Photoelectrochemical studies on earth abundant pentanickel polyoxometalates as co-catalysts for solar water oxidation†
Received
26th October 2017
, Accepted 20th January 2018
First published on 22nd January 2018
Abstract
The present contribution reports results from photoelectrochemical studies on anatase TiO2 photoanodes for solar water oxidation in the presence of an earth abundant homogeneous pentanickel silicotungstate molecular water oxidation catalyst K10H2[Ni5(OH)6(OH2)3(Si2W18O66)]·34H2O (Ni5-POM). Highly robust anatase TiO2 thin films were prepared in situ on fluorine-doped tin oxide glass (F![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SnO2 or FTO) by spin coating and thermal annealing. The as-prepared films were characterized by scanning electron microscopy, energy dispersive X-ray analysis, and UV-vis spectroscopy. Under simulated solar irradiation, a maximum photocurrent of 0.20 mA cm−2 was obtained from FTO/TiO2 photoanodes in the presence of 20 μM Ni5-POM-0.050 M pH 9.0 borate buffer electrolyte at 0.70 V vs. Ag/AgCl (3.0 M KCl), which was a 2.3 times increase in photocurrent as compared to that without using the Ni5-POM catalyst. The use of the catalyst also significantly shifted the onset potential of water oxidation to a much negative potential value with respect to −0.2 V vs. Ag/AgCl. As verified by electrochemical impedance spectroscopy, the above results could be attributed to the effect of the catalyst on decreasing electron–hole recombination and facilitating the hole utilization rate on the semiconductor TiO2–electrolyte interface upon light illumination. Dissolved oxygen produced from the FTO/TiO2–Ni5-POM system was detected with an oxygen sensor. This is the first report on the use of the homogeneous water oxidation catalyst Ni5-POM as a functional co-catalyst for photoelectrochemical water oxidation with semiconductor photoanodes.
:SnO2 or FTO) by spin coating and thermal annealing. The as-prepared films were characterized by scanning electron microscopy, energy dispersive X-ray analysis, and UV-vis spectroscopy. Under simulated solar irradiation, a maximum photocurrent of 0.20 mA cm−2 was obtained from FTO/TiO2 photoanodes in the presence of 20 μM Ni5-POM-0.050 M pH 9.0 borate buffer electrolyte at 0.70 V vs. Ag/AgCl (3.0 M KCl), which was a 2.3 times increase in photocurrent as compared to that without using the Ni5-POM catalyst. The use of the catalyst also significantly shifted the onset potential of water oxidation to a much negative potential value with respect to −0.2 V vs. Ag/AgCl. As verified by electrochemical impedance spectroscopy, the above results could be attributed to the effect of the catalyst on decreasing electron–hole recombination and facilitating the hole utilization rate on the semiconductor TiO2–electrolyte interface upon light illumination. Dissolved oxygen produced from the FTO/TiO2–Ni5-POM system was detected with an oxygen sensor. This is the first report on the use of the homogeneous water oxidation catalyst Ni5-POM as a functional co-catalyst for photoelectrochemical water oxidation with semiconductor photoanodes.
1. Introduction
The development of viable systems for harnessing energy from renewable sources has been an important scientific pursuit in recent years.1,2 Solar to fuel conversion is an efficient strategy to capture solar energy directly in the form of fuel. Photoelectrochemical (PEC) water splitting offers a safe, localized, and low temperature approach for production of hydrogen and oxygen from water, and has an enormous potential for facile integration with hydrogen fuel cells in the near future.1,3 This approach could eventually result in effective water electrolysis with sunlight in the absence of any external potential bias.
A wide spectrum of research has been carried out in the development of semiconductor based photoelectrodes for PEC water splitting and has also been documented in the literature.1,2,4–7 However, the high energy barrier required for water oxidation (1.23 V vs. SHE under standard conditions5), in addition to losses from ineffective light absorption and separation of photogenerated electrons and holes poses severe limitations on the ability of single absorber based solar water splitting systems under unbiased or low biased conditions.7,8 The prohibitive cost of these noble metal based catalysts necessitates the need for the development of alternate earth abundant non-noble metal based catalysts for efficient solar to hydrogen conversion by water splitting.8–10
Catalysts developed for PEC water oxidation processes must be capable of effectively capturing photogenerated holes and facilitating low energy pathways for oxidation of water, so as to increase the observed photocurrent while reducing the onset potential.8–11 In the last few years, there has been considerable research progress on transition metal based solution-phase12–15 and solid-state3,8–10,14 water oxidation catalysts, most of which have been used in photocatalytic8,16–24 or electrocatalytic9,13,25,26 water splitting devices. However, electrocatalytic water splitting still demands the application of significant external bias, and the use of photocatalytic methods for large scale device applications comes with engineering constraints for O2/H2 gas separation. PEC water splitting with the above-mentioned transition metal based catalysts could provide an efficient, localized system to split water and produce high purity O2/H2 under unbiased or low applied bias conditions.
Present research focuses upon the catalytic applications of transition metal substituted polyoxometalates (POMs).12,20,27–29 This is due to their ability for facile reversible multi-electron transport kinetics within the POM architecture. Moreover, POM anions are capable of stabilizing active catalytic sites in higher oxidation states, which is an essential requirement for efficient water oxidation and reduction.21,23,29–31 POMs are also able to coordinate multi-metal cores, which effectively increases the number of active sites for water oxidation and reduction catalysis. Previously, Ru,17,18,21,22,26 Co,16,19,24,25,32–34 and Ni35 based POMs have been employed for photochemical18–23,35 and electrocatalytic25,26 water oxidation. Recently, Hill et al. reported the use of dye sensitized TiO2 photoanodes treated with Ru based POMs for water oxidation, focusing on the effect of sensitizer design on the performance.36
The use of water oxidation catalysts in solution offers a variety of advantages for PEC applications, including fast reaction kinetics, low electric resistance, and facile photoelectrode fabrication. In the current study, we investigate the use of K10H2[Ni5(OH)6(OH2)3(Si2W18O66)]·34H2O (Ni5-POM) as an earth abundant water oxidation catalyst in solution for PEC water splitting using spin-coated TiO2 photoanodes. Ni5-POM has been reported to be a stable and active homogeneous water oxidation catalyst.35 The penta-nickel unit has multiple bridging oxo groups that act as acceptor bases for the water oxidation mechanism, and also stabilize the metal centre in higher oxidation states. This cluster of oxo-bridged multiple d-electrons resembles the structure of Mn4CaO4,37 the active oxygen evolution centre in Photosystem II.38 As it is carbon free, the complex is oxidatively stable within the applied operating potential range.35 Additionally, Ni5-POM is earth abundant, and has shown to be more active toward water oxidation than other Ni based polyoxometalates.39 In this study, we report for the first time that Ni5-POM can be a suitable co-catalyst in PEC water oxidation with TiO2 photoanodes.
2. Materials and methods
2.1 Chemicals
Unless otherwise stated, all chemicals were purchased from Aldrich and used as received. Titanium(IV) butoxide (97%) and hydrogen peroxide (50 wt% in H2O) were used in the preparation process for TiO2 films. Sodium metasilicate (>99%), nickel chloride hexahydrate (>99%), sodium tungstate dihydrate (>99%), sodium carbonate (>99%), and potassium chloride (Fischer, ACS grade) were the chief salts used for the preparation of the Ni5-POM catalyst. De-ionized water was used when performing all experiments, and all instruments were calibrated in accordance with accepted standards.
2.2 Preparation of FTO/TiO2 and Ni5-POM
The FTO/TiO2 photoanode and Ni5-POM catalyst were prepared according to the literature40 with some modifications. TiO2 thin films were prepared by the spin coating technique from a precursor solution consisting of titanium(IV) butoxide and H2O2 in a 1:3 volume ratio, and water–ethanol mixture was the solvent. Typically, 100 μL of 97% titanium(IV) butoxide and 300 μL of 50% H2O2 were added to 5.00 mL of solvent containing equal volumes of water and ethanol, followed by ultrasonic dispersion for 2 h. Cleaning of FTO substrates was done by sonication in deionized water, methanol, acetone, isopropanol, and deionized water, each for 15 min, respectively. Layers of dispersed precursor solution were spin coated onto a 1 cm2 active area of the dry FTO substrate by spinning 50 μL of the freshly prepared solution at 2000 rpm three times. This was followed by sintering at 773 K for 2 h and gradual cooling to produce the TiO2 photoelectrode for study. Synthesis of the Ni5-POM catalyst was done in accordance with procedures reported previously.35 The synthesized Ni5-POM was first characterized by FT-IR spectroscopy. A majority of principle peaks in the FT-IR spectrum, which was taken with a Nicolet Nexus 470 FT-IR spectrometer, matched closely with those shown in the previous report35 (see Fig. S1 in the ESI† for details). The slight shifts in FT-IR peak positions could be due to the fact that two different FT-IR instruments were used, where the FT-IR spectra in the literature were recorded with a KBr pellet method.35 Electrospray mass spectrometric analysis, which was carried out with a Thermo-Fischer LXQ ESI-ion trap mass spectrometer (Waltham, MA, USA), was used to further confirm the composition of the synthesized Ni5-POM. As revealed in Fig. S2 and Table S1 in the ESI,† a number of observed mass/charge (m/z) peaks, such as 194.75 (Fig. S2a†), 623.17 (Fig. S2b†), 718.00 (Fig. S2c†), 825.46 (Fig. S2d†), 1286.08 (Fig. S2e†), and 1664.67 (Fig. S2f†), are consistent with the POM related fragments, [K(H2O)4W18O66]23−, [K3HX]8−, [K4HX]7−, [K2H4X]6, [K7HX]4−, and [K3H6X]3−, respectively, where X = [Ni5(OH)6(OH2)3(Si2W18O66)].
2.3 Physico-chemical characterization of the FTO/TiO2 photoanode
A Zeiss Sigma VP FEG scanning electron microscope (SEM) was used for the SEM images of TiO2 photoelectrodes on the FTO substrate. The UV-vis spectrum of TiO2 photoelectrodes was recorded using an Evolution 300 Thermo-Scientific spectrophotometer.
2.4 Photoelectrochemical studies
PEC measurements were carried out with a three-electrode electrochemical setup using a 50 mL quartz beaker as the cell, Ag/AgCl (3.0 M KCl) as the reference electrode, a Pt wire as the counter electrode, and FTO/TiO2 (1 cm2) as the working electrode, respectively. Electrochemical impedance spectroscopic (EIS) measurements were carried out at open circuit DC potential with a 5 mV super-imposed AC signal. For unbiased potential studies, a two-electrode setup was used with a Pt wire as the reference/counter electrode. Experiments were performed in a 0.050 M pH 9.0 borate buffer solution at a scan rate of 0.020 V s−1, with and without the presence of 20 μM Ni5-POM catalyst. All electrochemical studies were performed using a CH instruments model 660A electrochemical workstation. A 150 W Xe lamp solar simulator (ABET technologies) with an AM 1.5 G filter providing a light intensity of 1000 W m−2 (1 sun) was used as the visible light source for PEC studies. Oxygen detection was done using dissolved oxygen measurements with an oxygen sensor (Hanna Instruments, Model HI9164).
3. Results and discussion
3.1 Physico-chemical characterization
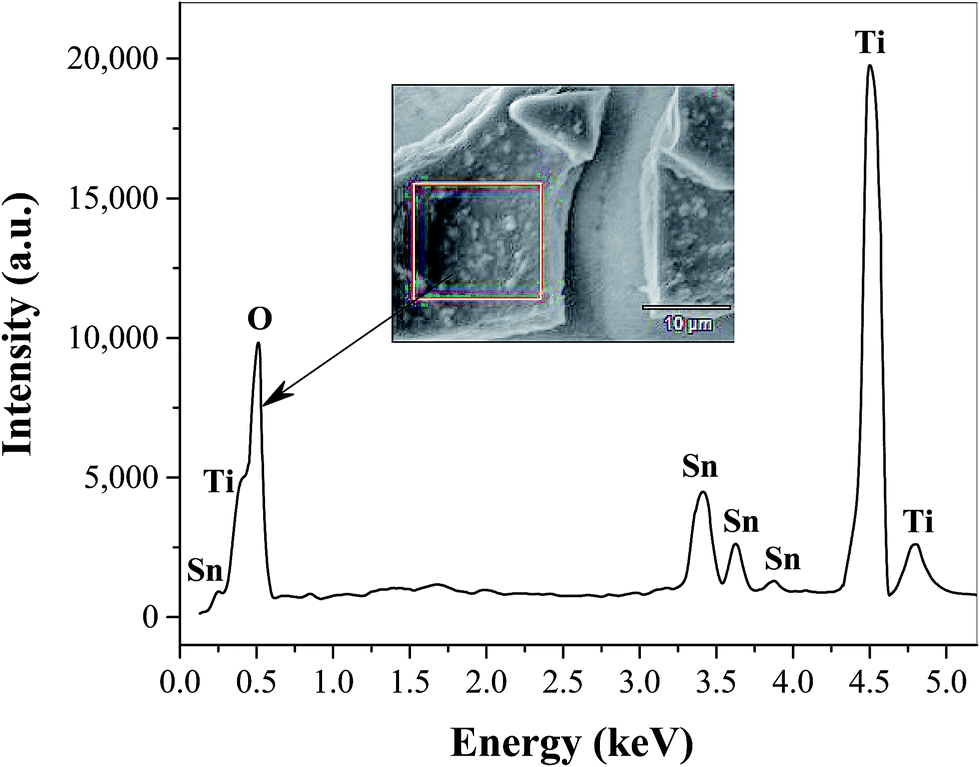
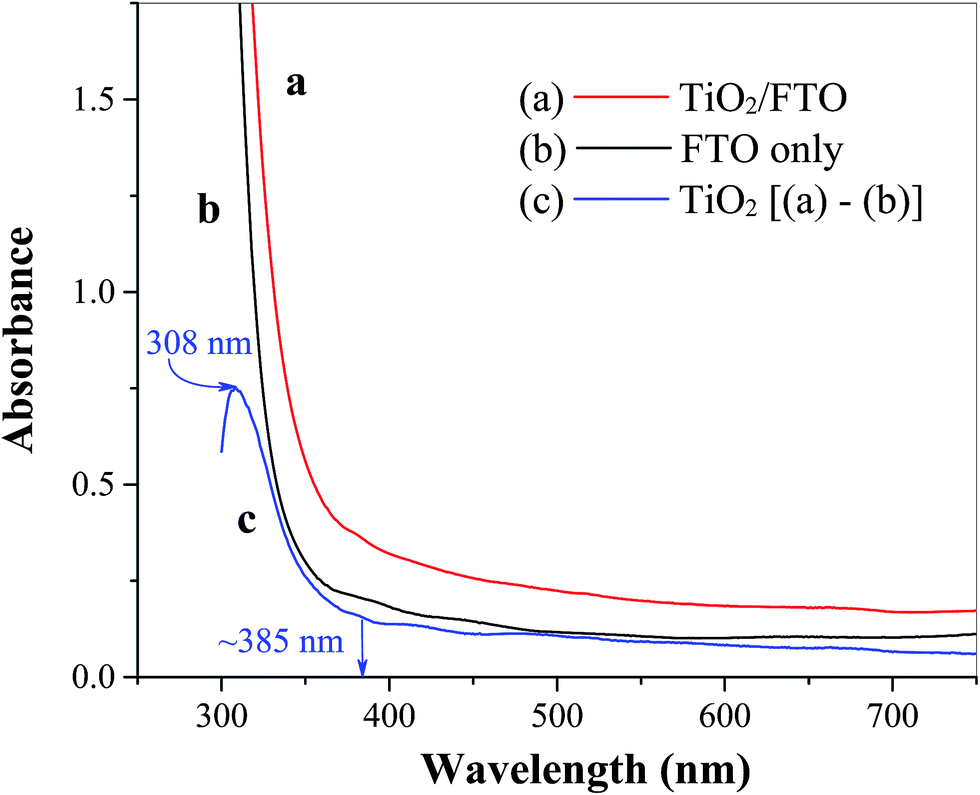
The surface morphology of TiO2 film coated FTO is shown in Fig. 1 at different magnifications, where even distribution of TiO2 particles in the form of micro-sized flakes (∼20 × 30 μm) is evident (Fig. 1a). The thickness of the cracked film can be estimated on the basis of SEM images at higher resolutions (Fig. 1b and c), which show an approximate value of 2–3 μm. These cracks are most likely formed during the annealing process probably due to the mismatch of the thermal expansion coefficients of the coating and FTO substrate, as observed previously for TiO2 on ITO41 and steel substrates.42 This as-prepared FTO/TiO2 photoanode could provide a large and uniform surface area for light absorption and charge generation, facilitate effective interaction between the TiO2 layer and solution-phase catalyst, as well as reduce the amount of recombination hotspots that result in efficiency losses. The energy dispersive X-ray (EDX) spectrum of TiO2 micro-flakes on the FTO substrate shown in Fig. 2 displays two major peaks from Ti and O, as well as few minor peaks from Sn. These data confirm the formation of TiO2 on the FTO (i.e., F:SnO2) substrate. Also note that no EDX peaks associated with impurities or dopants are observed. Fig. 3 shows the UV-vis spectra of FTO/TiO2 (Fig. 3a), FTO substrate (Fig. 3b), and TiO2 (Fig. 3c), respectively. An onset wavelength (λ) of ∼385 nm can be estimated from Fig. 3c for TiO2 absorption, which corresponds to a band gap energy (E) of 3.22 eV as E (in eV) = 1240/λ (in nm).1 Additionally, TiO2 shows a maximum absorption at ∼308 nm as seen from previous reports.43,44 In other words, the synthesized TiO2 nanoparticles are anatase type TiO2.40,45
 |
| | Fig. 1 SEM images of spin coated TiO2 films on the FTO substrate at (a) 206×, (b) 704×, and (c) 2.35k× magnification. | |
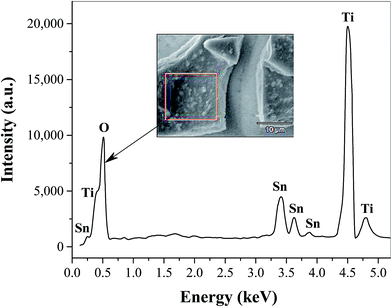 |
| | Fig. 2 Energy dispersive X-ray (EDX) spectrum of spin coated TiO2 films on the FTO substrate. The analysis was performed on the TiO2 microflake shown in the inset. | |
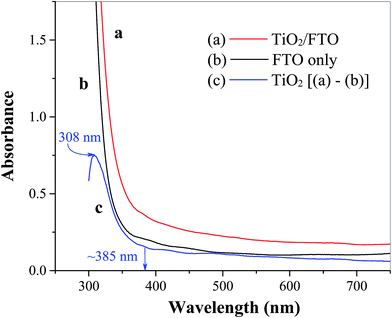 |
| | Fig. 3 UV-vis spectra of (a) spin coated TiO2 film on the FTO substrate, (b) FTO substrate only, and (c) TiO2 film obtained by subtracting (b) from (a). | |
3.2 Photoelectrochemical studies
3.2.1 Three-electrode photocurrent measurements and electrochemical impedance spectroscopy.
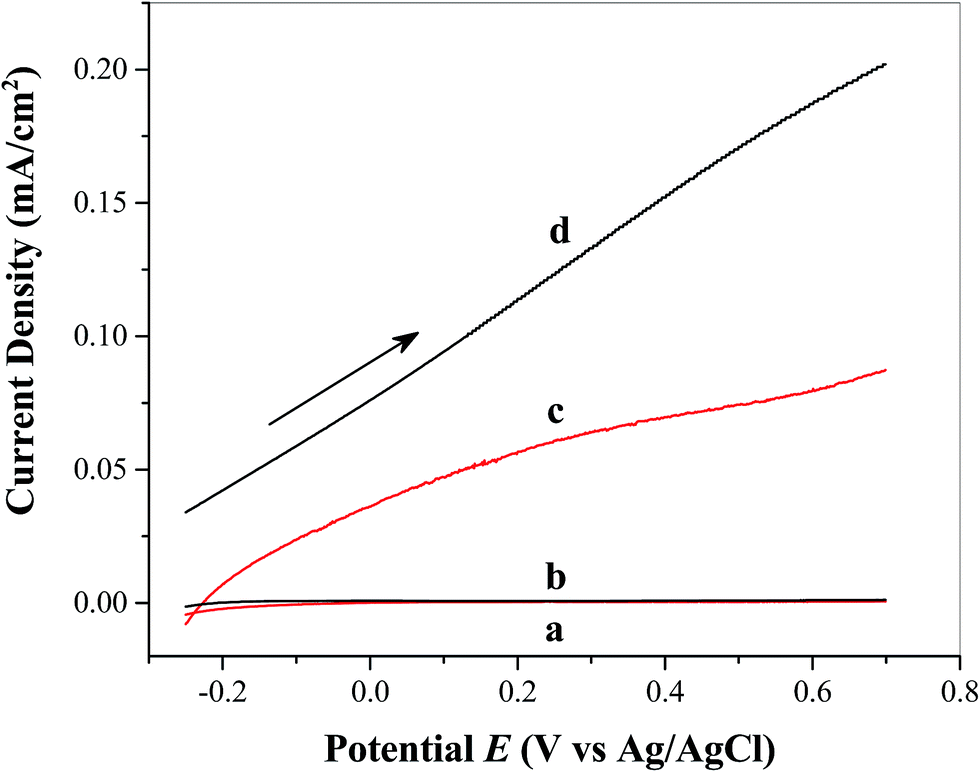
The study of photoelectrochemical properties is important for understanding of photoelectrode–catalyst interactions and determining the overall effectiveness of the catalyst in facilitating solar water oxidation. Fig. 4 shows the linear sweep voltammograms obtained from FTO/TiO2 photoanodes in the absence (Fig. 4a) and presence (Fig. 4b) of the water oxidation catalyst Ni5-POM (20 μM in 0.050 M pH 9.0 borate buffer) under dark conditions and the photoanodes in the absence (Fig. 4c) and presence (Fig. 4d) of Ni5-POM catalyst (20 μM in 0.050 M pH 9.0 borate buffer) under simulated solar radiation, respectively. Clearly, in the dark, negligible current is seen at the FTO/TiO2 electrode in the absence (Fig. 4a) or presence (Fig. 4b) of the Ni5-POM catalyst within the sweep potential window of −0.25 to +0.70 V vs. Ag/AgCl. However, the overall current density in the presence of Ni5-POM is marginally higher than that in its absence due to its electrocatalytic activity. This is better evidenced by dark current measurements on control FTO electrodes (ESI Fig. S3†), which indicate a significant increase in water oxidation current density and negative shift in onset potential in the presence of the Ni5-POM catalyst in solution. Under light irritation, a photocurrent density of 0.087 mA cm−2 at 0.70 V vs. Ag/AgCl at the FTO/TiO2 electrode is observed even in the absence of Ni5-POM in solution (Fig. 4c). This photocurrent density is increased by 2.3 times (i.e., 0.20 mA cm−2) upon the addition of 20 μM Ni5-POM to the borate buffer electrolyte (Fig. 4d). The use of Ni5-POM catalyst also significantly shifts the onset potential of water oxidation to a much negative potential value with respect to −0.2 V vs. Ag/AgCl (Fig. 4dvs.4c). The above findings suggest that under the present experimental conditions, light irradiation is a prerequisite for water oxidation and Ni5-POM can effectively facilitate the oxidation process. The increase in the observed photocurrent in the presence of the catalyst points its effectiveness in enhancing electron–hole separation by capturing holes from the conduction band of TiO2 for its mechanistic pathway to oxidize water. Moreover, the negative shift in onset potential with added Ni5-POM points at its catalytic activity for water oxidation through probably facile charge transfer kinetics and hole utilization. As listed in Table S2 in the ESI,† the obtained photocurrent, i.e., 0.20 mA cm−2 at 0.70 V vs. Ag/AgCl, is significantly larger than that produced from Ag nanoparticles,46 Co3O4,47 CdS,48 or glycerol49 modified TiO2 photoelectrode under similar experimental conditions. The present system also shows comparable photocurrent to that from a photoelectrode of TiO2 modified with a Ru(bpy)32+ derivative dye attached with a Ru(II)–POM water oxidation catalyst at ∼0 V vs. Ag/AgCl.36 Clearly, our system does not involve the use of any photosensitizers, noble metal based catalysts or sophisticated electrode processing techniques. Instead, it offers a straightforward path to water oxidation under simulated solar light under near-neutral pH conditions. Given the fact that reports on the use of POMs as co-catalysts are very limited, further research on POM based co-catalysts may offer a viable practical approach to single absorber and tandem solar water splitting.
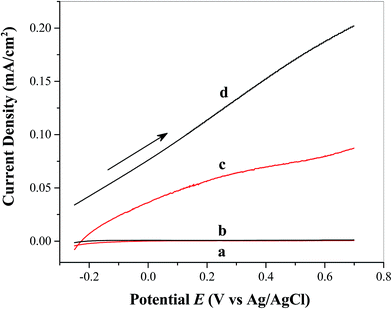 |
| | Fig. 4 Linear sweep voltammograms of FTO/TiO2 photoanodes in the (a) absence and (b) presence of the water oxidation catalyst Ni5-POM (20 μM in 0.050 M pH 9.0 borate buffer) under dark conditions and FTO/TiO2 photoanodes in the (c) absence and (d) presence of the water oxidation catalyst Ni5-POM (20 μM in 0.050 M pH 9.0 borate buffer) under simulated solar radiation at a scan rate 0.020 V s−1. | |
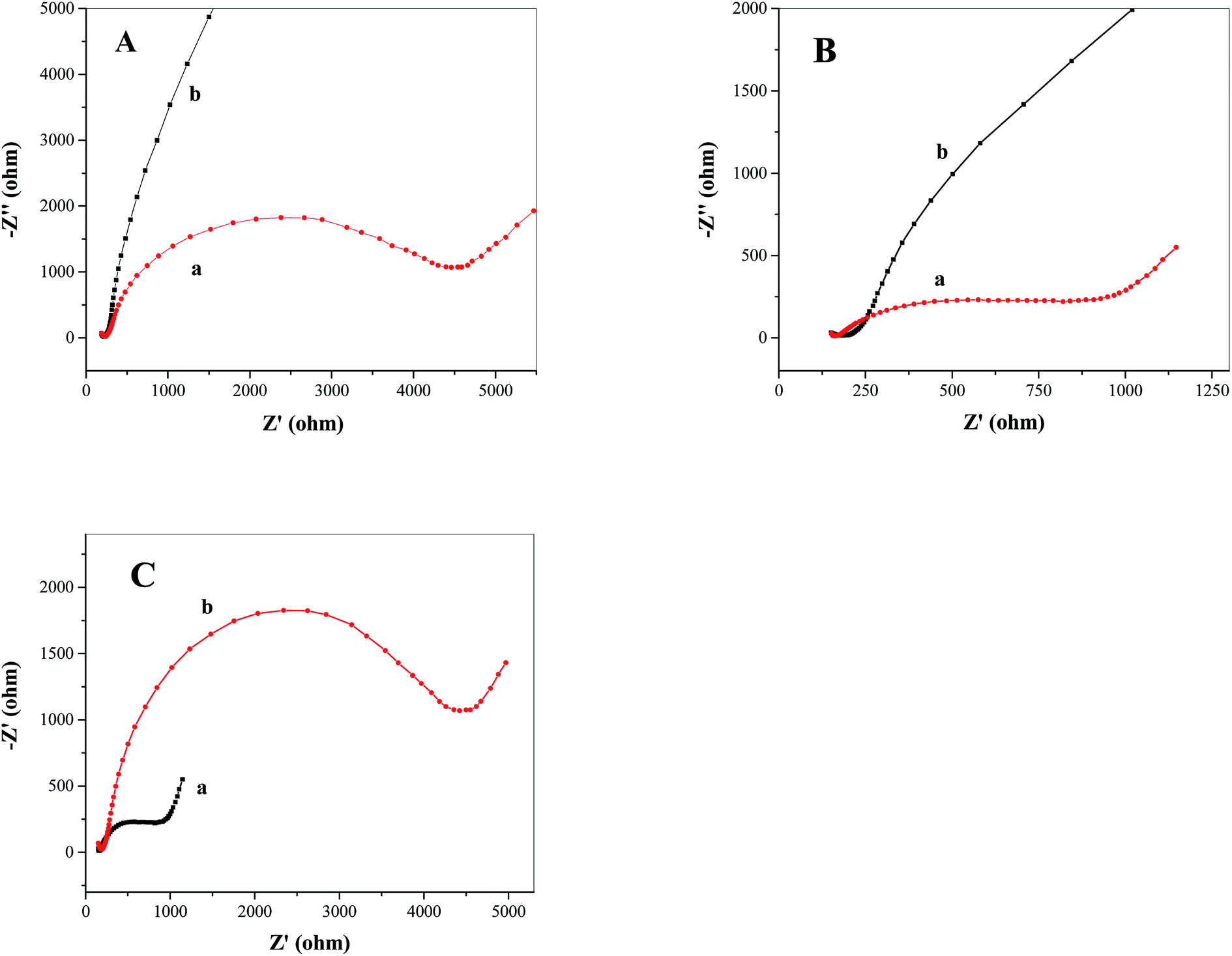
Electrochemical impedance spectroscopy (EIS) was used to verify the effect of catalyst on increasing electron–hole separation of TiO2 and on increasing the rate of photoelectrochemical water oxidation. Fig. 5A and B show the EIS spectra (Nyquist plots) obtained from FTO/TiO2 photoanodes under light (Fig. 5A(a) and B(a)) and dark (Fig. 5A(b) and B(b)) conditions in the absence (Fig. 5A) and presence (Fig. 5B) of the water oxidation catalyst Ni5-POM, respectively. As compared to the data obtained in the dark, with light irradiation, a much smaller semi-circle on the EIS spectra (Fig. 5A(a)vs.5A(b) as well as Fig. 5B(a)vs.5B(b)) corresponding to a higher double layer capacitance and a lower charge transfer resistance50–52 is observed, which confirms that the electron and hole are well separated on the FTO/TiO2 photoanode and that the charge transfer process at the electrode surface turns to be much faster in the presence of light illumination. Fig. 5C compares the Nyquist plots of FTO/TiO2 photoanodes in the presence (Fig. 5C(a)) and absence (Fig. 5C(b)) of the water oxidation catalyst Ni5-POM under light radiation. In the former case, a considerably lower charge transfer resistance and a higher double layer capacitance are evident. This observation complements data from voltammetric studies (Fig. 4), indicating increased electron–hole separation and capture of photogenerated holes for water oxidation by the catalyst. Hence, Ni5-POM acts as an efficient homogeneous co-catalyst for photoelectrochemical water oxidation. On the basis of the above observations and previously reported literature,8,11,28,29 a three-step mechanism associated with the photoelectrochemical water oxidation at the FTO/TiO2 electrode using Ni5-POM as the solution-phase catalyst is proposed as follows:
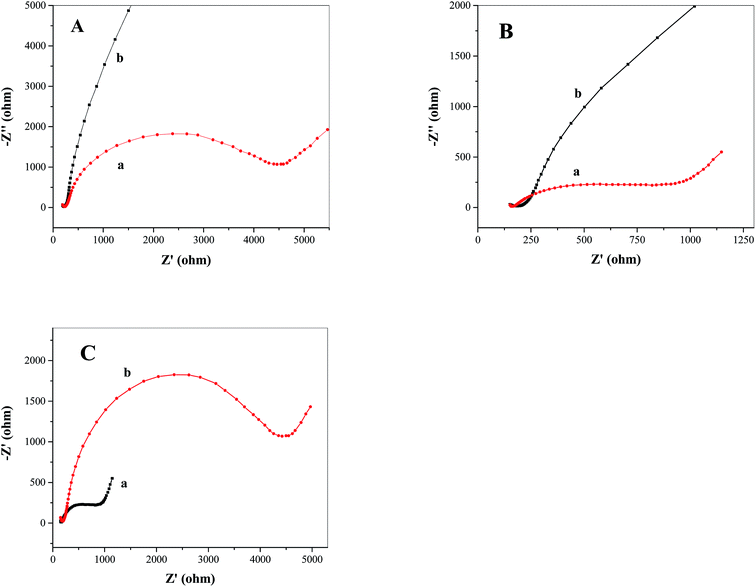 |
| | Fig. 5 Nyquist plots of FTO/TiO2 photoanodes in the (A) absence and (B) presence of the water oxidation catalyst Ni5-POM (20 μM in 0.050 M pH 9.0 borate buffer) under (a) simulated solar irradiation and (b) dark. (C) Nyquist plots of FTO/TiO2 photoanodes in the (a) presence and (b) absence of the water oxidation catalyst Ni5-POM (20 μM in 0.050 M pH 9.0 borate buffer) under simulated solar irradiation. | |
Step 1 – Absorption of light by the FTO/TiO2 photoelectrode and generation of electron–hole pairs:
Step 2 – Hole capture from TiO2 by the Ni5-POM water oxidation catalyst and redox levelling:
| | | Ni5-POM + 4h+ → Ni5-POM(4h+) | (2) |
Step 3 – Utilization of photogenerated holes for oxidation of water through catalytic pathways
| | | 2H2O + Ni5-POM(4h+) → O2 + 4H+ | (3) |
3.2.2 Unbiased two-electrode photocurrent measurements.
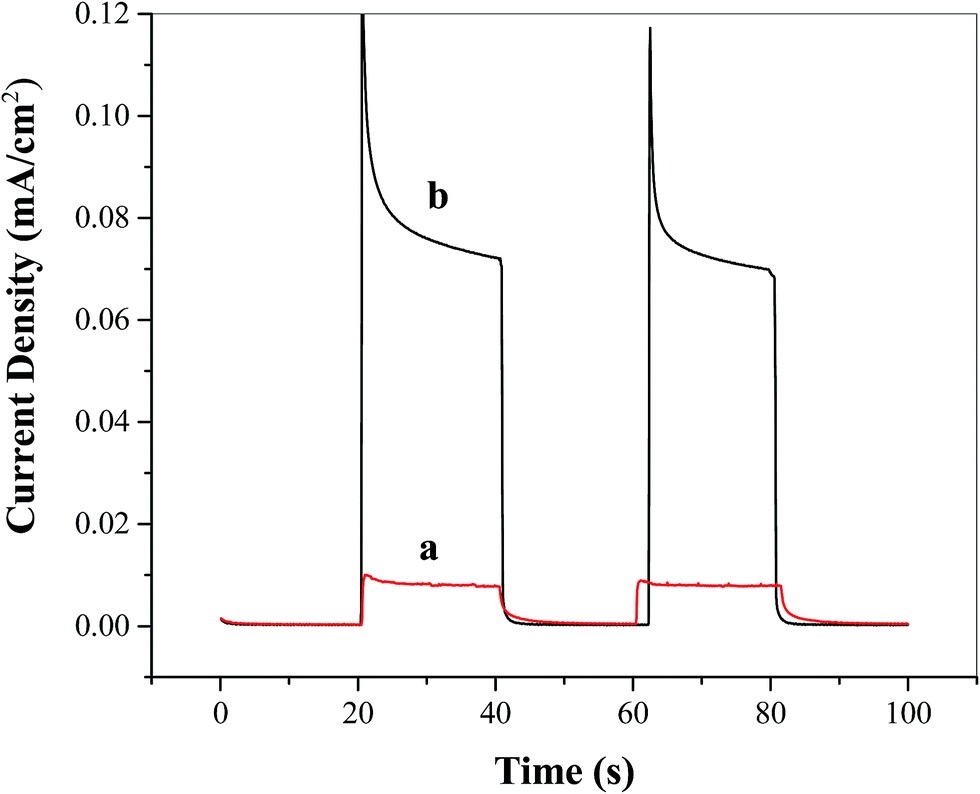
To study the effect of the Ni5-POM catalyst on facilitating overall water splitting, photocurrents were measured at zero applied bias under intermittent light with a two-electrode configuration. As shown in Fig. 6, after stabilization and decay of non-faradaic current, at the TiO2 photoelectrode a very small photocurrent of ∼0.008 mA cm−2 is observed in the absence of the catalyst (Fig. 6a), whereas an approximately 10 times increase in photocurrent (i.e., ∼0.08 mA cm−2) is obtained in the presence of 20 μM Ni5-POM water oxidation catalyst in solution (Fig. 6b). Although this unbiased photocurrent is relatively low, the present finding is significant, given the fact that TiO2 anatase has only weak solar absorption in the range of UV irradiation (see Fig. 3c) and that studies of water splitting under unbiased potential using the TiO2 photoelectrode are very limited. Hence, further investigation on the possibilities of incorporating POM based water oxidation catalysts for both single absorber and tandem photoelectrochemical water splitting processes could pave the way for the development of efficient solar to hydrogen conversion under no applied external bias.
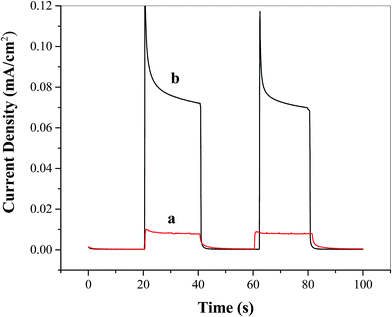 |
| | Fig. 6 Unbiased two-electrode photocurrent plots of FTO/TiO2 photoanodes under intermittent simulated solar light in the (a) absence and (b) presence of the water oxidation catalyst Ni5-POM (20 μM in 0.050 M pH 9.0 borate buffer). | |
3.2.3 Stability studies.
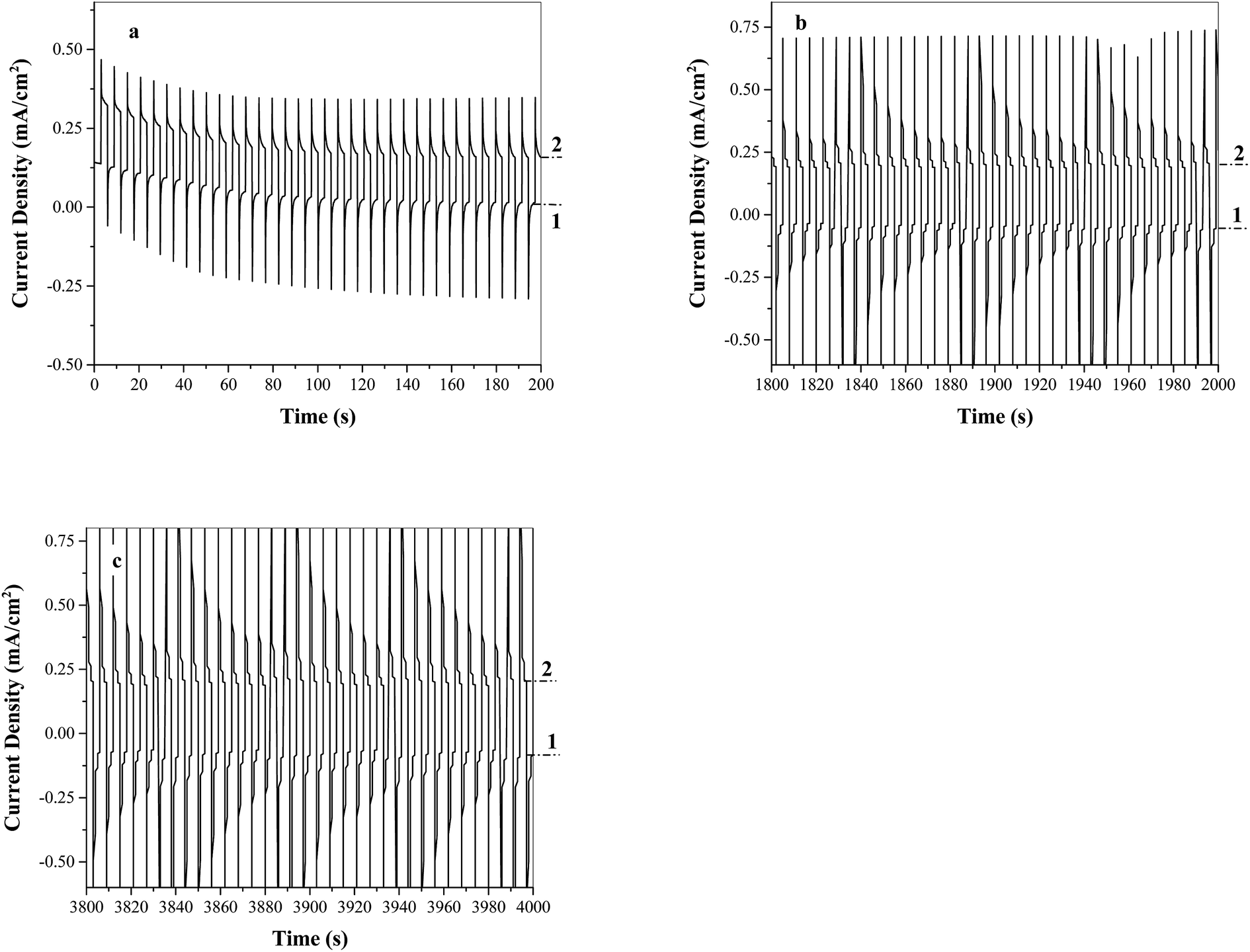
The stability of FTO/TiO2 electrodes for photoelectrochemical water oxidation in the presence of Ni5-POM was evaluated by multiple-potential step and constant potential experiments using a typical three-electrode system. The stability measurements for FTO/TiO2 under constant potential conditions are provided in the ESI (Fig. S4†). However, such a measurement may not actually reflect the stability of the electrode toward water oxidation, because the depletion of the electroactive species (i.e., OH−) near the surface of the electrode expectedly leads to an inverse time1/2 function as indicated by the Cottrell equation.53 In the presence of a co-catalyst, water oxidation becomes more effective, resulting in faster depletion of OH− hence quicker decay of the photocurrent vs. time profile. Another issue of such an experiment is that extensive O2 gas produced under constant potential accumulated on the electrode surface and inhibited further electro-oxidation of water. This is probably the reason why curves c and d in Fig. S4† are so “noisy”. Consequently, the stability study of the photoelectrode was focused on the data generated from the multi-potential step experiment, in which the applied potential was regularly shifted from 0 to 1.10 V vs. Ag/AgCl in 3 s intervals for a time period of 4000 s, and responses of photocurrent were recorded. The positive bias of 1.10 V was applied because photocurrent saturation and extensive gas evolution can be expected at this applied potential range. Hence, stability testing at 1.10 V could provide an adequate benchmark in terms of practical performance. Fig. 7a–c are the plots of photocurrent density vs. time from a FTO/TiO2 electrode under multi-potential step excitations in three different time regions, namely 0–200 s, 1800–2000 s, and 3800–4000 s, respectively. Immediately after each potential switch, an initial quick decay in photocurrent due to charging current is displayed. The photocurrent density is then gradually decreased to a relatively stable value within the entire test time window. At the end of this stability experiment, the surface of the photoelectrode exhibited the presence of minor pits owing to extensive gas evolution from the electrode surface. There was no visible appearance of film formation. On the other hand, when a step potential greater than 1.10 V was applied, a washable, grayish film of possibly nickel borate was noticed. As a result, an oxidation potential positive than 1.10 V vs. Ag/AgCl is not recommended when testing a system containing a POM based solution-phase catalyst. The present FTO/TiO2–Ni5-POM system has a low onset potential (≤−0.20 V vs. Ag/AgCl, Fig. 4c) and a high operating onset potential (∼1.10 V vs. Ag/AgCl) for water splitting, which provides a practical photoelectrolyser with a wide range of operating potential window of ∼1.3 V.
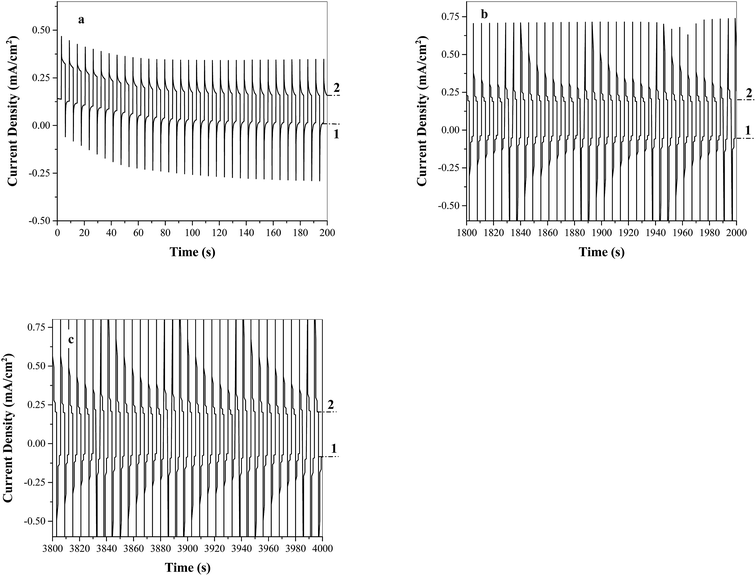 |
| | Fig. 7 Stability studies of FTO/TiO2 photoanodes under simulated solar light in the presence of the water oxidation catalyst Ni5-POM (20 μM in 0.050 M pH 9.0 borate buffer) for the time region of (a) 0–200 s, (b) 1800–2000 s, and (c) 3800–4000 s. Potentials applied at step 1: 0 V vs. Ag/AgCl and step 2: 1.10 V vs. Ag/AgCl. | |
3.2.4 Detection of oxygen.
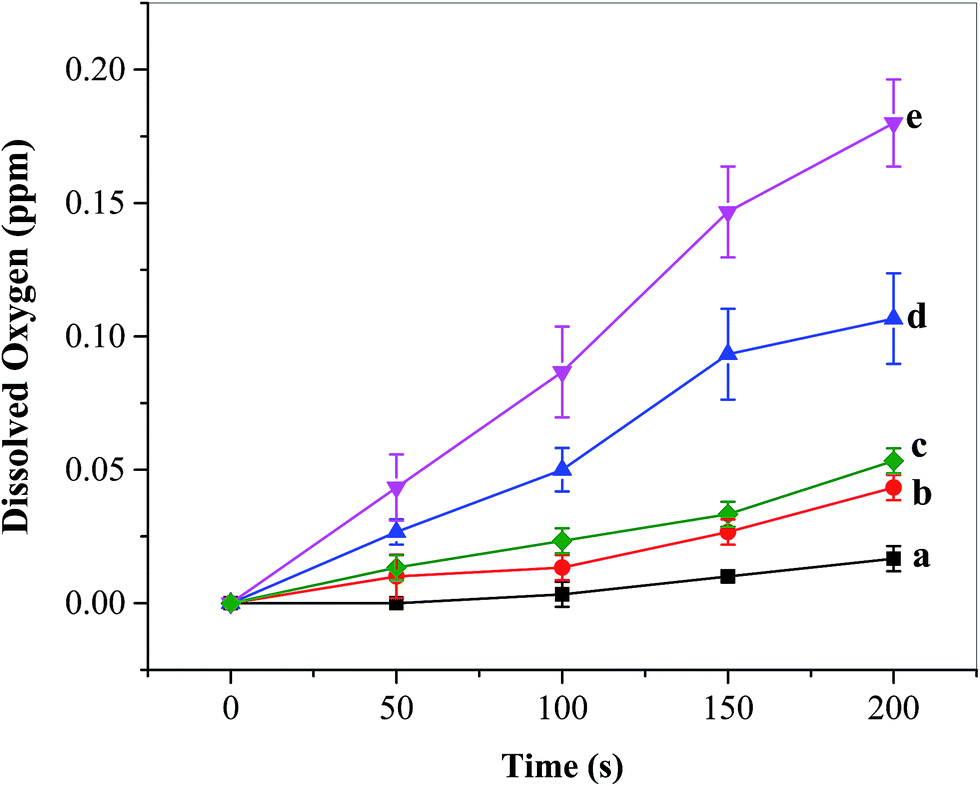
O2 detection and its concentration evaluation were carried out using a dissolved oxygen (DO) sensor during multi-potential pulse experiments as discussed in Section 3.2.3. The cell was degassed with ultrapure N2 (Airgas, Hattiesburg, MS, USA) for 15 min prior to the start of the experiment, and was placed under a N2 blanket throughout the course of measurements. As the oxygen sensor was placed near the photoelectrode, the recorded DO levels should be a reflection of instant local oxygen concentrations. It can be seen from Fig. 8 that under AM 1.5 G irradiation the DO concentration generated from the FTO/TiO2 electrode in the presence of Ni5-POM (Fig. 8e) is much higher than that in its absence (Fig. 8d). As expected, the corresponding DO concentration produced in the dark is remarkably lower than its counterpart (Fig. 8evs.8c and Fig. 8dvs.8b). Note that the oxygen produced from the present photoelectrochemical setup, for example, 0.18 ppm from the FTO/TiO2–Ni5-POM system after 200 s light irradiation (Fig. 8e), is limited as compared with the amount of DO at 100% saturation in water (8.68 ppm at 1 atm at 21 °C).54 Also note that precise quantification of O2 and the faradaic efficiency are found to be difficult due to practical constraints in our current photoelectrochemical cell setup and the charging current produced during the photoelectrochemical oxidation. Table S2 in the ESI† presents the faradaic efficiencies estimated on the basis of oxygen measured at different time intervals on the FTO/TiO2 electrode in the presence of Ni5-POM under simulated solar irradiation, where an average faradaic efficiency of ∼294% is obtained. Such a high faradaic efficiency probably results from the fact that the oxygen sensor was placed near the photoanode, leading to the reporting of a much higher local oxygen concentration rather than a well-spread average oxygen level. In other words, the measured oxygen concentration is approximately three times that of the actual concentration of oxygen in a well-mixed solution. Nevertheless, the overall DO data trends presented in Fig. 8 point at the fact that the obtained photocurrent is indeed due to oxygen evolution, and correlate well with the results obtained from photocurrent and impedance measurements that point at the role of Ni5-POM with respect to increased water oxidation photocurrent and reduced onset potential by means of enhanced charge separation and catalytic activity (see Sections 3.2.1 and 3.2.2). The turnover number (TON) and turnover frequency (TOF) for this system were calculated to be 0.085 and 1.5 h−1 based on the number of moles of oxygen evolved (250 nmol) within 200 s at the FTO/TiO2 photoanode in the presence of Ni5-POM (20 μM in 50.0 mL or 1000 nmol) under simulated solar irradiation, with the assumption of 100% faradaic efficiency and Ni5-POM as the sole catalytic contributor. Clearly, TOF is a better expression of the catalytic activity than TON, as the latter will increase with the increase of the experimental time. A number of factors, including the rate of diffusion of the homogeneous catalyst to and from the electrode surface, the deactivation of the catalyst during the course of electrolysis, as well as the overpotential, could significantly affect the TON and TOF of a homogeneous electrocatalyst system.55 Hence, the TON and TOF obtained from the above simplified calculations must be used and interpreted cautiously. On the other hand, from an industrial standpoint of view, the use of homogeneous catalysts has unique advantages over heterogeneous co-catalysts, because the stability of catalyst films in the face of prolonged gas evolution is not a factor, and it is more convenient to design flow systems that can replace inactivated homogeneous catalysts.
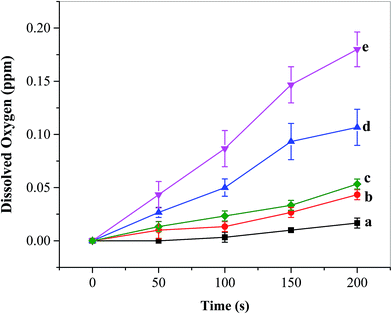 |
| | Fig. 8 Dissolved oxygen (DO) for (a) borate buffer blank, (b) during multi-potential step experiments in the absence and (c) presence of the water oxidation catalyst Ni5-POM (20 μM in 0.050 M pH 9.0 borate buffer) under dark conditions, (d) FTO/TiO2 photoanodes in the absence and (e) presence of the water oxidation catalyst Ni5-POM (20 μM in 0.050 M pH 9.0 borate buffer) under simulated solar radiation. Each datum dot in the figure was an average of three replicated measurements. | |
4. Conclusion
Photoelectrochemical studies on the earth abundant water oxidation catalyst Ni5-POM for solar water splitting using spin coated FTO/TiO2 photoanodes were reported. TiO2 film was characterized by SEM, EDX, and UV-vis spectroscopy. The three-electrode photocurrent measurements revealed significant increase in photocurrent and negative shift in the onset potential of water oxidation upon the addition of 20 μM Ni5-POM water oxidation catalyst. As verified by EIS data, these observations were attributed to increased electron–hole separation of TiO2 and catalysis of the water oxidation reaction resulting from the capture and utilization of photo-generated holes by the catalyst. At zero biased potential, the FTO/TiO2–Ni5-POM system displayed a notable photocurrent, which is significant considering that the unbiased photocurrent was produced from the anatase TiO2 photoelectrode under simulated solar light. Multi-potential step experiments revealed that the cracked TiO2 film on FTO was stable even under a harsh experimental parameter of 1.10 V vs. Ag/AgCl in the presence of the Ni5-POM catalyst and simulated solar irradiation. Oxygen produced from the FTO/TiO2–Ni5-POM system was detected. Moving forward with the results of this investigation, future studies on the use of homogeneous POM based catalysts are expected to focus on their integration with lower bandgap photoanodes and possible application as co-catalysts in tandem water splitting, with the objective of designing high efficiency solar fuel conversion devices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This investigation was partially supported by the National Science Foundation CAREER grant (CHE0955878). The School of Polymers and High Performance Materials at the University of Southern Mississippi is acknowledged for assistance in taking SEM images.
References
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
-
M. G. Roel van de Krol, Photoelectrochemical Hydrogen Production, Springer, 2012 Search PubMed.
-
C. A. Grimes, O. K. Varghese and S. Ranjan, Light, Water, Hydrogen, 2008 Search PubMed.
- J. R. Bolton, Sol. Energy, 1996, 57, 37–50 CrossRef CAS.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- L. M. Peter and K. G. U. Wijayantha, ChemPhysChem, 2014, 15, 1983–1995 CrossRef CAS PubMed.
- M. S. Prévot and K. Sivula, J. Phys. Chem. C, 2013, 117, 17879–17893 Search PubMed.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 CAS.
- D. K. Zhong, S. Choi and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 18370–18377 CrossRef CAS PubMed.
- F. Wen and C. Li, Acc. Chem. Res., 2013, 46, 2355–2364 CrossRef CAS PubMed.
- Y. V. Geletii, Q. Yin, Y. Hou, Z. Huang, H. Ma, J. Song, C. Besson, Z. Luo, R. Cao, K. P. O'Halloran, G. Zhu, C. Zhao, J. W. Vickers, Y. Ding, S. Mohebbi, A. E. Kuznetsov, D. G. Musaev, T. Lian and C. L. Hill, Isr. J. Chem., 2011, 51, 238–246 CrossRef CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 Search PubMed.
- B. Nepal and S. Das, Angew. Chem., Int. Ed., 2013, 52, 7224–7227 CrossRef CAS PubMed.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- I. Bazzan, A. Volpe, A. Dolbecq, M. Natali, A. Sartorel, P. Mialane and M. Bonchio, Catal. Today, 2017, 290, 39–50 CrossRef CAS.
- M. Burian, Z. Syrgiannis, G. La Ganga, F. Puntoriero, M. Natali, F. Scandola, S. Campagna, M. Prato, M. Bonchio, H. Amenitsch and A. Sartorel, Inorg. Chim. Acta, 2017, 454, 171–175 CrossRef CAS.
- P. Kar and P. J. S. Rana, Bull. Korean Chem. Soc., 2014, 35, 1931–1932 CrossRef CAS.
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Koegerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef CAS PubMed.
- H. Lv, J. Song, H. Zhu, Y. V. Geletii, J. Bacsa, C. Zhao, T. Lian, D. G. Musaev and C. L. Hill, J. Catal., 2013, 307, 48–54 CrossRef CAS.
- M. Orlandi, R. Argazzi, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and F. Scandola, Chem. Commun., 2010, 46, 3152–3154 RSC.
- A. Sartorel, P. Miro, E. Salvadori, S. Romain, M. Carraro, G. Scorrano, M. Di Valentin, A. Llobet, C. Bo and M. Bonchio, J. Am. Chem. Soc., 2009, 131, 16051–16053 CrossRef CAS PubMed.
- Y. Yang, B. Zhang, Y. Wang, L. Yue, W. Li and L. Wu, J. Am. Chem. Soc., 2013, 135, 14500–14503 CrossRef CAS PubMed.
- C. Zhao, C. S. Kambara, J. Song, H. Lv, Y. V. Geletii, G. Zhu, J. Vickers and C. L. Hill, Abstr. Pap. Am. Chem. Soc., 2011, 242, 491-INOR Search PubMed.
- S. J. Folkman and R. G. Finke, ACS Catal., 2017, 7, 7–16 CrossRef CAS.
- S.-X. Guo, Y. Liu, C.-Y. Lee, A. M. Bond, J. Zhang, Y. V. Geletii and C. L. Hill, Energy Environ. Sci., 2013, 6, 2654–2663 CAS.
- Z. Han, A. M. Bond and C. Zhao, Sci. China: Chem., 2011, 54, 1877–1887 CrossRef CAS.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- F. Song, Y. Ding and C. Zhao, Acta Chim. Sin., 2014, 72, 133–144 CrossRef CAS.
- J. P. Falkenhagen, B. Braun, E. Bill, D. Sattler and C. Limberg, Inorg. Chem., 2014, 53, 7294–7308 CrossRef CAS PubMed.
- J. M. Sumliner, H. Lv, J. Fielden, Y. V. Geletii and C. L. Hill, Eur. J. Inorg. Chem., 2014, 635–644, DOI:10.1002/ejic.201301573.
- Z. Huang, Z. Luo, Y. V. Geletii, J. W. Vickers, Q. Yin, D. Wu, Y. Hou, Y. Ding, J. Song, D. G. Musaev, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2011, 133, 2068–2071 CrossRef CAS PubMed.
- J. Soriano-Lopez, S. Goberna-Ferron, L. Vigara, J. J. Carbo, J. M. Poblet and J. Ramon Galan-Mascaros, Inorg. Chem., 2013, 52, 4753–4755 CrossRef CAS PubMed.
- S. K. Cho, H. S. Park, H. C. Lee, K. M. Nam and A. J. Bard, J. Phys. Chem. C, 2013, 117, 23048–23056 CAS.
- G. Zhu, E. N. Glass, C. Zhao, H. Lv, J. W. Vickers, Y. V. Geletii, D. G. Musaev, J. Song and C. L. Hill, Dalton Trans., 2012, 41, 13043–13049 RSC.
- J. Fielden, J. M. Sumliner, N. Han, Y. V. Geletii, X. Xiang, D. G. Musaev, T. Lian and C. L. Hill, Chem. Sci., 2015, 6, 5531–5543 RSC.
- J. Yano, J. Kern, Y. Pushkar, K. Sauer, P. Glatzel, U. Bergmann, J. Messinger, A. Zouni and V. K. Yachandra, Philos. Trans. R. Soc., B, 2008, 363, 1139–1147 CrossRef CAS PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed.
- P.-E. Car, M. Guttentag, K. K. Baldridge, R. Alberto and G. R. Patzke, Green Chem., 2012, 14, 1680–1688 RSC.
- Z. Wang, U. Helmersson and P.-O. Käll, Thin Solid Films, 2002, 405, 50–54 CrossRef CAS.
-
R. Jeba Beula, S. Devadason and V. Mahesh Kumar, in Recent Trends in Materials Science and Applications: Nanomaterials, Crystal Growth, Thin Films, Quantum Dots, & Spectroscopy (Proceedings ICRTMSA 2016), ed. J. Ebenezar, Springer International Publishing, Cham, 2017, pp. 437–449, DOI:10.1007/978-3-319-44890-9_40.
- M. Momeni, F. Golestani-Fard, H. Saghafian, N. barati and A. Khanahmadi, Appl. Surf. Sci., 2015, 357, 1902–1910 CrossRef CAS.
-
M. Stefan, O. Pana, C. Leostean, C. Bele, D. Silipas, M. Senila and E. Gautron, Synthesis and Characterization of Fe3O4–TiO2 Core–Shell Nanoparticles, 2014 Search PubMed.
- G. Yang, Z. Jiang, H. Shi, T. Xiao and Z. Yan, J. Mater. Chem., 2010, 20, 5301–5309 RSC.
- C. Dette, M. A. Pérez-Osorio, C. S. Kley, P. Punke, C. E. Patrick, P. Jacobson, F. Giustino, S. J. Jung and K. Kern, Nano Lett., 2014, 14, 6533–6538 CrossRef CAS PubMed.
- A. Sreedhar, T. V. M. Sreekanth, J. H. Kwon, J. Yi, Y. Sohn and J. S. Gwag, Ceram. Int., 2017, 43, 12814–12821 CrossRef CAS.
- C. Cao, C. Hu, W. Shen, S. Wang, J. Wang and Y. Tian, J. Alloys Compd., 2013, 550, 137–143 CrossRef CAS.
- A. Meng, B. Zhu, B. Zhong, L. Zhang and B. Cheng, Appl. Surf. Sci., 2017, 422, 518–527 CrossRef CAS.
- S. Palmas, A. M. Polcaro, J. R. Ruiz, A. Da Pozzo, M. Mascia and A. Vacca, Int. J. Hydrogen Energy, 2010, 35, 6561–6570 CrossRef CAS.
- D. D. Macdonald, Electrochim. Acta, 2006, 51, 1376–1388 CrossRef CAS.
- B.-Y. Chang and S.-M. Park, Annu. Rev. Anal. Chem., 2010, 3, 207–229 CrossRef CAS PubMed.
-
Electrochemical Impedance Spectroscopy, ed. M. E. Orazem and B. Tribollet, John Wiley & Sons, Inc., 2008 Search PubMed.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2nd edn, 2001 Search PubMed.
-
M. Langland and T. Cronin, A Summary Report of Sediment Processes in Chesapeake Bay and Watershed, in Water-Resources Investigations Report 03-4123, New Cumberland, PA, 2003 Search PubMed.
- C. Costentin, G. Passard and J.-M. Savéant, J. Am. Chem. Soc., 2015, 137, 5461–5467 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00523g |
|
| This journal is © The Royal Society of Chemistry 2018 |

 and
Wujian
Miao
and
Wujian
Miao