015, 51, 9265–9268 RSC.
- C. Xu, Y. Chen, S. Shi, J. Li, F. Kang and D. Su, Secondary batteries with multivalent ions for energy storage, Sci. Rep., 2015, 5, 14120 CrossRef CAS PubMed.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, A layered δ-MnO2 nanoflake cathode with high zinc-storage capacities for eco-friendly battery applications, Electrochem. Commun., 2015, 50, 121–125 CrossRef.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Reversible aqueous zinc/manganese oxide energy storage from conversion reactions, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- X. Wu, Y. Li, Y. Xiang, Z. Liu, Z. He, X. Wu, Y. Li, L. Xiong, C. Li and J. Chen, The electrochemical performance of aqueous rechargeable battery of Zn/Na0.44MnO2 based on hybrid electrolyte, J. Power Sources, 2016, 336, 35–39 CrossRef CAS.
- S. Bai, J. Song, Y. Wen, J. Cheng, G. Cao, Y. Yang and D. Li, Effects of zinc and manganese ions in aqueous electrolytes on structure and electrochemical performance of Na0.44MnO2 cathode material, RSC Adv., 2016, 6, 40793–40798 RSC.
- B. Jiang, C. Xu, C. Wu, L. Dong, J. Li and F. Kang, Manganese sesquioxide as cathode material for multivalent zinc ion battery with high capacity and long cycle life, Electrochim. Acta, 2017, 229, 422–428 CrossRef CAS.
- W. Qiu, Y. Li, A. You, Z. Zhang, G. Li, X. Lu and Y. Tong, High-performance flexible quasi-solid-state Zn-MnO2 battery based on MnO2 nanorod arrays coated 3D porous nitrogen-doped carbon cloth, J. Mater. Chem. A, 2017, 5, 14838 CAS.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, Zn/MnO2 battery chemistry with H+ and Zn2+ coinsertion, J. Am. Chem. Soc., 2017, 29, 9775–9778 CrossRef PubMed.
- A. E. Fischer, K. A. Pettigrew, D. R. Rolison, R. M. Stroud and J. W. Long, Incorporation of homogeneous, nanoscale MnO2 within ultraporous carbon structures via self-limiting electroless deposition: Implications for electrochemical capacitors, Nano Lett., 2007, 7, 281–286 CrossRef CAS PubMed.
- A. E. Fischer, M. P. Saunders, K. A. Pettigrew, D. R. Rolison and J. W. Long, Electroless deposition of nanoscale MnO2 on ultraporous carbon nanoarchitectures: Correlation of evolving pore-solid structure and electrochemical performance, J. Electrochem. Soc., 2008, 155, A246–A252 CrossRef CAS.
- M. B. Sassin, C. P. Hoag, B. T. Willis, N. W. Kucko, D. R. Rolison and J. W. Long, Designing high-performance electrochemical energy-storage nanoarchitectures to balance rate and capacity, Nanoscale, 2013, 5, 1649–1657 RSC.
- J. W. Long, M. B. Sassin, A. E. Fischer, A. N. Mansour, V. S. Johnson, P. E. Stallworth, S. G. Greenbaum and D. R. Rolison, Multifunctional MnO2−carbon nanoarchitectures exhibit battery and capacitor characteristics in alkaline electrolytes, J. Phys. Chem. C, 2009, 113, 17595–17598 CAS.
- M. B. Sassin, S. G. Greenbaum, P. E. Stallworth, A. N. Mansour, B. P. Hahn, K. A. Pettigrew, D. R. Rolison and J. W. Long, Achieving electrochemical capacitor functionality from nanoscale LiMn2O4 coatings on 3-D carbon nanoarchitectures, J. Mater. Chem. A, 2013, 1, 2431–2440 CAS.
- M. D. Donakowski, J. M. Wallace, M. B. Sassin, K. W. Chapman, J. F. Parker, J. W. Long and D. R. Rolison, Aerogel architectures boost oxygen-evolution performance of NiFe2Ox spinels to activities commensurate with nickel-rich oxides, CrystEngComm, 2016, 18, 6035–6048 RSC.
- J. C. Lytle, J. M. Wallace, M. B. Sassin, A. J. Barrow, J. W. Long, J. L. Dysart, C. H. Renninger, M. P. Saunders, N. L. Brandell and D. R. Rolison, The right kind of interior for multifunctional electrode architectures: Carbon nanofoam papers with aperiodic submicrometre pore networks interconnected in 3D, Energy Environ. Sci., 2011, 4, 1913–1925 CAS.
- B. Lee, H. R. Seo, H. R. Lee, C. S. Yoon, J. H. Kim, K. Y. Chung, B. W. Cho and S. H. Oh, Critical role of pH evolution of electrolyte in the reaction mechanism for rechargeable zinc batteries, ChemSusChem, 2016, 9, 2948–2956 CrossRef CAS PubMed.
- C. A. Beasley, M. B. Sassin and J. W. Long, Extending electrochemical quartz crystal microbalance techniques to macroscale electrodes: Insights on pseudocapacitance mechanisms in MnOx-coated carbon nanofoams, J. Electrochem. Soc., 2015, 162, A5060–A5064 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Charge storage mechanism of MnO2 electrode used in aqueous electrochemical capacitor, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS.
- O. Ghodbane, J.-L. Pascal and F. Favier, Microstructural effects on charge-storage properties in MnO2-based electrochemical supercapacitors, ACS Appl. Mater. Interfaces, 2009, 1, 1130–1139 CAS.
- L. Liu, Y. Lao, W. Tan, F. Liu, S. L. Suib, Y. Zhang and G. Qiu, Zinc removal from aqueous solution using a deionization pseudocapacitor with a high-performance nanostructured birnessite electrode, Environ. Sci.: Nano, 2017, 4, 811–823 RSC.
- F. Yin, Z. Liu, S. Yang, Z. Shan, Y. Zhao, Y. Feng, C. Zhang and Z. Bakenov, Na4Mn9O18/carbon nanotube composite as a high electrochemical performance material for aqueous sodium-ion batteries, Nanoscale Res. Lett., 2017, 12, 569 CrossRef PubMed.
- H. Lindström, S. Södergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt and S.-E. Lindquist, Li+ ion insertion in TiO2 (anatase). 2. Voltammetry on nanoporous films, J. Phys. Chem. B, 1997, 101, 7717–7722 CrossRef.
- K. Aoki, K. Tokuda and H. Matsuda, Theory of linear sweep voltammetry with finite diffusion space, J. Electroanal. Chem., 1983, 146, 417–424 CrossRef CAS.
- K. Aoki, K. Tokuda and H. Matsuda, Theory of linear sweep voltammetry with finite diffusion part II. Totally irreversible and quasi-reversible cases, J. Electroanal. Chem., 1984, 160, 33–45 CrossRef CAS.
- L. Bai and B. E. Conway, AC impedance of faradaic reactions involving electrosorbed intermediates: Examination of conditions leading to pseudoinductive behavior represented in three-dimensional impedance spectroscopy diagrams, J. Electrochem. Soc., 1991, 138, 2897–2907 CrossRef CAS.
- L. Bai and B. E. Conway, Three-dimensional impedance spectroscopy diagrams for processes involving electrosorbed intermediates, introducing the third electrode-potential variable—Examination of conditions leading to pseudo-inductive behavior, Electrochim. Acta, 1993, 38, 1803–1815 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Pseudocapacitive oxide materials for high-rate electrochemical energy storage, Energy Environ. Sci., 2014, 7, 1597–1614 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00540g |
| This journal is © The Royal Society of Chemistry 2018 |