
Efficient nanomaterials for harvesting clean fuels from electrochemical and photoelectrochemical CO2 reduction
Yang
Yang†
,
Saira
Ajmal†
,
Xiuzhen
Zheng
and
Liwu
Zhang
 *
*
Department of Environmental Science and Engineering, Fudan University, Shanghai, 200433, P. R. China. E-mail: zhanglw@fudan.edu.cn
First published on 24th November 2017
Abstract
The excessive utilization of fossil fuels accompanied by large amounts of anthropogenic CO2 emissions have led to adverse global environmental changes and a growing global energy crisis. Hence, converting CO2 into high-value chemical fuels, such as CO, CH4, HCOOH, and CH3OH, through catalysis is one of the most attractive topics in energy conversion. Among various approaches, electrochemical (EC) and photoelectrochemical (PEC) reduction are considered to be promising methods. Over the past decades, research in the area of CO2 EC and PEC reduction has been growing quickly. Herein, highly efficient nanostructured metals, metal alloys, metal oxides, metal-free electrodes, and photoelectrodes developed in recent years are discussed in detail in this review. Additionally, the strategies and mechanisms for improving the faradaic efficiency (FE), current density, and stability of catalysts are also discussed. The challenges and future perspectives for CO2 EC and PEC reduction are also discussed.
 Yang Yang | Yang yang is a PhD candidate under the supervision of Prof. Liwu Zhang in the Department of Environmental Science and Engineering at Fudan University. He obtained his Master's degree from the School of Environmental Science and Engineering at Nanjing University of Information Science & Technology in 2017. His research is focused on environmental materials development and CO2 reduction. |
 Saira Ajmal | Saira Ajmal is a PhD candidate under the supervision of Prof. Liwu Zhang in the School of Environmental Science and Engineering at Fudan University. She obtained her Master's degree from the School of Chemistry at the University of Sargodha Pakistan in 2015. Her research is focused on CO2 electrochemical reduction. |
 Xiuzhen Zheng | Dr Xiuzhen Zheng is a postdoctoral research fellow in the Department of Environmental Science and Engineering at Fudan University. She received her PhD degree from the State Key Laboratory of Photocatalysis on Energy and Environment from the Fuzhou University in 2015. Currently, she is conducting her postdoctoral studies under the supervision of Prof. Liwu Zhang, with a focus on enhancing the performance of artificial photosynthesis with the incorporation of photonic nanostructures, and further studying the impact of light manipulation in these systems. |
 Liwu Zhang | Prof. Liwu Zhang joined the Department of Environmental Science and Engineering at Fudan University in November 2014. Prior to his faculty appointment, he did his postdoctoral work under the supervision of Prof. Jeremy J. Baumberg (FRS) at the Nanophotonics Centre in the Cavendish Laboratory and collaborated with Dr Erwin Reisner from the Department of Chemistry at the University of Cambridge. Before that, he was an Alexander von Humboldt Research Fellow under the supervision of Prof. Detlef Bahnemann at Hannover University. He received his PhD in Chemistry from Tsinghua University under the supervision of Prof. Yongfa Zhu. He is a recipient of the Marie Curie Intra-European Research Fellow award. He has over a decade of experience in environmental nanotechnology and a strong background in photocatalysis and has published more than 50 highly cited papers in this field. His work has been cited >3000 times, and he has an h-index of 23. |
1 Introduction
Due to the increasing global population, the demand for energy resources is also growing quickly. As early as 2012, researchers reported that a large portion of the worldwide energy requirement (80%) is still fulfilled by fossil fuels, including petroleum, nature gases, and coal.1 However, the use of such resources results in an increased concentration of greenhouse gases (GHS) and many associated environmental problems, including climate change, acid rain, depletion of the ozone layer, sea level rises, and global warming.2,3 According to the studies of the Intergovernmental Panel on Climate Change (IPCC), the CO2 gas emitted by the burning of fossils fuel among all other anthropogenic gases is supposed to be the chief source of climate change and global warming.4,5 Thus in an ideal scenario, there should be a balance between CO2 production and consumption, but unfortunately this has been disrupted.6 It has been reported that the atmospheric level of CO2 reached up to ∼403.95 ppm by July 2017 (Data from the NOAA research) and was predicted to rise and reach nearly 600 ppm by 2100.7,8How to remove the excessive CO2 is one of the biggest current challenges for mankind. CO2 also has wide applications in agriculture, food, as an inert gas, in fire extinguishers, in the medical field, as a refrigerant, and in other fields, but these cannot remove CO2 from the air.9 In order to solve this problem, converting CO2 into another useful fuel is an attractive proposition.10,11 Up to now, a number of routes have been established for CO2 conversion, such as biological,12,13 chemical reorganizing,14–17 mineralization,18–20 photochemical, EC methods,16,21,22 and PEC methods.23–26
Also, effective energy storage of non-fossil fuel sources is required as part of the long-term solution of mankind's fuel requirements.27,28 With this in mind, various fuels have been projected as alternative fuels. One of the emerging fuels proposed as the next generation energy source is hydrogen (H2). However, the production of H2 fuel requires a high amount of energy intensive labor along with the need for special materials to avoid H2 diffusion and leakage. The use of H2 gas as a fuel also carries a risk of explosion, so wide safety precautions are usually required.29,30 Bioelectrochemical systems (BESs) have recently been proposed as a new and sustainable technology for CO2 reduction.31 However, these often lack effective bacteria, and furthermore, the bacteria are easily broken by the reduction production. Additionally, the membrane is easily polluted, which also increases the cost. Photochemical (PC), electrochemical (EC), and photoelectrochemical (PEC) methods have attracted great attention for CO2 reduction due to their energy-saving benefits and wide range of product, including carbon monoxide (CO), methane (CH4), ethylene (C2H4), methanol (CH3OH), formic acid (HCOOH), formaldehyde (HCHO), and other high alcohols, hydrocarbons, and oxygenates.14,16,21,22,32–35 The driving force of the photochemical reduction of CO2 is only solar energy, and consequently it is regarded as an artificial model for photosynthesis. However, the current low solar energy conversion efficiency, charge separation, and transportation efficiency have limited its application. For the EC reduction of CO2, the driving force is an external applied bias, which consumes a lot of energy, and so this is a limiting factor to its wider take up. However, the reduction products and process can be tuned by the applied external bias, which are considered benefits. For the PEC reduction of CO2, the driving force is the synergy of solar energy and an external bias, which thus combines the advantages of both the EC and PC processes.32,36 However, the CO2 molecule is very stable due to its linear and centrosymmetric structure.32,37 Additionally, the C–O bond length in CO2 is about 116.3 pm, which is shorter than that of a C–O single bond and most other C–O multiply-bonded functional groups. It is thus difficult to break the C–O bond to form new carbon-based materials. Although a number of photocatalysis systems, electrodes, and photoelectrodes have been developed for CO2 reduction, most of them still suffer from the low energy conversion efficiency, low selectivity, a low FE, poor stability, and incapability to suppress the competing hydrogen evolution reaction (HER). Therefore, how to design and fabricate the required photocatalysts, electrodes, and photoelectrodes with high selectivity, high efficiency, and good stability is still a big challenge for CO2 reduction.
Up to now, several excellent reviews about CO2 photochemical reduction have been published.38–42 These reviews involve the design and fabrication of various photocatalysts as well as descriptions of the CO2 reduction process. Chang et al. summarized the strategies for enhancing the adsorption and activation of CO2 as well as the factors determining the reaction pathways and selectivity for the reduction products.42 There are also some reviews in the literature about EC reduction covering various metal electrodes, metal oxide electrodes, sulfide catalysts, and carbon catalysts, etc., as well as the crucial factors affecting the electroreduction performance.10,43,44 There are also some reviews available on CO2 PC, and EC and PEC reduction reactions employing metal complex catalysts.45–50 In addition, there are a few reviews involving PEC reduction.49,51 However, as far as we know, there are hardly any reviews entirely devoted to both EC and PEC reduction of CO2 by metals, metallic alloys, and inorganic semiconductors in inorganic salt aqueous solutions. Therefore, herein we specifically focus on emerging nanomaterials and nanoarchitectures as efficient EC and PEC electrodes with a high FE of the desired CO2 reduction products. Moreover, the current review summarizes the variety of electrocatalysts that have been commonly employed in the past 5 years for CO2 reduction through either the EC or PEC method. We summarize different groups of electrodes, including metal nanostructures (Cu, Au, Ag), metal alloys (Cu–Ni, Cu–Pd, Cu–Sn, Cu–Zn, Cu–Ag, Cu–In, and others), metal oxides (CuOx, SnO2, CeO2, etc.), chalcogenides (MoS2, WS2, etc.), and metal-free electrodes (C, Si, etc.). Besides, we divided the photoelectrodes into three categories: (1) phosphides (GaP, InP, etc.); (2) metal oxides (CuxOy, TiO2, ZnO, etc.); and (3) metal-free (C3N4, Si) photoelectrodes for CO2 reduction. The reaction mechanisms and catalysis pathways are also discussed in this review.
2 Basic concepts and mechanism of CO2 reduction
The CO2 molecule has a linear structure and is highly stable thermodynamically as well as kinetically. It exhibits a highest oxidation state of +4 and is the ultimate product of many biological and chemical processes.37 For the reduction of CO2 into other useful compounds, a high amount of energy is required because of its low energy levels. As CO2 is a linear molecule and centrosymmetric, the molecule has no electrical dipole. The length of carbon–oxygen bonds is 116.3 pm, which is smaller than that of a C–O single bond, and even shorter than most other C–O multiply-bonded functional groups. Its particular molecular structure and weak electrophile nature makes it difficult to transfer to other carbon compounds. The reduction of CO2 into renewable lower products involves multistep electron pathways.24 Among the various methods of CO2 reduction, the EC and PEC methods have advantages as the reaction rate and products could be controlled by tuning the external bias overpotential.52 The reduction of CO2 forms various products, such as carbon monoxide (CO), methanol (CH3OH), ethanol (C2H5OH), ethylene (C2H4), methane (CH4), formate (HCOO−), and formic acid (HCOOH).53,54 The production of final products is greatly influenced by the nature of the catalyst used and the applied potential.55CO2 adsorption and activation are the key steps in both EC and PEC reduction processes. The pathways of EC and PEC reduction of CO2 are also significantly affected by the applied potential. According to the different reaction mechanisms and pathways, we can get various products, such as CO, CH4, CH3OH, HCOOH, HCHO, C2H6, other carbohydrates. One of the challenges is to suppress the evolution of H2 (HER), which always occurs during the CO2 reduction process. The equilibrium potentials of the different products are shown in Table 1. For the pathway involving 2 electrons, the possible reduction products are H2, CO, and HCOOH. However, the standard redox potentials for CO and HCOOH are more negative than that of H2, which means that the HER will compete with the CO2 reduction. Also, 6 and 8 electrons are needed for the formation of CH3OH and CH4, respectively, which would be more difficult than that of H2 evolution. Additionally, the Gibbs free energy for the formation of CH3OH and CH4 by the reaction of CO2 and H2O is about 702 and 818 kJ mol−1, respectively.56 Meanwhile, the Gibbs free energy change for converting H2O to H2 and 1/2O2 is about 273 kJ mol−1, which will occur more easily than CO2 reduction.57 The solubility of CO2 in H2O is about 0.33 mol L−1 at 25 °C under 1 atm, which would further make it difficult to adsorb and activate CO2.42,58 It has been found in some research studies that the nature of the products formed is mainly governed by the kinetics. Furthermore, that the kinetics greatly depend on the reaction temperature, pressure, reactant concentration, electrolyte, and so on. For enhancing the surface-adsorbed CO2 concentration, researchers usually design some porous materials, including a 3D inverse opal structure, nanotubes, hollow structures and so forth. Designing suitable devices can also enhance the catalyst surface CO2 concentration and enable forming different products. Ampelli et al. designed a novel EC setup by using metal-doped conjugated microporous polymers for CO2 reduction, where their design had the ability to assemble the electrode materials to form a multilayered composite to guarantee good proton mobility and electron conductivity.59,60 Compared with conventional cells, this electrolyte-less configuration exhibited differences in the productivity and product types, which may indicate a different reaction mechanism. More discussion of this can be found in the following sections. Hence, how to suppress the evolution of H2 and improve the selectivity is still the biggest challenge in PEC and EC CO2 reduction.
| Products | Reaction | E0 (V vs. NHE) | Eqn |
|---|---|---|---|
| H2 | 2H+ + 2e− → H2 | −0.41 | (1) |
| CO | CO2 + 2H+ + 2e− → CO + H2O | −0.51 | (2) |
| HCOOH | CO2 + 2H+ + 2e− → HCOOH | −0.58 | (3) |
| HCHO | CO2 + 4H+ + 4e− → HCHO + H2O | −0.48 | (4) |
| CH3OH | CO2 + 6H+ + 6e− → CH3OH + H2O | −0.38 | (5) |
| CH4 | CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 | (6) |
| CH3CH2OH | 2CO2 + 12H+ + 12e− → CH3CH2OH + 3H2O | −0.33 | (7) |
| CH3CH3 | 2CO2 + 14H+ + 14e− → CH3CH3 + 4H2O | −0.27 | (8) |
| — | CO2 + e− → CO2˙− | −1.9 V (pH = 0) | (9) |
| O2 | H2O + 2h+ → 1/2O2 + 2H+ | 0.82 | (10) |
For photocatalytic CO2 reduction, the oxidation and reduction reactions often occur on different sites of the catalyst; hence, the products are mixed. In addition, the driving force for the photocatalytic reduction of CO2 is only from solar energy, while the solar-to-fuel energy conversion efficiency is very low, which has limited its application to date. For the EC reduction of CO2, the driving force is from an external applied bias, and the reaction can thus be more easily manipulated. For the PEC reduction of CO2, the driving force is from the synergy between solar energy and an external bias, which thus combines the advantages of PC and EC methods. However, their reaction pathways are similar, and are usually affected by the experiment conditions, including the properties of the catalysts, electrolyte species, reaction temperature, and pressure, etc.42 Among the various factors, the property of the catalysts (electrodes and photoelectrodes) often plays a key role in the products distribution. For EC CO2 reduction, taking the metal electrodes as an example, the reaction pathways may be different by using different types of electrodes, which could then lead to a distinct distribution of products. According to the previous investigations,61 In, Sn, Pb, and Cd have a high overpotential for H2 evolution, but have a negligible ability to adsorb the CO intermediate, resulting in the preferred formation of HCOOH on these electrodes; while, Au, Ag, and Zn have a medium overpotential for H2 evolution and a weak ability to adsorb the CO intermediate, resulting in the preferred formation of CO; and Al, Si, V, Fe, Mn, Co, Mo, and Pt, etc. have a low overpotential for H2 evolution and a strong ability to adsorb CO, resulting in the formation of H2. The distribution of products on the Cu electrode is very wide, including CO, H2, CH4, CH3OH, HCOOH, and C2H4 and other hydrocarbons. The external bias applied on the electrode also plays an important role in the products distribution, which has been widely investigated by many groups, especially on the Cu electrode.62–64 At the initial potential (around 0.8 to 0.95 V vs. SHE), HER is the main reaction. Until the applied potential reaches −0.95 V vs. the standard hydrogen electrode (SHE), the rate of HER start to decrease, and meanwhile CO and formate begin to be generated. The generation rate of CO, HCOO, C2H4, and CH4 increase significantly with the negative shift of the applied potential (from −1.12 V to −1.35 V vs. SHE). When the applied external bias is more negative than −1.35 V, C2H4 and CH4 become the predominant products. However, it is worth noting that different electrodes may show different responses to the applied potential during CO2 reduction.42 More detailed influencing factors in the EC reduction of CO2 have been discussed by Gong's group and by Zhong et al.10,42,43Additionally, Centi and coauthors reported that a suitable device also can change the reaction pathway and products.65–67 They designed a new strategy for enhancing the electrode surface concentration of CO2 by adding a CO2 adsorption component to a gas diffusion layer (GDL) type electrode, which showed a significantly enhanced activity and selectively for >C1 products. This report provided a new approach to generate >C1 carbon products.65
For the PEC reduction of CO2, the conduction and valence band potential, exposed facets, external optical properties, and potential, etc. play important roles in the products distribution, FE, and conversation efficiency. The band-edge position of some typical semiconductor and redox potentials versus (vs.) NHE of CO2 reduction at pH = 7 are shown in Fig. 1.38 From Table 1 and Fig. 1, we can see that all the candidate semiconductors listed here cannot carry out the one electron reduction of CO2 to CO2˙− (about −1.9 V vs. NHE, pH = 0), because they do not have enough driving force, as their conduction band edge are below the potential of CO2 to CO2˙−. Therefore, another mechanistic route has been proposed, which is through a series of multiple proton-coupled electron transfers to form CO2˙− radicals. However, it still needs to overcome high activation barriers. It has been reported that the higher concentration of available protons and the partial electron density on the surface of catalysts would promote the reaction.42 In addition, a suitable heterojunction will benefit the CO2 reduction because the heterojunction can facilitate the transportation of electrons and holes, which can lower the overpotential toward CO2 reduction and improve the charge-carrier separation. Moreover, the surface state may also be modified due to the formation of a heterojunction, which may promote the adsorption and activation of CO2. Except for the driving force for CO2 reduction, the mechanism and pathways of CO2 reduction are generally similar for EC reduction and PEC reduction processes.
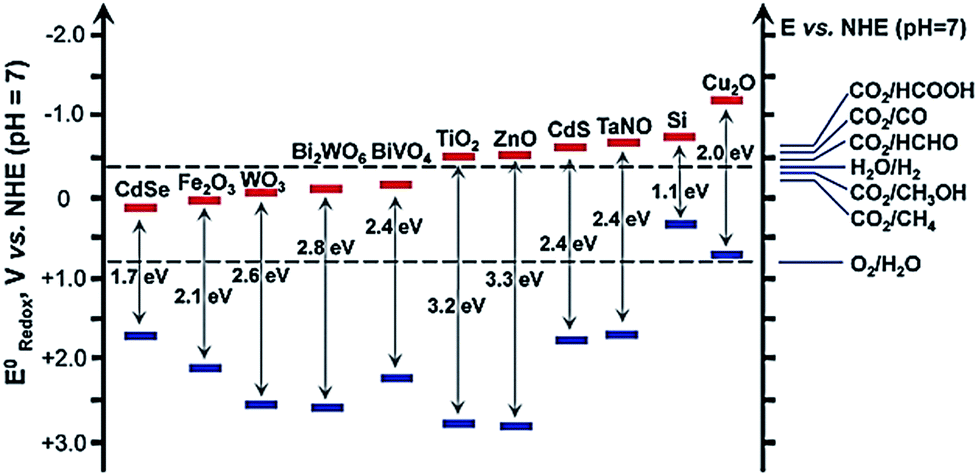 | ||
| Fig. 1 Band-edge positions of some typical semiconductor photocatalysts relative to the energy levels of the redox couples involved in the reduction of CO2.38 Copyright 2016 Royal Society of Chemistry. | ||
3 Electrochemical reduction of CO2
Among all the various approaches for reducing CO2 into low carbon chemicals for fuel purposes, the methodology of EC reduction is the simplest one that can work on a huge scale under green conditions.50,68 The electricity produced from renewable sources, such as from solar and wind power, is used to drive the EC reduction processes.69 CO2 has high stability and very complicated kinetics from multiple electron transformations, which are a hindrance to overcoming the high energy barrier.70 For CO2 EC reduction, a variety of electrocatalysts, including metals, metals oxides, metallic nanostructures, and molecular complexes, have been explored.71–75 As their activities for huge-scale application are still not active enough, the biggest challenge during CO2 EC reduction reaction is to improve the low activity, poor efficiency, selectivity, and yield of products.32,76 In the field of CO2 EC reduction, a lot of the research efforts have focused on the development of the best electrocatalyst in order to overcome the above challenges. So in the following section of the review, we summarize several groups of electrocatalysts, such as metallic nanostructures, metallic alloys, metal oxides, and metal-free electrodes, for CO2 reduction.3.1 Metallic nanostructures
In the early 1980s, introductory research in the field of CO2 reduction was carried out with the catalytic performance of bulky metallic catalysts. For this, some single metallic elements were examined. The products obtained after the CO2 EC reduction depend upon various factors, like the kind of materials used as the cathode, the electrolyte, and also on the voltage, temperature, and pressure. In order to improve the CO2 EC reduction efficiency, nanostructured metallic materials have attracted much attention because of their versatile properties. These metallic nanomaterials have a high surface area and contain a large portion of edges or low coordinated sites. Due to these properties, they show different catalytic behaviors compared to the bulk materials.43,77,78 This section describes the recent progress in various metallic nanostructures for CO2 EC reduction. | ||
| Fig. 2 (a) The highly internal surface morphology of a nanopore of a Ag electrocatalyst (b), (c) SEM and TEM images of nanoporous Ag (for b and c, the scale bar is 500 and 2 nm, respectively). (e)–(g) Electrocatalytic activity of nanoporous Ag and polycrystalline Ag at −0.60 V, and np–Ag at −0.50 V, and at −0.40 V verses RHE, total current density and FE verses time on the left axis and right axis, respectively.79 Copyright 2014 Nature Publishing Group. | ||
Very recently, Daiyan et al. fabricated a three-dimensional (3D) nanoporous Ag electrode termed Ag foam by using a simple electrodeposition method.85 Ag foam has the capability toward the selective reduction of CO2 into CO with a high FE of 94.7% and a current density of 10.8 mA cm−2 at −0.99 V vs. RHE. Due to its unique 3D nanoporous structure, Ag foam has shown amazing activities with a higher FE and current density. In this particular work, the electrocatalytic activity of Ag foam was investigated in a 0.1 M solution of KHCO3 for CO2 EC reduction, with the Ag foam directly used as the working electrode without any further treatment. The comparison between Ag foam and other high performance Ag electrodes showed that it had the highest FE efficiency among the others, including a high-cost organometallic Ag electrode (90% FE at −1.1), polycrystalline Ag foil (90% FE at −1.1 V), immobilized Ag nanoparticle (80% FE at −0.75 V), and nanostructured Ag foil (83.5% FE at −1.04 V).85 From this research, it was concluded that the 3D porous Ag foam electrode has more EC active sites, and that these dramatically increase the contact between the electrolyte and electrode. We can thus conclude that the highly porous structure leads to the more effective EC reduction of CO2.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratio can be tailored by changing the size of the catalyst particles. Koh et al., for the first time, synthesized Au nanostructured islands by a single-step oxygen plasma-induced method, in which thermal annealing simultaneously occurred to prevent the formation of gold oxide.93 The physical and chemical properties, including the crystal structure and oxidative state of the nanostructured Au electrode, was explored by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). The CO2 EC reduction was performed in an acetonitrile–water electrolyte solution. They showed a higher selectivity for CO with a 95% FE and enhanced current density. As compared to polycrystalline Au, the production rate, onset potential, and FE were significantly improved by the nanostructured Au island electrode. The authors further demonstrated that, in the presence of 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM] BF4), the CO2 reduction activity was improved on Au islands as compared to that without [BMIM] BF4. CO and H2 were confirmed as the main products by gas chromatography, and the formic acid concentration was found to be below the detection limited. For bare Au, Au islands, and Au with ([BMIM] BF4), the FE and CO rate of production plotted against voltage (vs. NHE) are illustrated in Fig. 3.
:CO ratio can be tailored by changing the size of the catalyst particles. Koh et al., for the first time, synthesized Au nanostructured islands by a single-step oxygen plasma-induced method, in which thermal annealing simultaneously occurred to prevent the formation of gold oxide.93 The physical and chemical properties, including the crystal structure and oxidative state of the nanostructured Au electrode, was explored by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). The CO2 EC reduction was performed in an acetonitrile–water electrolyte solution. They showed a higher selectivity for CO with a 95% FE and enhanced current density. As compared to polycrystalline Au, the production rate, onset potential, and FE were significantly improved by the nanostructured Au island electrode. The authors further demonstrated that, in the presence of 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM] BF4), the CO2 reduction activity was improved on Au islands as compared to that without [BMIM] BF4. CO and H2 were confirmed as the main products by gas chromatography, and the formic acid concentration was found to be below the detection limited. For bare Au, Au islands, and Au with ([BMIM] BF4), the FE and CO rate of production plotted against voltage (vs. NHE) are illustrated in Fig. 3.
 | ||
| Fig. 3 (a) Schematic representation of the fabrication of Au islands prepared by oxygen plasma treatment (b) (c) linear sweep voltammograms (LSV) for the bare Au and Au islands in acetonitrile with or without 0.1 M [BMIM]BF4 and of bare Au and a Au islands electrode prepared under various oxygen plasma treatment times (d), (e) FE and products rate of CO measured on the bare Au and Au islands (60 min) in acetonitrile with or without 0.1 M [BMIM]BF4.93 Copyright 2015 American Chemical Society. | ||
Sargent et al. described Au nanoneedles with a high current density for CO2 EC reduction,21,94 and further predicted, by utilizing DFT calculations, that the berries could be reduced by the adsorption of K+ ions during CO2 reduction at various Au facets. The authors explored the CO2 reduction activity of Au needles, particles, and rods in 0.5 M KHCO3. In the range of −0.30 V to −0.50 V, they found that on Au needles, the voltammetry curve displayed a distinct reduction peak, while Au rods and particles showed plane current–voltage curves. The FE for CO was approximately >95% on Au needles, while on Au rods and particles it was about 25% and 3%. The Au needles showed great stability during an 8 h electrocatalytic process. The Tafel analysis showed a slope of 96 mV dec−1 for Au particles, 80 mV dec−1 for Au rods, and 42 mV dec−1 for Au needles. The significantly smaller Tafel slope indicated a faster kinetics of first electron transfer and confirmed that the Au needles have a higher activity for CO2 reduction. Additionally, Hall et al. designed a Au inverse opal (Au–IO) film for CO2 reduction, which showed high activity for CO at modest overpotentials and a high selectivity relative to hydrogen evolution (HER).95 They demonstrated that the origin of hydrogen suppression in Au–IO films was derived from the generation of diffusional gradients within the pores of the electrodes rather than from changes in surface faceting or in the Au grain size. As a result, the activity for HER decreased by 10-fold with increasing the porous film thickness, while the CO evolution activity was largely unchanged. For the optimal mesostructured electrodes, 99% selectivity for CO could be obtained at an overpotential as low as 0.4 V.
In order to improve the catalytic activity of the Cu electrode, different kinds of Cu nanostructured have been fabricated, including nanoparticles of zero-dimensional (0D), one-dimensional (1D), and two-dimensional (2D) overlayers and three-dimensional (3D) foams. Their effects on CO2 EC reduction have been explored systematically. An enhanced catalytic activity and selectivity were observed on 0D, 2D, and 3D nanoparticles.43 Recently, Wang et al. fabricated a more innovative electrocatalyst of Cu nanowires for CO2 reduction.100 The EC reduction of CO2 was performed in an electrocatalytic cell with a separating membrane between the cathode and anode in 0.1 M KHCO3.100 Different characterization techniques were used for gas and liquid phase samples, including gas chromatography (GC) with a mass spectrometer (MS) detector and nuclear magnetic resonance (NMR) spectroscopy. The catalytic activity of Cu nanowires was reported in comparison with different previously reported Cu nanomaterials. The authors demonstrated that Cu nanowires produced by EC reduction processes have a highly advanced catalytic ability for CO2 reduction. Under 0.3 V overpotential, the current density could reach 1 mA cm−2 with an FE of 60% for CO production. Recently, Yang et al. reported ultrathin (diameter ∼20 nm) 5-fold twinned copper nanowires (Cu NWs) for CO2 reduction.101 They showed a higher selectivity for CH4 with a 55% FE over other hydrocarbons at −1.25 V vs. RHE, and this was attributed to the presence of a high density of wedge edge sites owing to the twin boundaries. The authors further explored wrapping the wires with graphene oxide, which was found to be a useful strategy for preserving both the morphology and reaction selectivity of the Cu NWs. Fig. 4 illustrates the morphological and electrocatalytic characterization of a Cu NWs electrocatalyst.
 | ||
| Fig. 4 TEM images of Cu NWs: (a) bare, (b) loaded with carbon black. (c) SEM image of Cu NWs/CB dispersed on glassy carbon black. Electrocatalytic and morphological characterization of Cu NW catalysts: (d) rapid formation of C2H4 with a concurrent decrease in CH4 activity after the first Coulomb of electrolysis, (e) TEM images of Cu NWs after passing a given amount of charge (f), (g) analogous morphological and electrocatalytic characterization for the RGO–Cu NW catalysts showing preservation of the selectivity and morphology.101 Copyright 2017 American Chemical Society. | ||
Robert and coworkers fabricated copper nanocubes for CO2 electrochemical reduction.102 The Cu nanocubes showed a higher selectivity for ethylene at a very low overpotential. CO2 could be reduced into multiple hydrocarbon over a Cu nanocube electrode by favoring C–C combination instead of completely reducing it into methane. This could be an important step toward developing catalysts with selectivity for multicarbon products. Very recently, Buonsanti et al. explored the activity and product selectivity of Cu nanocrystals (NCs) toward CO2 reduction with assessing also the effects of size and shape simultaneously.103 Through colloidal-based chemistry, Cu NCs of sphere and cube shaped were fabricated. They found that cube shaped Cu NCs were intrinsically more active as compared to sphere shaped Cu NCs. Fig. 5(a) illustrates the TEM images and XRD pattern of Cu cubes and Cu spheres. The FE of Cu NCs for different products in comparison with Cu foil is shown in Fig. 5(b). It was found that Cu cubes with sizes of 24 nm and 44 nm and the smallest sphere sized 7.5 nm exhibited 80% higher selectivity toward the CO2 reduction reaction and 41% FE for ethylene.
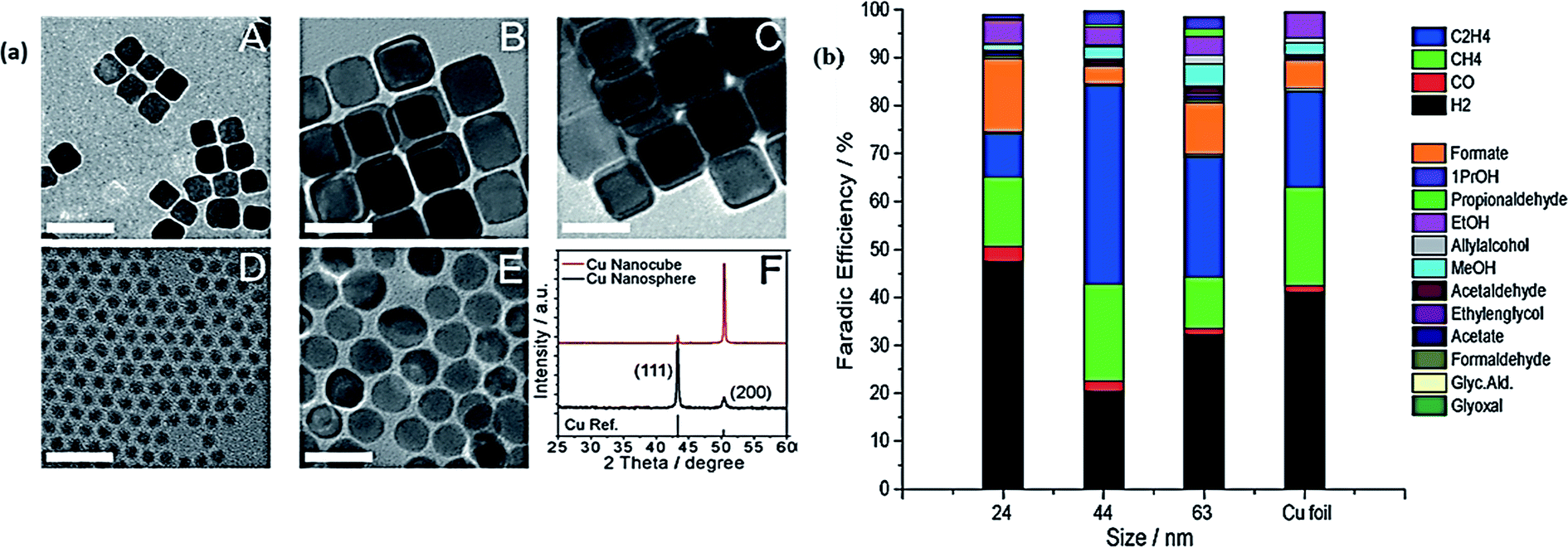 | ||
| Fig. 5 (a) TEM images of Cu cubes (A–C) and Cu spheres (D–E). (F) Typical XRD patterns of Cu cubes and spheres. (b) The bar graph exhibits the FE at −1.1 V vs. RHE for each product in the different sizes of Cu NC cubes, and on Cu foil, with glassy carbon subtracted.103 Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Moreover, for enhancing the CO2 reduction activity, in addition to controlling the size of the NP grains, the boundary density has also been proposed as a useful approach for increasing the low coordination sites. A large atomic disorder has been suggested to enhance the catalytic activity of many reactions, particularly in the field of organic chemistry and thermal processes.104 Kanan et al.105 fabricated Cu NPs on carbon nanotubes (Cu/CNTs) with different average grain boundaries (GBs), which were quantified by transmission electron microscopy (TEM). At potentials ranging from −0.3 V to −0.5 V vs. RHE, the specific reduction activity of CO was linearly proportional to the fraction of surfaces comprising GBs of NPs. The products were ethanol (25–35% FE), acetate (10–30% FE), and small amounts of C2H4, C2H6, and propanol (<10% FE). Their work showed that at a moderate overpotential, the CO reduction activity on Cu NPs was correlated to the density of GBs. Bell et al. attributed the higher selectivity of CuNPs for C2H4 due to the high density of Cu grain boundaries.106 It has also been revealed by several other research groups that the EC activity is directly related to the grain boundary density on the electrode surface.107–109
3.2 Metal alloys
For the EC reduction of CO2, the biggest challenge is the fabrication of a catalyst with high activity and selectivity at low cost. During the reduction processes of CO2, the binding strength of the intermediates play a vital role in the reaction activity and product selectivity.55 Nowadays, metal alloys have drawn great attention because they can boost the electrocatalytic activity of metallic catalysts. They were also found to have the ability to improve the stability of the key intermediates.117,118 Cu has been demonstrated as one of the most promising candidates for CO2 reduction. However, the products selectivity and durability still need to be improved.119 Alloying is a promising approach to enhance the products selectivity and the FE and to lessen the overpotential. Various alloys of Cu have been reported, such as Cu–Ni, Cu–Sn, Cu–Pb, Cu–Zn, Cu–Cd, Cu–In, and Cu–Ag, for the reduction of CO2 in 0.05 M KHCO3 aqueous solution.120Recently, Cu–Pt monodispersed nanocrystals (NCs) with different molar ratios were fabricated by Guo et al.121 The change in the molar concentration of the precursor Cu and Pt had no noticeable influence on the morphology of the Cu–Pt NCs. The authors observed that the composition-dependent activity of Cu–Pt NCs with increasing the Cu content in the Cu–Pt NCs facilitated the reduction of CO2, leading to enhanced catalytic activity. Due to the composition-dependent catalytic activity, the Cu–Pt (3:1) NCs exhibited the highest activity and FE for CH4 at room temperature in 0.5 M KHCO3. On the basis of experimental analysis, the authors proposed a possible mechanism for CO2 EC reduction at the Cu–Pt NCs. Also, DFT calculations for the reduction of CO2 to CH4 have been clearly reported by Peterson et al.122 According to the proposed reaction mechanism and DFT calculations, the key step is the protonation of adsorbed CO* to form HCO* because of Pt, which shows high activity for HER. The equation: CO* + H+ + e− → HCO* was proposed for the reduction of CO to CHO. This step shows the selectivity of CO2 electroreduction on Cu, which ultimately leads to the formation of CH4 due to the synergistic effect between Cu and Pt. The probable reaction mechanism can be expressed by the equation: CO* + H+/H2O + e− → HCO* → CH4.
Zhao and coworkers fabricated Au3Cu alloy NCs by a phase stabilization method,123 which showed enhanced catalytic activity and selectivity toward CO2 EC reduction as compared to nanocrystals of monometallic Au. The Au3Cu alloy NCs endorsed the production of other hydrocarbons too, like CH4 and C2H4, with a 30% FE, as Au NPs favored the production of CO. The authors proposed that this methodology of fabricating alloys could provide a more convenient route to obtaining different alloy NCs. Very recently, Sarfraz et al. reported an electrocatalyst comprising Cu and Sn with a great potential for CO2 EC reduction124 and showing high activity and selectivity over a wide range of potentials. This bimetallic electrode was fabricated by the deposition of Sn on oxide-derived copper (OD–Cu). It showed a maximum selectivity for CO with an FE greater than 90% at a potential range from −0.4 to −0.8 V vs. RHE. The current density, FE comparison, and SEM images of the Cu–Sn before and after CO2 reduction are shown in Fig. 6. The elemental analysis showed that the presence of Sn with Cu enhanced its selectivity for CO, as monometallic Cu can promote H2 production. Wang et al.118 reported carbon-based palladium–copper (PdCu/C) alloy nanoparticles (<5 nm) as an efficient electrocatalyst for CO2 EC reduction. Due to the synergistic effects, CuPd exhibited excellent activity toward CO2 reduction and a higher selectivity for CO. Among the different catalysts of PdCu/C and Pd/C, Pd85Cu15/C showed the highest catalytic activity in CO2-saturated 0.1 M KHCO3 solution and the highest FE of 86% with a current density 6.9 mA cm−2 and mass activity of 4.5 A g−1 for CO production at −0.89 V vs. RHE. X-ray absorption fine structure (EXAFS) analysis and temperature programmed desorption-mass spectrometry (TPD-MS) results revealed that the higher selectivity of Pd85Cu15/C for CO is due to it having low coordination sites and an optimum ratio of copper element. The authors demonstrated that bimetallic Pd–Cu alloys have a great potential for selective CO2 EC reduction, and that a variety of bimetallic alloy nanoparticle catalysts can be fabricated in order to achieve a higher selectivity for CO2 reduction. The Cu alloys exhibited a distinct behavior for CO2 EC reduction as compared to pure metals. The results obtained utilizing different combinations of Cu with various metals opened up a new direction of research for developing Cu-based electrocatalysts.
 | ||
| Fig. 6 (a) FE representation of 0D-Cu (b) Cu–Sn, and (c) Sn deposited on the Sn sheet at applied potentials; the FE is indicated as an average and calculated at the steady-state current and product concentration. (d), (e) SEM images of Cu–Sn before and after 1 h CO2 reduction at −0.6 V vs. RHE.124 Copyright 2016 American Chemical Society. | ||
Besides Cu-based alloys, there are some other metallic alloys applied in CO2 reduction. Recently, Kim et al.125 fabricated Sn–Zn alloys with various compositions by an electrodeposition method. The Sn–Zn electrocatalyst showed the highest selectivity for formic acid (HCOOH) formation. Among all the studied alloys in that work, Sn0.91Zn0.09 exhibited the maximum production and conversion efficiencies for HCOOH. The authors demonstrated that the alloying of Zn on Sn resulted in a modification of its surface morphology and electronic surface, which led to an enhanced activity and selectivity for CO2 EC reduction. It was hypothesized by the authors that Zn–Sn can be further enhanced by increasing the surface area, but this requires new strategies. Very recently, Park et al.126 reported the CO2 EC reduction to formate by using tin-lead (Sn–Pb) alloys can lead to a novel electrocatalyst. Such Sn–Pb alloy electrocatalysts were prepared with different compositions by electrodepositing on the surface of carbon paper. The results obtained by performing electroreduction in an H-type cell at −0.2 V vs. Ag/AgCl showed a 16% FE and 25% partial current density for formate production, which are both higher values than for the pure metallic electrode. The maximum FE (79.8%) and partial current density (45.7 mA cm−2) was exhibited by an alloy with the surface composition of Sn56.3Pb43. The results of cyclic voltammetry (CV) and X-ray photoelectron spectroscopy (XPS) analyses revealed that, on the surface of the alloys, Sn was oxidized to SnOx, which enhanced the catalytic activity and electrical conductivity of the Sn–Pb alloys toward CO2 EC reduction to HCOO−.
Jedidi et al. successfully prepared Cu–In alloy surfaces by the reduction of mixed metal oxides of CuInO2, which showed good activity for electrocatalytic CO2 reduction.127 They combined experimental results and density functional theory calculations (DFT) to demonstrate that In site occupation occurs on Cu step sites and that these In sites could hinder the adsorption of H and CO. These synergistic effects accounted for an improved selectivity for CO and HCOOH products. Some differences with the research reported by Jedidi et al. and Rasul et al. showed that the In was preferentially located on the edge sites rather than on the corner or flat sites.128 Additionally, DFT calculations also showed that the d-electron nature of Cu remained intact, but the adsorption properties of neighboring Cu was perturbed by the presence of In. Moreover, the prepared Cu–In bimetallic electrocatalyst showed nearly 95% FE of CO under a −0.7 V vs. RHE condition. Recently, Larrazábal and coauthors prepared Cu–In nanoalloy electrocatalysts by an in situ reduction of CuInO2− and In2O3-supported Cu nanoparticles.129 This study showed that In(OH)3 played a key role in CO formation, which indicated that catalysts with controlled interfaces would be beneficial for the desired reaction.
3.3 Chalcogenides
 | ||
| Fig. 7 (a), (b) SEM images of SnS2 nanosheets on carbon cloth (CC) and porous SnO2 nanosheets on CC. (c) XRD patterns of SnS2 and SnO2 nanosheets. (d) Current density of the SnO2/CC electrode in CO2 saturated 0.5 M NaHCO3 solution at different applied potentials. (e) The corresponding FE for CO, H2, and formate (f) Tafel slope of the SnO2/CC electrode.135 Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Another transition metal oxide, MoO2, has drawn attention for the EC reduction of CO2. Oh et al.136 reported the highest catalytic activity of MoO2 toward CO2 EC reduction in acetonitrile with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as an organic solvent. The highest activity was observed at −20 °C and the addition of a small amount of water promoted the performance. After that, the same group reported an enhanced catalytic activity of MoO2 in several ionic liquids based on imidazolium acting as cocatalysts and an electrolyte simultaneously. The catalytic activity of MoO2 toward CO2 reduction was enhanced by the addition of BMIN-PF6 in acetonitrile. The improvement in catalytic activity and the change in product selectivity proved the combined effect of the ionic liquid acting as both an electrolyte and as a cocatalyst.137
Some other metal oxide electrodes have been reported by various research groups. Kim et al.138 reported a cuprous oxide (Cu2O) electrode for the highly effective conversion of CO2 to lower hydrocarbon fuels and also reported its higher selectivity for ethylene (C2H4). In this study, the authors explored the formation of oxygen vacant structures during the electrolysis processes, which led to a higher production of C2H4. For the selective conversion of CO2 into larger oxygenate molecules, the oxide-derived metallic layer and surface oxide coverage were the key factors. Great efforts have been made in order to obtain higher carbon chain products.139–142 Centi and coworkers investigated the behavior of Fe, Co, Cu (mono-, bi-, and trimetallic) carbon nanotube (CNT) electrocatalysts for CO2 reduction and obtained C1–C3 products. The research showed that the inhibition of surface reactivity toward H2 formation plays a key role in improving the performance and for realizing selective electrocatalysts.142 Recently, they deposited ultrafine Cu NPs (less than 3 nm) on the CNTs, which showed a high activity for C2–C3 products (ethanol, acetic acid, isopropanol, and acetone) due to the interfacial catalytic sites between Cu NPs and CNTs.139 Genovese et al. investigated the critical step of C–C bond formation by comparing the reactivity in tests with CO2 and CO, in which formic acid or formaldehyde was initially added.140 The results demonstrated that dissolved CO2 was needed for acetic acid formation, which showed a different pathway toward formic acid.
Sekimato et al. developed Sn- or Si-doped gallium oxide (Ga2O3) cathodes for the selective conversion of CO2 into HCOOH and showed a 80% FE for HCOOH.143 The DFT results showed that the higher selectivity of Ga2O3 for HCOOH production occurred because of the stable intermediate on the surface. This study revealed that compound catalysts could be more efficient catalysts for CO2 EC reduction. Ramesha et al.144 used TiO2 nanostructures for CO2 EC reduction, with methanol observed as the primary product. Ullah et al.145 reported an indium/ruthenium oxide (Ir/Ru oxide) electrocatalyst for CO2 reduction into various hydrocarbons with FEs of 85% and 96% for ethanol at 295 K and 277 K, respectively. It was demonstrated that for CO2 conversion into hydrocarbons, it can be considered as a highly efficient electrode.
:50 vol% ionic liquid/water electrolyte. At η = 0.65 V, the current density for WSe2 reached 330 mA cm−2 with an FE of ≈85%. The DFT calculations revealed that on TMD edges, the CO was stabilized, and the formation of COOH was exergonic unlike on transition metals surfaces, indicating that the conversion of CO2 into CO was kinetically favorable.148 Recently, the same research group reported that the catalytic activity of TMD materials can be further enhanced by adding an appropriate dopant.149 MoS2 doped with 5% Nb exhibited the lowest onset potential of 31 mV. These catalysts showed a TOF for CO one order of magnitude higher compared to the pristine form at an overpotential range of 50−150 mV and two orders of higher magnitude than that of Ag nanoparticles at the potential range of 100−650 mV. The DFT calculations revealed that low concentrations of Nb near the Mo edge atoms could enhance the TOF of CO formation by modifying the binding energies of the intermediates to MoS2 edge atoms. This work indicated that embedding a dopant into the atomic structure of the catalysts could open a promising route to enhance the catalytic performance of TMD materials.
Nørskov et al.150 explored the active edge sites of MoS2, MoSe2, and Ni-doped MoS2 by using DFT calculations. These edges showed the unique property of the selective binding of COOH and CHO to S or Se atoms and CO to the metal atom. The DFT calculations suggested a significant improvement in CO2 reduction activity over the transition metals. Furthermore, the same authors found that the metal-doped MoS2 provided two different linear scaling relationships. This was due to a combination of dopant metal sites for *CO binding and S binding for *COOH, *CHO, and *COH. This synergistic result enhanced the CO2 reduction performance compared to pristine Mo metal.151 Han et al.152 reported a Mo–Bi bimetallic chalcogenide (BMC) as an efficient electrocatalyst for CO2 reduction into methanol. The Mo–Bi BMC showed a higher FE of 71.2% with a current density of 12.1 mA cm−2 at −0.7 V vs. SHE for methanol. The higher catalytic activity of the electrode resulted from the excellent synergistic effect of Mo and Bi, where the formation of CO from CO2 was favored by Bi and Mo, which further promoted the production of methanol by the hydrogenation of CO. In the future research into CO2 fixation, more efforts should be devoted to metal chalcogenides.
3.4 Metal-free catalysts
Renewable metal-free electrodes could be a next generation catalyst for CO2 EC reduction because of their distinct properties, such as low cost and flexibility.153 Recently, nanostructured carbon materials, like S, N-doped carbon nanofibers, carbon nanotubes, fullerenes, and graphene, have been studied for CO2 EC reduction.55 These nanostructured carbon materials have different properties, including high stability, outstanding conductivity, and mechanical strength, as well as all being cheap and environmentally friendly. Kumar et al.154 reported the catalytic activity of polyacrylonitrile-based heteroatomic carbon nanofibers (CNFs) as a low-cost metal-free electrocatalyst for CO2 EC reduction. This metal-free electrocatalyst showed a higher efficiency for CO2 EC reduction as compared to noble metal catalysts (e.g., Ag) at a low overpotential of 0.17 V, and also exhibited remarkable selectivity for CO production. Fig. 8 illustrates the structural characterization and electrocatalytic performance of a CNFs electrocatalyst.154 The authors found that the nitrogen (N) in the CNFs did not directly participate in the CO2 reduction processes, and that the oxidized carbon atoms were responsible for CO2 reduction. The authors proposed a possible reaction mechanism for the CO2 electroreduction in the electrolyte 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIM-BF4) ionic liquid; whereby during the redox cycling process, the oxidized carbon atoms are initially reduced. Then, an intermediate EMIM–CO2 complex is formed, which when dissolved the CO2 molecules coordinate with EMIM cations. These further adsorb on the reduced carbon atoms, which is accompanied by CO2 reduction to CO. Finally, the reduced carbon atoms are reoxidized to their initial state and the produced CO is released. So the metal-free catalyst CNFs possess a high catalytic proficiency due to redistribution of the spin density and charge between the doped atoms of nitrogen with the nearby carbon atoms. Recently, N-doped carbon nanotubes (NCNTs) as a metal-free electrocatalyst were developed by Wu et al.155 These NCNTs showed a high efficiency, stability, and selectivity for the reduction of CO2 into CO. The authors used liquid chemical vapor deposition (CVD) for fabrication of the NCNTs, which had a multiwall bamboo-shaped structure with a uniform distribution of N atoms of almost 5 atom%. The N content with the configuration of graphitic, pyrrolic, and pyridine are 1.24, 1.1, and 1.5 atom%, respectively. To understand the mechanism for CO2 reduction and the role of the N dopant, the binding energies of CO and COOH were calculated by density field theory (DFT) simulations and a computational hydrogen electrode model. The pyridine N has strong binding with the COOH and at the same time eases the desorption of CO, so the higher selectivity toward CO is due to the N of pyridine. The NCNTs showed an 80% FE toward CO at −0.26 V. The authors proposed that for the commercial reduction of CO2, NCNTs could be a good alternative to high-cost noble metallic electrocatalysts.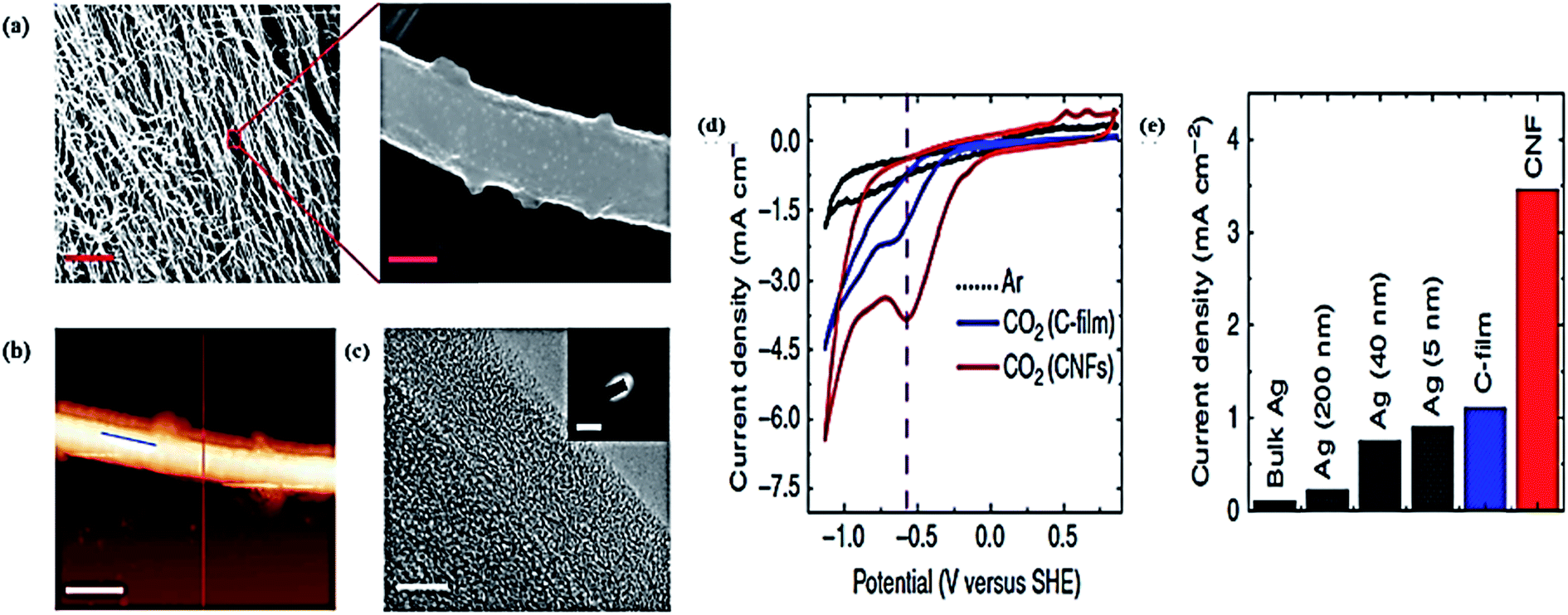 | ||
| Fig. 8 (a) SEM image of a CNFs mat-entangled fibers and high resolution SEM images of an individual fiber (scale bar, 5 nm and 200 nm). (b) Atomic force microscopic image of an individual fiber (scale bar, 500 nm). (c) TEM image (scale bar, 5 nm) of the edge of the fiber surface. (d) Electrocatalytic performance of the CNF CVs for CO2 reduction. (e) The absolute current density for CO2 reduction at different electrodes (bulk Ag, Ag nanoparticles, and CNF) in pure EMIM-BF4 electrolyte.154 Copyright 2013 Nature Publishing Group. | ||
More recently, Wu and coworkers introduced N-doped graphene quantum dots (NGQDs) as a metal-free electrocatalyst for the reduction of CO2 into more useful products.156 The NGQDs were fabricated from graphene oxide (GO) cutting and exfoliating, followed by N doping at elevated temperature and pressure in dimethylformamide (DMF) solvent. The CO2 EC reduction was performed in 1 M KOH electrolyte, and it was shown that the overall FE efficiency increased from 18% to 90% at potentials varying from −0.26 to −0.75 V. At −0.26 V, the reduction products of CO and HCOO− were observed and other different hydrocarbons and multicarbon oxygenates, including methane (CH4), C2H4, C2H5OH, n-propanol (n-C3H7OH), and acetate (CH3COO), were also formed at more negative potentials with a maximum FE of 31% for C2H4 as the major hydrocarbon product and 26% for multicarbon oxygenates. The structure characterization revealed that the doping of heteroatom N was responsible for the improved selectivity and activity of NGQDs. Xu et al. synthesized a series of N-doped carbon nanotube materials with tailored nitrogen contents and varying fractions of pyrrolic, pyridinic, and quaternary nitrogen species. They also studied the CO2 reduction performance on these catalysts, which showed excellent performances with a 90% current efficiency for CO and stability over 60 h. Their research showed that the most active nitrogen chemical state for CO2 reduction is quaternary nitrogen, followed by pyridinic and pyrrolic nitrogen, which provide researchers a strategy for designing nanocarbon catalysts for CO2 reduction.157 Recently, Ju et al. further investigated the metal (containing Mn, Fe, Co, Ni, and Cu)-nitrogen-doped carbon catalysts for CO2 reduction. They studied their intrinsic catalytic reactivity, CO turnover frequencies, and CO faradaic efficiencies, which show that the Ni–N–C catalysts can rival Au- and Ag-based catalysts. They also provided reactivity–selectivity descriptors combining density functional theory (DFT) calculations and experimental data, which led to a greater atomic scale understanding of potential-dependent CO and hydrocarbon selectivity from the M–Nx moieties (M = Mn, Fe, Co, Ni, and Cu).158
Bandosz et al. investigated polymer-derived S-doped and dual S, N-doped nanoporous carbon materials as metal-free electrocatalysts for CO2 EC reduction.159 The S, N-doped electrocatalyst showed a higher selectivity for CO and CH4 production with FEs of 13.8% and 0.18% at −0.99 V vs. RHE, respectively. The authors demonstrated that the high catalytic performances of the heteroatom-doped carbon materials were due to the positively charged sites of the carbon matrix next to the electronegative nitrogen atoms of pyridine and sulfur atoms in the thiophene configuration. The CO production was promoted by stabilization of the intermediates. The catalytic selectivity for the production of CH4 was improved due to the basicity of the surface and ultra-microspores. Jhong et al. developed a metal-free N-doped carbon catalyst for CO2 EC reduction comprised of carbon nitride and multiwall carbon nanotube composites.160 The morphology and electrocatalytic performance are shown in Fig. 9. This material exhibited a higher activity and selectivity for CO and H2 production, with FEs of 98% and 2%, respectively. Also, compared to Ag nanoparticles-based catalysts, this material showed a maximum current density of 90 mA cm−2. The mass activity and energy efficiency of up to 48% were also higher than the Ag nanoparticles reference.
 | ||
| Fig. 9 (a) Reconstructed 3D view of the electrode obtained from MicroCT data. (b) SEM cross-sectional image of the CN/MWCNT catalyst layer on the microporous layer of the gas diffusion electrode. (c) SEM images of the CN/MWCNT electrocatalyst layer (loading 2.39 mg cm−2). Electrocatalytic performance of CN/MWCNT and Ag catalysts: (d) partial current density for CO and H2versus the cathode potential, (e) FE efficiency for CO and H2versus the cathode potential.160 Copyright 2017 John Wiley and Sons. | ||
4 PEC reduction of CO2
The PEC reduction of CO2 has attracted extensive interest due to its high efficiency compared to the PC and EC approaches. However, reduction of the protons or H2O leads to a decrease in the CO2 conversion efficiency, production selectivity, and FE during the CO2 reduction process. Moreover, the stability and improper band gap of the semiconductor are also potential problems, making it suffer from a limited light irradiation range and high overpotential. Therefore, various semiconductors have been designed and employed as photocathodes to convert CO2 to other useful fuels. The PEC cells for CO2 reduction can be categorized into three typical types, which are shown in Fig. 10.38 The semiconductors can be used as photocathodes, photoanodes, or both photocathodes and photoanodes. In this review, we mainly divide semiconductor photoelectrodes into the following three categories: (1) phosphides; (2) metal oxides; and (3) metal-free photoelectrodes. | ||
| Fig. 10 Schematics of three typical two-compartment PEC cells separated by proton-exchange membranes for CO2 reduction: (a) semiconductor as a photocathode; (b) semiconductor as a photoanode; (c) semiconductor as both a photocathode and photoanode.38 Copyright 2016 Royal Society of Chemistry. | ||
4.1 Phosphides
Since Halmann161 first reported P-GaP as an efficient photocathode for the reduction of CO2, the reduction of CO2 by the PEC method has been investigated by many researchers. Many utilized P-GaP under 365 nm wavelength illumination with an applied overpotential of −1.4 V vs. SCE, but the current efficiency was very low. Later, an FE of 80% was obtained with a P-GaP cathode at −1.0 V (vs. a standard silver electrode, NSE) in 0.5 M Na2CO3 solution under 8.5 atm pressure of CO2, with a 150 W Xe lamp, as reported by Aurian-Blajeni et al.162 After many efforts, Barton and coauthors designed a program and used P-GaP to reduce CO2 to methanol, achieving nearly a 100% FE in acidified aqueous pyridine solution, which was greater than 300 mV below the standard potential of −0.52 V vs. SCE at a pH of 5.2.163 In 2015, Kronawitter et al. uncovered the mechanism of the high FE reported by Aurian-Blajeni etc. through experimental and theoretical calculations.164 They demonstrated that a partially dissociated surface layer containing Ga–OH, H2O and P–H species was produced when it was exposed to water, and the strongly surface hydrides contributed greatly to the low H2 evolution efficiency. Recently, GaP as an attractive photoelectrode has been investigated by other researchers as well.165–167 Carter's group did a lot of work on pyridine-catalyzed CO2 reduction with GaP photoelectrodes,166–171 and also clarified the role of pyridinium in the pyridine-catalyzed CO2 reduction on GaP photocathodes.168,169 In their research system, they proposed an energetically viable pathway for pyridinium reduction on the P-GaP photocathode, which led to the formation of adsorbed species that react to form adsorbed dihydropyridine, which played the role of the active catalyst. Recently, they used density function theory (DFT) calculations and experiments to demonstrate that surface hydrides were transferred to adsorbed pyridine, accompanied by aqueous proton addition, to form the active catalyst adsorbed dihydropyridine, which in turn transfers hydrides to CO2, leading to its reduction.171Another phosphide, InP, has a nearly ideal band gap for the efficient conversion of solar energy to electrical or chemical energy, and also has a good photoelectric stability.172 In 1984, Parkinson et al. combined the P–InP photoelectrode with a biological catalyst to influence the first step: a two-electron reduction of CO2 to formic acid.172 Kaneco et al. reported that the maximum current efficiency for CO (41.5%) was obtained at −2.4 V for the P–InP photocathode in a methanol-based electrolyte, which was thus more effective than P–Si and P-Ga-As.173 Later, their group also investigated metal-modified P–InP photoelectrodes in a LiOH/methanol-based electrolyte and improved the current efficiency for CO to 80.4%.174 Arai et al. modified the P–InP with an electropolymerized ruthenium complex in water and successfully obtained HCOO− during the CO2 reduction process.175 Sato et al. combined p-type InP with a Ru complex polymer hybrid photocatalyst for the PEC reduction of CO2 to HCOO−.176
Some other efforts to further improve the production selectively, FE, and materials stability of phosphides have also been reported.163,177–179 Zeng et al. used a TiO2-passivated GaP photocathode for CO2 reduction under 532 nm wavelength illumination, which showed a high FE for methanol.179 They also investigated the influence of the TiO2 layer thickness on the PEC performance, where no enhancement was observed for TiO2 layer thicknesses above 10 nm. Optical microscopy and atomic force microscopy images of bare GaP and TiO2-passivated GaP are shown in Fig. 11. Recently, Qiu et al. reported that the TiO2-passivated InP photocathode's FE for methanol could be improved to 4.79%, which was 5.7 times higher than that for the bare InP at −0.6 V vs. NHE.180 In addition, copper was also deposited on TiO2, and acted as a cocatalyst to further improve the selectivity and yield of methanol production by up to 8-fold with an FE of 8.7%.180 Zeng et al. deposited TiO2 on the InP surface and used this for CO2 reduction. The FE for CO reached 99% with a potential of +0.78 V in the ionic liquid electrolyte (1-ethyl-3-methylimidazolium tetrafluoroborate, [EMIM]BF4).181
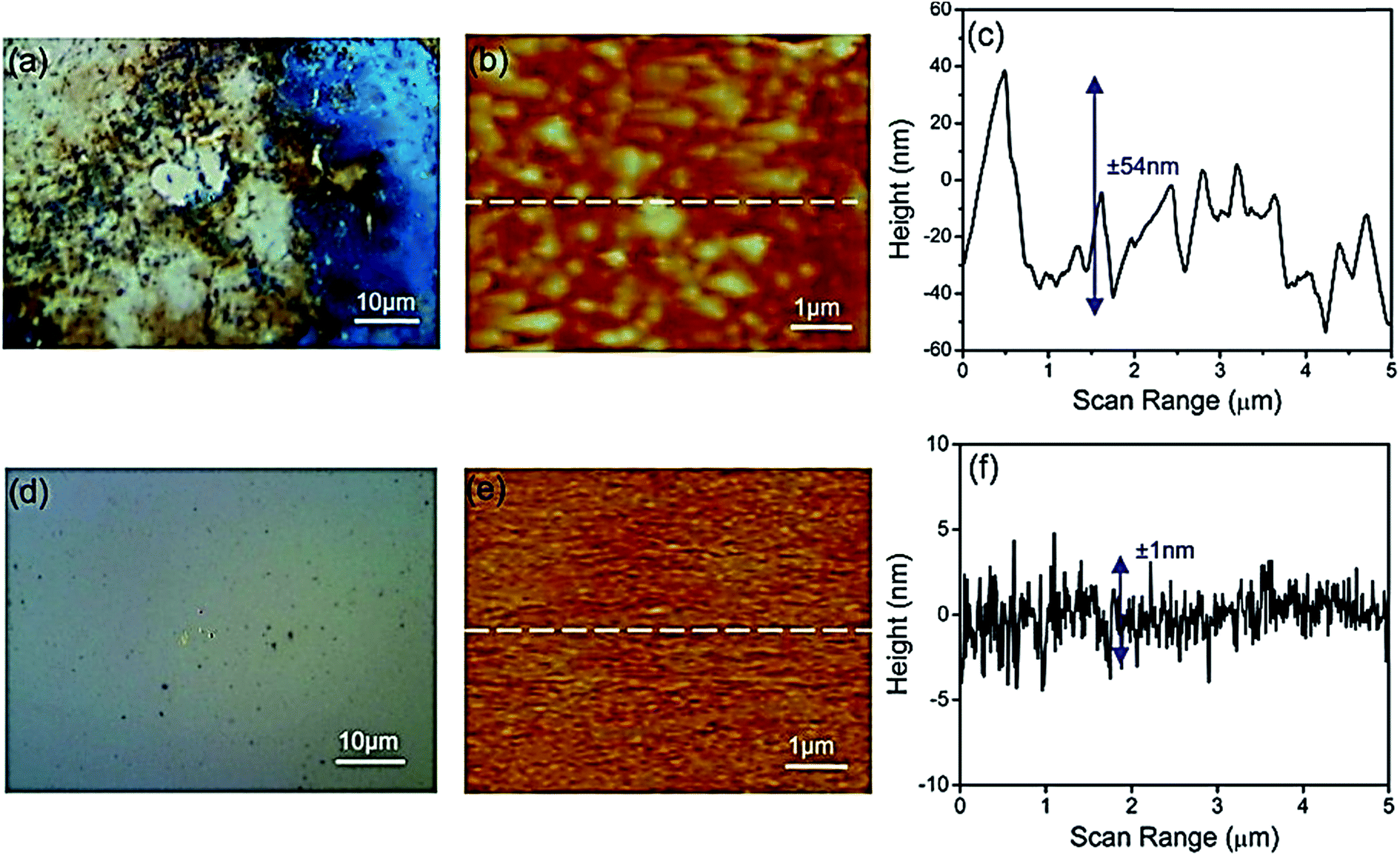 | ||
| Fig. 11 (a) Optical microscope image, (b) atomic force microscope image, and (c) surface topography of the bare GaP surface after an 8 h reaction at −0.5 V overpotential. (d) Optical microscope image, (e) atomic force microscope image, and (f) surface topography of 5 nm TiO2 on the GaP surface after an 8 h reaction.179 Copyright 2014 American Chemical Society. | ||
4.2 Metal oxides
Most metal oxides can be used as a semiconductor, and have thus attracted great attention and been widely studied in the photocatalysis, electrocatalysis, and photoelectrocatalysis research fields. Among these, TiO2, ZnO, and CuxOy, have been widely used as photoelectrodes for the PEC reduction of CO2. | ||
| Fig. 12 (a) View and schematic diagram of the PEC system (a1, view of the lab-scale PEC device; a2, image of a photo/electrocatalytic disc; a3, scheme of a PEC device for CO2 reduction), (b) SEM image of TiO2 nanotubes, (c) SEM cross-sectional image of TiO2, (d) SEM image of Pt-modified TiO2 nanotubes (Pt-TNT), (e) and (f) AFM images and section analysis of graphene oxide (GO): (e) single layer and (f) double layers, (g) and (h) AFM images and section analysis of Pt-modified reduced graphene oxide (Pt-RGO): (g) single layer and (h) double layers. (a).189 Copyright 2010 Royal Society of Chemistry; (e), (f), (g), and (h).188 Copyright 2014 American Chemical Society; (b), (c), and (d).185 Copyright 2014 Elsevier. | ||
In most cases, TiO2 is used as the photoanode, while CO2 can only be reduced on the cathode, and thus the products greatly depend on the material used for the cathode. Yamamoto et al. reported TiO2 as a photoanode in a methanol electrolyte for the PEC reduction of CO2, where the main product on the Pb cathode was methyl formate. When Ag was used as the cathode, the main product was carbon monoxide.190 Zhou et al.191 fabricated a solar-driven CO2 reduction cell, consisting of a GaAs/InGaP/TiO2/Ni photoanode for the oxygen-evolution reaction (OER) and a Pd/C/Ti mesh cathode for CO2 reduction in 2.8 M KHCO3 solution. At the operational current density of 8.5 mA cm−2, the cathode exhibited <100 mV overpotential and >94% FE for the reduction of CO2 (g) to formate. The anode exhibited a 320 ± 7 mV overpotential for the OER in 1.0 M KOH (aq), while the bipolar membrane exhibited a ∼480 mV voltage loss with minimal product crossovers and >90% and >95% selectivity for protons and hydroxide ions, respectively.
There are also some reports using TiO2 as a photocathode. Xu et al. designed a PEC cell containing a multifunctionalized TiO2 photocathode and Co–Pi/W:BiVO4 anode.192 On the surface of the TiO2 film, Pd nanoparticles were introduced to catch protons, while molecules of Eosin Y disodium salt were used to absorb solar energy, and four different amine ligands were immobilized to capture and activate the CO2.192 On the optimized sample, the maximum formation rate and highest selectivity for methanol were 43.6 μM cm−2 h−1 and 100%, respectively. Some methods to further improve its efficiency are also available.192–196 A heterojunction structure and cocatalysts are efficient methods to improve the CO2 conversion efficiency and the products FE.
 | ||
| Fig. 13 Time-profiled production of CO and H2 for the ZnTe/ZnO–NWs (ZOZT) (a and b, respectively) and the ZnTe/ZnO–NWs (ZOZT–Au) (c and d, respectively) in CO2-purged 0.5 M KHCO3 electrolytes under simulated 1 sun illumination at various applied potentials: −0.1, −0.3, −0.5, and −0.7 VRHE; (e) schematic illustration of the Au coupled ZOZT; (f) faradaic efficiencies of CO and H2 for the bare ZOZT (A) and Au coupled ZOZT (B) at different applied potentials in CO2-saturated KHCO3 electrolyte for 3 h. The values above the top of each column present the selectivity of CO from CO2 reduction relative to H2 from H2O.198 Copyright 2015 Royal Society of Chemistry. | ||
 | ||
| Fig. 14 (a) Schematic model of a PEC device for the selective conversation of CO2 to CO; cathode: mesoporous Pd7Cu3 alloy; photoanode: ternary hierarchical CNTs-ZnO-Co3O4 NW composite; (b): FEs for CO over the samples; (c) IPCE spectra of M-PdCu/T-ZnO at a high FECO bias region.201 Copyright 2016 Royal Society of Chemistry. | ||
Although the coating of a protective layer can improve the stability and CO2 conversion efficiency, the active sites for the preferential reduction of CO2 on the Cu2O surface may be covered by the protective layers. As a result, a large amount of H2 may be generated, resulting in a decreased selectivity for carbonaceous products. Chang et al. used Cu2O as the dark cathode and TiO2 as the typical photoanode for CO2 reduction, which showed an 87.45% FE and 92.65% selectivity for all carbonaceous products at a low potential of 0.75 V vs. RHE.210 This strategy could significantly improve the stability of Cu2O and its selectivity for carbonaceous products.
CuO has also been reported as a photoelectrode for PEC CO2 reduction, where it has been frequently combined with other materials to enhance the performance.209,211–215 Kang et al. used a CuFeO2/CuO electrode to convert CO2 to formate, with over 90% selectivity under simulated solar light (100 mW cm−2) at +0.9 V vs. RHE.215 Moreover, a strategy connecting the CuFeO2/CuO photocathode to a Pt anode was further developed, which could produce formate over 1 week at a solar-to-formate energy conversion efficiency of about 1% (selectivity of >90%) without any external bias. The PEC cell and performances are shown in Fig. 15. Yang et al. also fabricated a CuFeO2/CuO photoelectrode for the PEC reduction of CO2, where the FE for acetate reached 80% at −0.4 V vs. Ag/AgCl under visible light irradiation.214 Meanwhile, the selectivity for CO2 production could be tuned by controlling the composition ratio of mixed Fe–Cu oxide catalysts. This article reported a material with the ability to efficiently couple C–C bonds and generate acetate on such a low-cost, earth-abundant catalyst during CO2 reduction.
 | ||
| Fig. 15 (a) Comparison of linear sweep voltammograms for CuFeO2/CuO, CuFeO2, and CuO electrodes. (b) Time-profiled formate production with CuFeO2/CuO electrodes at +0.15 VRHE in bicarbonate (0.1 M) solutions (PCE-1). (c) Time-profiled formate production with open-circuited CuFeO2/CuO, CuFeO2, and CuO electrodes in CO2-purged bicarbonate solution (PEC-2). No potential biases were applied. (d) Time-profiled changes in cell voltage (Ecell), simultaneous production of formate, and solar-to-formate (STF) energy conversion efficiencies (PEC-3 and 4). (e) Changes in Ecell and formate production with a wired CuFeO2/CuO and Pt couple in CO2-purged bicarbonate (0.1 M) solution (PEC-4).215 Copyright 2015 Royal Society of Chemistry. | ||
4.3 Metal-free
Metal-free photoelectrodes have attracted great attention due to their many advantages, such as low cost, environmental friendliness and high charge-carrier mobility. Graphitic, silicon (Si), and C3N4 as typical metal-free photoelectrodes have been widely used in various fields. Tahir et al. reported a review on graphene and its unique structure, band structure engineering, physical and chemical properties, applications in solar energy conversation (including the PEC reduction of CO2), energy storage, environment purification, and biosensors.216 However herein, we do not discuss the PEC performance for CO2 reduction by using a graphene photoelectrode.Graphene-like carbon nitride (C3N4), as a metal-free semiconductor, has also attracted great attention for CO2 reduction. However, most of the reports about C3N4 are about its use as a photocatalyst, and there are few works on C3N4 film as a photoelectrode for CO2 reduction.217–220 Recently, Azofra and coauthors investigated the role of corrugation in C3N4 during CO2 conversion by a DFT method.221 Besides, Sagara et al. fabricated B-doped g-C3N4 as a photoelectrocatalyst for CO2 reduction, whose photocurrent response was about 5 times higher than that of pure g-C3N4.222 In addition, they also investigated the effects of a cocatalyst (including Au, Ag, and Rh) on the CO2 reduction performance. Although they have done lots of work toward improving the CO2 reduction performance, the selectivity, FE, and productivity are still unsatisfactory. Hence, efforts are still needed to further improve the performance of C3N4 for CO2 reduction.
Si is another good candidate as a photoelectrode for CO2 reduction. Recently, a mini review of silicon materials for solar-to-chemical conversion have involved some PEC CO2 reduction processes.223 Zhang et al. designed a PEC cell composed of a nanoporous Ag cathode and a Ni-coated Si photoanode for CO2 conversion to CO.224 The photocurrent density could reach up to about 10 mA cm−2 with a 70% FE for CO at 2.0 V vs. RHE. Song et al. coated a nanoporous Au thin film on Si photoelectrodes for CO2 reduction, which exhibited an FE of 96% for CO production at 480 mV overpotential.225,226 The schematic of electron transfer, CO2 reduction, and the FE of CO are shown in Fig. 16. Additionally, Wang et al. designed a monolithically integrated GaN nanowire/Si solar cell photocathode for CO2 reduction, where the FE for CH4 (19%) production was more than 30 times higher than that for CO.227 Urbain et al. recently designed a bias-free reactor device for CO2 conversion. In this system, a Si heterojunction solar cell with Ni foam catalyst was used as a photoanode to facilitate the OER, while a Cu foam coated with nanosized Zn flakes was used as a cathode for the CO2 reduction reaction.228 A stable and tunable H2-to-CO ratio between 5 and 0.5 along with an 85% FE and 39.4 mA cm−2 current density for CO was obtained in their work. In order to understand the reaction mechanism of CO2 reduction, Sekimoto and coauthors carried out 13CO2-labeling experiments on a GaN-Si-based tandem photoelectrode in KCl and KHCO3 solutions, respectively.229 The results showed that almost all the products (including HCOOH, CH4, and C2H4) were converted form 13CO2 when KCl was used as the electrolyte. However, HCOOH from 12CO2 was observed when KHCO3 was used, indicating that HCO3− partly contributed to CO2 reduction.
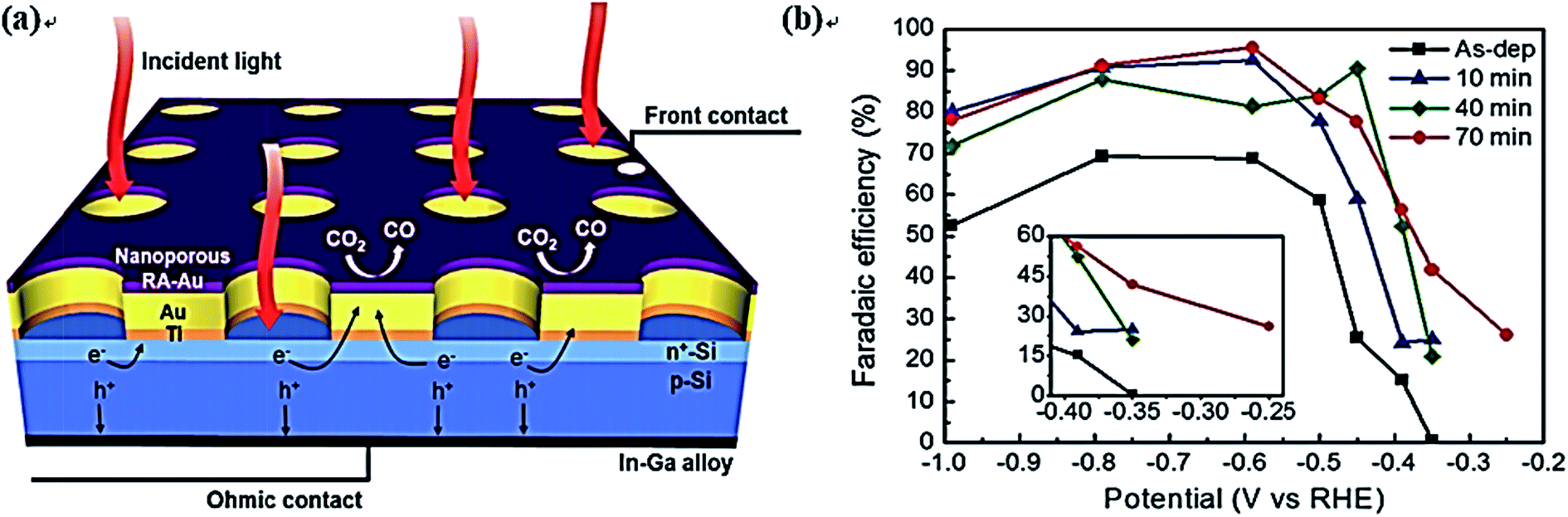 | ||
| Fig. 16 (a) Si photoelectrode with Au mesh cocatalysts: schematic of the photoelectrode design for CO2 reduction, where mesh-type cocatalysts form on the Si photoelectrode with a buried p–n junction. The mesh-type cocatalysts allow independent wiring on top of the catalyst surface for the RA treatment. Ohmic contact was made on the backside of the photoelectrode to collect photoexcited holes; (b) faradaic efficiency for CO production from the RA-treated Au thin film. Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
5 Summary and perspectives
To date, many efforts have been made to design highly selective, highly conversation efficient, and energy-saving electrocatalysts and photoelectrocatalysts for CO2 reduction. The main products and FE for the EC and PEC reduction of CO2 are summarized and shown in Tables 2 and 3.| Electrocatalyst | Electrolyte | Overpotential | Major products | FE | Ref. |
|---|---|---|---|---|---|
| Ag nanoporous | 0.5 M KHCO3 | <500 mV vs. RHE | CO | 92% | 79 |
| Ag nanocorals | 0.1 M KHCO3 | 0.37 V vs. RHE | CO | 95% | 83 |
| Ag nanoparticles | 0.5 M KHCO3 | −0.75 and −1.0 V vs. RHE | CO | 94.2% | 84 |
| Ag foam | 0.1 M KHCO3 | −0.99 V vs. RHE | CO | 94.7% | 85 |
| Au nanoparticles (4 nm, 8 nm) | 0.5 M KHCO3 | −0.9 V vs. RHE | CO, H2 | Up to 90% | 89 |
| Au nanowires (500 nm) | 0.5 M KHCO3 | −0.35 V vs. RHE | CO | 94% | 91 |
| Au nanoneedles | 0.5 M KHCO3 | −0.30 V to −0.50 V vs. RHE | CO | ≥95% | 21 and 94 |
| Cu nanoparticles | 0.1 M KHCO3 | −1.1 V vs. RHE | CO, H2 | 60–70%, 20–30% | 92 |
| Cu nanowires | 0.1 M KHCO3 | 0.3 V vs. RHE | CO | 60% | 100 |
| Ultrathin Cu nanowires (diameter 20 nm) | 0.1 M KHCO3 | −1.25 V vs. RHE | CH4 | 55% | 101 |
| Cu nanocrystals | 0.1 M KHCO3 | −1.1 V vs. RHE | H2, CO, CH4, C2H4 | 41% (ethylene) | 102 |
| Zn nanoparticles | 0.5 M NaHCO3 | −0.93 V to −2.13 V vs. RHE | HCOO− | 87% | 115 |
| Zn dendrite nanostructures | 0.5 M NaHCO3 | 0.9 V to −1.1 V vs. RHE | ∼80% | 75 | |
| Au3Cu alloys | 0.1 M KH2PO4/0.1 M K2HPO4 | −1.6 V vs. Ag/AgCl | CO | 30% | 123 |
| Cu–Sn bimetallic alloys | 0.1 M KHCO3 | −0.6 V vs. RHE | H2, CO HCOOH, CH3COOH | ≥90% (CO) | 124 |
| PdCu/C nanoparticle alloys | 0.1 M KHCO3 | −0.89 V vs. RHE | CO | 86% | 118 |
| Sn–Pb alloys | 0.5 M KHCO3 | −0.2 V vs. Ag/AgCl | HCOO− | 79.8% | 126 |
| Graphene (GN)/Cu2O | 0.5 M NaHCO3 | −1.7 V vs. Ag/AgCl | Ethanol (predominant product) | 9.95% | 130 |
| SnO2 nanoparticles | 0.1 M NaOH | −1 to −1.8 V vs. SCE | HCOO− | 93% | 133 |
| 3D SnO2 nanoparticles | 0.5 M NaHCO3 | −1.6 V vs. Ag/AgCl | H2, HCOO− | 87% | 135 |
| Ga2O3 | 3.0 M KCl & 5.0 M NaOH | −1.6 to −2.0 V vs. Ag/AgCl | HCOOH | 80% | 143 |
| N-doped graphene quantum dots (NGQDs) | 1 M KOH | −0.20 to −1.10 V vs. RHE | CO, HCOO− | 90% | 156 |
| Carbon nanoporous (CNF), (CPNS) | 0.1 M KHCO3 | −0.99 V vs. RHE | CO, CH4 | 13.8%, 0.18% | 159 |
| N-doped carbon | 1.0 M KCl | −1.46 V vs. Ag/AgCl | CO, H2 | 90%, 2% | 160 |
| Photoelectrocatalyst | Electrolyte | Conditions | Products | FE | Ref. |
|---|---|---|---|---|---|
| P-GaP | 0.1 M acetate buffer containing 10 mM pyridinium | 200 W Hg–Xe arc light source, pH = 5.2, External bias: −0.52 V vs. SCE | Methanol | ∼100% | 163 |
| InP | Methanol | Xenon lamp: 1.5 mA cm−2, External bias: −2.4 V vs. Ag/AgCl | CO | ∼41.5% | 173 |
| Metal-modified P–InP | LiOH/methanol | Xenon lamp: 1.5 mA cm−2 | CO | 80.4% | 174 |
| InP/TiO2/Cu | 0.5 M KCl | Under 532 nm illumination, External bias: −0.6 V vs. NHE | Methanol, H2 | 8.7%, 76.9% | 180 |
| CdSeTe NSs/TiO2 | 0.1 M KHCO3 | 500 W xenon lamp (λ ≥ 420 nm, 100 mW cm−2), External bias: −0.8 V vs. NHE | Methanol, Methane | 87.95% (selectivity: 74.2%), 40.79%, (Selectivity: 25.81%) | 182 |
| Si/TiO2/Pt | 0.1 M NaHCO3 | 125 W (250–600 nm, 21 mW cm−2), External bias: −0.8 V | Methanol, Methane, Acetone | Total FE: 96% | 183 |
| Cathode: Pt-RGO/Cu foam, Photoanode: TiO2 nanotube | Catholyte: 0.5 M NaHCO3, Anolyte: 0.5 M H2SO4 | UV Xe lamp, Potential between cathode and anode is about 2 V | Ethanol, Acetic acid, Propanoic acid | Total conversation rate: 4340 nm (h−1 cm−2) | 184 |
| Cathode: Pb or Ag, Photoanode TiO2 | Catholyte: 0.3 M KOH in methanol, Anolyte: 3.0 M KHCO3 in water | UV LED 365 nm, Potential: 2 V vs. Ag QRE | Pb: formic acid, CO and H2, Ag: formic acid, CO and H2 | Pb: 80%, 9.5%, 5.1%, Ag: 13%, 61%, 3.3% | 190 |
| Cathode: Pd/C/Ti mesh, Photoanode: GaAs/InGaP/TiO2/Ni | Catholyte: 2.8 M KHCO3, Anolyte: 1.0 M KOH | <100 mV overpotential, 1 atm CO2 (g) | Formate | >94% | 191 |
| ZnTe–ZnO/Zn | 0.5 M KHCO3 | 500 W Hg lamp (>420 nm, 400 mW cm−2), Potential: −0.7 V vs. RHE | CO, H2 | CO: 22.9%, H2: 60.7% | 197 |
| Au–ZnTe/ZnO nanowire array | 0.5 M KHCO3 | 100 mW, Potential: −0.7 V vs. RHE | CO, H2 | CO: 58%, H2: 31.8% | 198 |
| Cathode: PdCu, Photoanode: CNTs–ZnO–Co3O4 nanowire | AM 1.5, Potential: −1.2 V vs. RHE | CO | 75% | 201 | |
| Cu2O/CuO nanorode array | 0.1 M NaHCO3 | AM 1.5 (70 mW cm−2), Potential: −0.2 V vs. SHE | Methanol | 95% | 202 and 203 |
| p-type CuO/Cu2O nanorode array | 0.1 M NaHCO3 | AM 1.5 (70 mW cm−2), Potential: −0.3 V vs. SHE | Alcohols | 2 h, Ethanol: 52%, Isopropanol: 40%, Methanol: 4% | 204 |
| Cu/Cu2O | 0.1 M KHCO3 | Blue LED, 435–450 nm, Potential: −2.0 V vs. Ag/AgCl | C2H4 | Cu/Cu2O (p-type, nanobelt arrays): 32.69%, Cu/Cu2O (p-type, stone-like): 30.60%, Cu/Cu2O (n-type, stone-like): 26.55% | 205 |
| Cu/Cu2O | 0.1 M NaCO3/NaHCO3 with different PH | AM 1.5, 100 mW cm−2, +0.2 V vs. Ag/AgCl, PH = 9 | Methanol, Ethanol, Formaldehyde, Acetaldehyde, Acetone | 80% of CO2 is converted | 206 |
| CuFeO2/CuO | 0.1 M KHCO3 | AM 1.5, 100 mW cm−2, Potential: +0.9 V vs. RHE | Formate | 90% | 215 |
| CuO/CuFeO2 (Fe:Cu = 1.3) |
0.1 M NaHCO3 | AM 1.5, 100 mW cm−2, Potential: −0.4 V vs. Ag/AgCl | Acetate | 80% | 214 |
| Cathode: Cu2O, Photoanode: TiO2 nanorod array | 0.1 M KHCO3 | AM 1.5, 100 mW cm−2, Potential: +0.75 V vs. RHE | CH4, CO, CH3OH, H2 | CH4: 54.63%, CO: 30.03%, CH3OH: 2.79%, H2: 6.94% | 210 |
| B-doped C3N4 | 0.5 M NaHCO3 | AM 1.5, 100 mW cm−2, Potential: −0.4 V vs. Ag/AgCl | Ethanol, CO | Ethanol: 78%, CO: 1.2% | 222 |
| Cathode: Ag porous, Photoanode: Ni-coated Si | Catholyte: 0.5 M KHCO3, Anolyte: boric acid, potassium chloride and potassium hydroxide in DI water | 300 W Xe lamp with 400 nm filter, Potential: 2.0 V vs. RHE | CO | 70% | 224 |
| Si photoelectrode with RA-treated Au mesh cocatalyst | 0.2 M KHCO3 | 1 sun, 100 mW cm−2, Potential: −0.59 V vs. RHE | CO | 96% | 226 |
| Cu/GaN/n+–p Si | 0.5 M KHCO3 | 300 W Xe lamp, Potential: −1.4 V vs. Ag/AgCl | CH4 | 19% | 227 |
Although great success has been achieved for CO2 reduction, it is still a big challenge to design and fabricate efficient catalysts. In particular, the low energy conversation efficiency has limited its practical application significantly. Generally, the CO2 adsorption and activation ability of catalysts are the crucial factors for the subsequent reduction steps and suppress the H2 evolution. In order to improve the adsorption and activation ability of electrodes and photoelectrodes, various strategies have been developed, such as controlling the structure and morphology (exposing the active facets), improving the surface area (using porous materials), the active sites (surface defects), cocatalysts, and heterojunctions. In this review, we classified and presented a detailed discussion about the typical electrodes and photoelectrodes for CO2 reduction, which could help the investigators to design a strategy for improving the CO2 conversation efficiency and product selectivity by selecting suitable materials. For developing EC or PEC electrodes with a high energy conversion efficiency, high FE, and good stability for CO2 reduction, whereby several suggestions are proposed:
(1) For selecting the suitable electrodes: the overpotential, CO2 adsorption and activating ability, conductivity, stability, economics, etc. should be carefully evaluated.
(2) To enhance the adsorption and activation performances of CO2: designing a porous structure or inverse opal structures may be a good strategy for improving the products selectivity and CO2 adsorption. Additionally, introducing surface defects or cocatalysts may be another option to maximize the adsorption and activation of CO2 molecules.
(3) For further understanding the mechanism and pathways of CO2 reduction: in situ characterization techniques are very urgently needed and are necessary to understand the reaction mechanism and pathways of CO2 reduction at the molecular level, such as in situ attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR),230 attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS),231in situ Raman,232in situ scanning transmission electron microscopy (STEM), and in situ X-ray photoelectron spectroscopy (XPS). In addition, density functional theory (DFT) calculations are also helpful to understand the pathway of CO2 reduction, especially in guiding researchers to finding the most suitable materials.
Specifically, for PEC electrodes, because light-active semiconductors are involved in this system, several more suggestions are proposed:
(1) For PEC electrodes, the suitable band gap and band-edge potential, are important factors for CO2 reduction. As the photoconversion efficiency of most semiconductors is very low; hence, more efficient semiconductors with superior light-harvesting ability, good charge generation ability, and excellent charge separation and transportation efficiency should be designed.
(2) Enhancing the semiconductor stability and selectivity of CO2 reduction products are urgent and necessary. As we have mentioned in this review, introducing surface defects, surface functional group, a cocatalyst, or doping will benefit the CO2 reduction reaction and semiconductor stability in some way.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from Ministry of Science and Technology of the People's Republic of China (2016YFE0112200), National Natural Science Foundation of China (No. 21507011 and No. 21677037).References
- L. Suganthi and A. A. Samuel, Renewable Sustainable Energy Rev., 2012, 16, 1223–1240 CrossRef.
- P. Nejat, F. Jomehzadeh, M. M. Taheri, M. Gohari and M. Z. A. Majid, Renewable Sustainable Energy Rev., 2015, 43, 843–862 CrossRef CAS.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. R. MacFarlane, G. F. Moore, P. D. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 Search PubMed.
- C. L. A. D. L. Hartmann, S. Brönnimann, F. J. Dentener, E. J. Dlugokencky, D. R. Easterling, A. Kaplan and B. J. Soden, IPCC (2013), Climate Change 2013, in The Physical Science Basis, Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, WMO/UNEP, Cambridge, 2014 Search PubMed.
- R. K. Pachauri, M. R. Allen, V. R. Barros, J. Broome, W. Cramer, R. Christ, J. A. Church, L. Clarke, Q. Dahe and P. Dasgupta, Contribution of working groups I, II and III to the fifth assessment report of the intergovernmental panel on climate change, Climate change 2014: synthesis report, IPCC, 2014 Search PubMed.
- G. A. Olah, A. Goeppert and G. K. Prakash, J. Org. Chem., 2016, 74, 487 CrossRef PubMed.
- X. D. Xu and J. A. Moulijn, Energy Fuels, 1996, 10, 305–325 CrossRef.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
- A. Guais, G. Brand, L. Jacquot, M. Karrer, S. Dukan, G. Grevillot, T. J. Molina, J. Bonte, M. Regnier and L. Schwartz, Chem. Res. Toxicol., 2011, 24, 2061–2070 CrossRef CAS PubMed.
- J. Gong, L. Zhang and Z. J. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef PubMed.
- S. A. Montzka, E. J. Dlugokencky and J. H. Butler, Nature, 2011, 476, 43–50 CrossRef CAS PubMed.
- M. Mondal, S. Khanra, O. N. Tiwari, K. Gayen and G. N. Halder, Environ. Prog. Sustainable Energy, 2016, 35, 1605–1615 CrossRef CAS.
- T. S. Wong, J. Bioprocess. Biotech., 2014, 4, 100155–100170 Search PubMed.
- X. Chang, T. Wang, P. Zhang, Y. Wei, J. Zhao and J. Gong, Angew. Chem., Int. Ed. Engl., 2016, 55, 8840–8845 CrossRef CAS PubMed.
- S. Tan, S. J. Kim, J. S. Moore, Y. Liu, R. S. Dixit, J. G. Pendergast, D. S. Sholl, S. Nair and C. W. Jones, ChemCatChem, 2016, 8, 214–221 CrossRef CAS.
- A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479–4484 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjaer, J. S. Hummelshoj, S. Dahl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- J. Highfield, H. Lim, J. Fagerlund and R. Zevenhoven, RSC Adv., 2012, 2, 6535–6541 RSC.
- J. W. Ko, S. W. Kim, J. Hong, J. Ryu, K. Kang and C. B. Park, Green Chem., 2012, 14, 2391–2394 RSC.
- W. J. Bao, H. Q. Li and Y. Zhang, Ind. Eng. Chem. Res., 2010, 49, 2055–2063 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, Catal. Today, 2014, 224, 3–12 CrossRef CAS.
- R. J. Lim, M. Xie, M. A. Sk, J. M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2013, 233, 169–180 CrossRef.
- A. J. Martin, G. O. Larrazabal and J. Perez-Ramirez, Green Chem., 2015, 17, 5114–5130 RSC.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. Schouten, G. Mul and M. T. Koper, Nat. Commun., 2015, 6, 8177–8185 CrossRef PubMed.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- P. L. Staddon and M. H. Depledge, Environ. Sci. Technol., 2015, 49, 8269–8270 CrossRef CAS PubMed.
- J. Baxter, Z. X. Bian, G. Chen, D. Danielson, M. S. Dresselhaus, A. G. Fedorov, T. S. Fisher, C. W. Jones, E. Maginn, U. Kortshagen, A. Manthiram, A. Nozik, D. R. Rolison, T. Sands, L. Shi, D. Sholl and Y. Y. Wu, Energy Environ. Sci., 2009, 2, 559–588 CAS.
- N. Hazari, E. Iglesia, J. A. Labinger and D. A. Simonetti, Acc. Chem. Res., 2012, 45, 653–662 CrossRef CAS PubMed.
- M. Villano, F. Aulenta, C. Ciucci, T. Ferri, A. Giuliano and M. Majone, Bioresour. Technol., 2010, 101, 3085–3090 CrossRef CAS PubMed.
- A. S. Agarwal, Y. Zhai, D. Hill and N. Sridhar, ChemSusChem, 2011, 4, 1301–1310 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- C. Liu, J. J. Gallagher, K. K. Sakimoto, E. M. Nichols, C. J. Chang, M. C. Chang and P. Yang, Nano Lett., 2015, 15, 3634–3639 CrossRef CAS PubMed.
- O. Martin, A. J. Martin, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferre and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- B. Mondal, F. Neese and S. Ye, Inorg. Chem., 2015, 54, 7192–7198 CrossRef CAS PubMed.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC.
- W. G. Tu, Y. Zhou and Z. G. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- X. X. Chang, T. Wang and J. L. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 CAS.
- Z. L. Wang, C. L. Li and Y. Yamauchi, Nano Today, 2016, 11, 373–391 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2016, 7, 70–88 CrossRef.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- W. Kim, B. A. McClure, E. Edri and H. Frei, Chem. Soc. Rev., 2016, 45, 3221–3243 RSC.
- W. H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- E. S. Sanzpérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194–1700222 CrossRef PubMed.
- J. Kim, D. Summers and K. Frese, J. Electroanal. Chem. Interfacial Electrochem., 1988, 245, 223–244 CrossRef CAS.
- H.-R. M. Jhong, S. Ma and P. J. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- X. Li, J. Q. Wen, J. X. Low, Y. P. Fang and J. G. Yu, Sci. China Mater., 2014, 57, 70–100 CrossRef.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- K. Hara, A. Kudo, T. Sakata and M. Watanabe, J. Electrochem. Soc., 1995, 142, L57–L59 CrossRef CAS.
- C. Ampelli, C. Genovese, M. Errahali, G. Gatti, L. Marchese, S. Perathoner and G. Centi, J. Appl. Electrochem., 2015, 45, 701–713 CrossRef CAS.
- C. Ampelli, C. Genovese, B. Marepally, G. Papanikolaou, S. Perathoner and G. Centi, Faraday Discuss., 2015, 183, 125–145 RSC.
- M. Azuma, K. Hashimoto, M. Hiramoto, M. Watanabe and T. Sakata, J. Electroanal. Chem., 1989, 260, 441–445 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- J. J. Kim, D. P. Summers and K. W. Frese Jr, J. Electroanal. Chem., 1988, 245, 223–244 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, ChemInform, 1990, 21, 2309–2326 Search PubMed.
- B. C. Marepally, C. Ampelli, C. Genovese, T. Saboo, S. Perathoner, F. M. Wisser, L. Veyre, J. Canivet, E. A. Quadrelli and G. Centi, ChemSusChem, 2017, 10, 4442–4446 CrossRef CAS PubMed.
- C. Ampelli, G. Centi, R. Passalacqua and S. Perathoner, Catal. Today, 2016, 259, 246–258 CrossRef CAS.
- C. Ampelli, C. Genovese, B. C. Marepally, G. Papanikolaou, S. Perathoner and G. Centi, Faraday Discuss., 2015, 183, 125 RSC.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2017, 136, 14107–14113 CrossRef PubMed.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 44, 2423–2436 RSC.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- Q. Lu, J. Rosen and F. Jiao, ChemCatChem, 2015, 46, 2 CrossRef.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS.
- J. M. Saveant, Chem. Rev., 2008, 108, 2348–2378 CrossRef CAS PubMed.
- J. H. Koh, H. S. Jeon, M. S. Jee, E. B. Nursanto, H. Lee, J. H. Yun and B. K. Min, J. Phys. Chem. C, 2015, 119, 883–889 CAS.
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 Search PubMed.
- B. A. Rosen, A. Salehikhojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- N. Hoshi, M. Kato and Y. Hori, J. Electroanal. Chem., 1997, 440, 283–286 CrossRef CAS.
- A. Salehi-Khojin, H. R. M. Jhong, B. A. Rosen, W. Zhu, S. C. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627–1632 CAS.
- Y. C. Hsieh, S. D. Senanayake, Y. Zhang, W. Q. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2017, 7, 779–785 CrossRef CAS.
- R. Daiyan, X. Y. Lu, Y. H. Ng and R. Amal, ChemistrySelect, 2017, 2, 879–884 CrossRef CAS.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, J. Am. Chem. Soc., 2012, 134, 10237–10243 CrossRef CAS PubMed.
- D. Gao, J. Wang, H. Wu, X. Jiang, S. Miao, G. Wang and X. Bao, Electrochem. Commun., 2015, 55, 1–5 CrossRef CAS.
- H. Kim, H. S. Jeon, M. S. Jee, E. B. Nursanto, J. P. Singh, K. Chae, Y. J. Hwang and B. K. Min, ChemSusChem, 2016, 9, 2097–2102 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, O. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- H. J. Feng, Y. M. Yang, Y. M. You, G. P. Li, J. Guo, T. Yu, Z. X. Shen, T. Wu and B. G. Xing, Chem. Commun., 2009, 15, 1984–1986 RSC.
- W. Zhu, Y. J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- H. Mistry, R. Reske, Z. Zeng, Z. J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- J. H. Koh, H. S. Jeon, M. S. Jee, E. B. Nursanto, H. Lee, Y. J. Hwang and B. K. Min, J. Phys. Chem. C, 2015, 119, 883–889 CAS.
- T. Saberi Safaei, A. Mepham, X. Zheng, Y. Pang, C. T. Dinh, M. Liu, D. Sinton, S. O. Kelley and E. H. Sargent, Nano Lett., 2016, 16, 7224–7228 CrossRef CAS PubMed.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- Y. Kwon, Y. W. Lum, E. L. Clark, J. W. Ager and A. T. Bell, ChemElectroChem, 2016, 3, 1012–1019 CrossRef CAS.
- Y. Shen, Q. Fang and B. Chen, Environ. Sci. Technol., 2015, 49, 67–84 CrossRef CAS PubMed.
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20–24 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. R. Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- D. Raciti, K. J. Livi and C. Wang, Nano Lett., 2015, 15, 6829–6835 CrossRef CAS PubMed.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 CrossRef CAS PubMed.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- K. D. Yang, C. W. Lee, J. H. Jang, T. R. Ha and K. T. Nam, Nanotechnology, 2017, 28, 352001–352010 CrossRef PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2, 169–174 CrossRef CAS PubMed.
- Y. Kwon, Y. Lum, E. L. Clark, J. W. Ager and A. T. Bell, ChemElectroChem, 2016, 3, 1012–1019 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- Z. M. Detweiler, J. L. White, S. L. Bernasek and A. B. Bocarsly, Langmuir, 2014, 30, 7593–7600 CrossRef CAS PubMed.
- L. Sun, G. K. Ramesha, P. V. Kamat and J. F. Brennecke, Langmuir, 2014, 30, 6302–6308 CrossRef CAS PubMed.
- K. Dhar and C. Cavallotti, J. Phys. Chem. A, 2014, 118, 8676–8688 CrossRef CAS PubMed.
- C. Shi, C. P. O'Grady, A. A. Peterson, H. A. Hansen and J. K. Norskov, Phys. Chem. Chem. Phys., 2013, 15, 7114–7122 RSC.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed.
- T. T. Zhang, H. X. Zhong, Y. L. Qiu, X. F. Li and H. M. Zhang, J. Mater. Chem. A, 2016, 4, 16670–16676 CAS.
- M. Baghayeri, Sens. Actuators, B, 2017, 240, 255–263 CrossRef CAS.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson, B. S. Brunschwig, M. P. Soriaga and N. S. Lewis, ACS Catal., 2016, 6, 2100–2104 CrossRef CAS.
- Z. Yin, D. Gao, S. Yao, B. Zhao, F. Cai, L. Lin, P. Tang, P. Zhai, G. Wang and D. Ma, Nano Energy, 2016, 27, 35–43 CrossRef CAS.
- C. S. Chen, A. D. Handoko, J. H. Wan, L. Ma, D. Ren and B. S. Yeo, Catal. Sci. Technol., 2014, 5, 161–168 Search PubMed.
- M. Watanabe, M. Shibata and A. Kato, J. Electrochem. Soc., 1991, 138, 3382–3389 CrossRef CAS.
- X. Guo, Y. Zhang, C. Deng, X. Li, Y. Xue, Y. M. Yan and K. Sun, Chem. Commun., 2015, 51, 1345–1348 RSC.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 CAS.
- W. G. Zhao, L. N. Yang, Y. D. Yin and M. S. Jin, J. Mater. Chem. A, 2014, 2, 902–906 CAS.
- S. Sarfraz, A. T. Garcia-Esparza, A. Jedidi, L. Cavallo and K. Takanabe, ACS Catal., 2016, 6, 2842–2851 CrossRef CAS.
- I. Choi, H.-Y. Kim, S. H. Ahn, S. J. Hwang, S. J. Yoo, H. Y. Kim, J. Choi, H. J. Park, J. H. Jang and S.-K. Kim, J. Nanosci. Nanotechnol., 2016, 16, 10470–10474 CrossRef CAS.
- S. Y. Choi, S. K. Jeong, H. J. Kim, I.-H. Baek and K. T. Park, ACS Sustainable Chem. Eng., 2016, 4, 1311–1318 CrossRef CAS.
- A. Jedidi, S. Rasul, D. Masih, L. Cavallo and K. Takanabe, J. Mater. Chem. A, 2015, 3, 19085–19092 CAS.
- S. Rasul, D. H. Anjum, A. Jedidi, Y. Minenkov, L. Cavallo and K. Takanabe, Angew. Chem., Int. Ed., 2015, 127, 2174–2178 CrossRef.
- Z. B. Hoffman, T. S. Gray, K. B. Moraveck, T. B. Gunnoe and G. Zangari, ACS Catal., 2017, 7, 5381–5390 CrossRef CAS.
- R. A. Geioushy, M. M. Khaled, A. S. Hakeem, K. Alhooshani and C. Basheer, J. Electroanal. Chem., 2017, 785, 138–143 CrossRef CAS.
- N. Kumari, N. Sinha, M. A. Haider and S. Basu, Electrochim. Acta, 2015, 177, 21–29 CrossRef CAS.
- Y. F. Li, W. P. Zhang, X. Shen, P. F. Peng, L. B. Xiong and Y. Yu, Chin. J. Catal., 2015, 36, 2229–2236 CrossRef CAS.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- Y. Oh, H. Vrubel, S. Guidoux and X. Hu, Chem. Commun., 2014, 50, 3878–3881 RSC.
- Y. Oh and X. Hu, Chem. Commun., 2015, 51, 13698–13701 RSC.
- D. Kim, S. Lee, J. D. Ocon, B. Jeong, J. K. Lee and J. Lee, Phys. Chem. Chem. Phys., 2015, 17, 824–830 RSC.
- B. C. Marepally, C. Ampelli, C. Genovese, F. Tavella, L. Veyre, E. A. Quadrelli, S. Perathoner and G. Centi, J. CO2 Util., 2017, 21, 534–542 CrossRef CAS.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, Green Chem., 2017, 19, 2406–2415 RSC.
- C. Ampelli, C. Genovese, M. Errahali, G. Gatti, L. Marchese, S. Perathoner and G. Centi, J. Appl. Electrochem., 2015, 45, 701–713 CrossRef CAS.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, J. Catal., 2013, 308, 237–249 CrossRef CAS.
- T. Sekimoto, M. Deguchi, S. Yotsuhashi, Y. Yamada, T. Masui, A. Kuramata and S. Yamakoshi, Electrochem. Commun., 2014, 43, 95–97 CrossRef CAS.
- G. K. Ramesha, J. F. Brennecke and P. V. Kamat, ACS Catal., 2014, 4, 3249–3254 CrossRef CAS.
- N. Ullah, I. Ali, M. Jansen and S. Omanovic, Can. J. Chem. Eng., 2015, 93, 55–62 CrossRef CAS.
- X. Cui, G.-H. Lee, Y. D. Kim, G. Arefe, P. Y. Huang, C.-H. Lee, D. A. Chenet, X. Zhang, L. Wang and F. Ye, Nat. Nanotechnol., 2015, 10, 534–540 CrossRef CAS PubMed.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Král, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 4470–4477 CAS.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato and R. Haasch, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- P. Abbasi, M. Asadi, C. Liu, S. Sharifi-Asl, B. Sayahpour, A. Behranginia, P. Zapol, R. Shahbazian-Yassar, L. A. Curtiss and A. Salehi-Khojin, ACS Nano, 2016, 11, 453–460 CrossRef PubMed.
- K. Chan, C. Tsai, H. A. Hansen and J. K. Nørskov, ChemCatChem, 2014, 6, 1899–1905 CrossRef CAS.
- X. Hong, K. Chan, C. Tsai and J. K. Nørskov, ACS Catal., 2016, 6, 4428–4437 CrossRef CAS.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 6771–6775 CrossRef CAS PubMed.
- X. W. Mao and T. A. Hatton, Ind. Eng. Chem. Res., 2015, 54, 4033–4042 CrossRef CAS.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819–2826 Search PubMed.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X. D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869–13875 CrossRef CAS PubMed.
- J. Xu, Y. Kan, R. Huang, B. Zhang, B. Wang, K. H. Wu, Y. Lin, X. Sun, Q. Li and G. Centi, ChemSusChem, 2016, 9, 1085–1089 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. R. Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944–953 CrossRef PubMed.
- W. L. Li, M. Seredych, E. Rodriguez-Castellon and T. J. Bandosz, ChemSusChem, 2016, 9, 606–616 CrossRef CAS PubMed.
- H. M. Jhong, C. E. Tornow, B. Smid, A. A. Gewirth, S. M. Lyth and P. J. Kenis, ChemSusChem, 2017, 10, 1094–1099 CrossRef CAS PubMed.
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS.
- B. Aurian-Blajeni, M. Halmann and J. Manassen, Sol. Energy Mater. Sol. Cells, 1983, 8, 425–440 CrossRef CAS.
- E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef CAS PubMed.
- C. X. Kronawitter, M. Lessio, P. Zhao, C. Riplinger, A. Boscoboinik, D. E. Starr, P. Sutter, E. A. Carter and B. E. Koel, J. Phys. Chem. C, 2015, 119, 17762–17772 CAS.
- E. S. Brown, S. L. Peczonczyk, Z. J. Wang and S. Maldonado, J. Phys. Chem. C, 2014, 118, 11593–11600 CAS.
- M. Lessio, C. Riplinger and E. A. Carter, Phys. Chem. Chem. Phys., 2016, 18, 26434–26443 RSC.
- T. P. Senftle, M. Lessio and E. A. Carter, Chem. Mater., 2016, 28, 5799–5810 CrossRef CAS.
- M. Lessio and E. A. Carter, J. Am. Chem. Soc., 2015, 137, 13248–13251 CrossRef CAS PubMed.
- M. Lessio, T. P. Senftle and E. A. Carter, ACS Energy Lett., 2016, 1, 464–468 CrossRef CAS.
- T. P. Senftle, M. Lessio and E. A. Carter, Chem. Mater., 2016, 28, 5799–5810 CrossRef CAS.
- M. Lessio, J. M. Dieterich and E. A. Carter, J. Phys. Chem. C, 2017, 121, 17321–17331 CAS.
- B. A. Parkinson and P. F. Weaver, Nature, 1984, 309, 148–149 CrossRef CAS.
- S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Chem. Eng. J., 2006, 116, 227–231 CrossRef CAS.
- S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Appl. Catal., B, 2006, 64, 139–145 CrossRef CAS.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944–6946 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- S. Kaneco, Y. Ueno, H. Katsumata, T. Suzuki and K. Ohta, Chem. Eng. J., 2009, 148, 57–62 CrossRef CAS.
- E. Barton Cole, P. S. Lakkaraju, D. M. Rampulla, A. J. Morris, E. Abelev and A. B. Bocarsly, J. Am. Chem. Soc., 2010, 132, 11539–11551 CrossRef CAS PubMed.
- G. T. Zeng, J. Qiu, Z. Li, P. Pavaskar and S. B. Cronin, ACS Catal., 2014, 4, 3512–3516 CrossRef CAS.
- J. Qiu, G. T. Zeng, M. A. Ha, M. Y. Ge, Y. J. Lin, M. Hettick, B. Y. Hou, A. N. Alexandrova, A. Javey and S. B. Cronin, Nano Lett., 2015, 15, 6177–6181 CrossRef CAS PubMed.
- G. Zeng, J. Qiu, B. Hou, H. Shi, Y. Lin, M. Hettick, A. Javey and S. B. Cronin, Chem.–Eur. J., 2015, 21, 13502–13507 CrossRef CAS PubMed.
- P. Li, J. Zhang, H. Wang, H. Jing, J. Xu, X. Sui, H. Hu and H. Yin, Catal. Sci. Technol., 2014, 4, 1070–1077 CAS.
- T. T. Guaraldo, J. F. de Brito, D. Wood and M. V. B. Zanoni, Electrochim. Acta, 2015, 185, 117–124 CrossRef CAS.
- J. Cheng, M. Zhang, J. Z. Liu, J. H. Zhou and K. F. Cen, J. Mater. Chem. A, 2015, 3, 12947–12957 CAS.
- J. Cheng, M. Zhang, G. Wu, X. Wang, J. Zhou and K. Cen, Sol. Energy Mater. Sol. Cells, 2015, 132, 606–614 CrossRef CAS.
- P. Li, J. Xu, H. Jing, C. Wu, H. Peng, J. Lu and H. Yin, Appl. Catal., B, 2014, 156, 134–140 CrossRef.
- P. Li, H. Jing, J. Xu, C. Wu, H. Peng, J. Lu and F. Lu, Nanoscale, 2014, 6, 11380–11386 RSC.
- J. Cheng, M. Zhang, G. Wu, X. Wang, J. H. Zhou and K. F. Cen, Environ. Sci. Technol., 2014, 48, 7076–7084 CrossRef CAS PubMed.
- C. Ampelli, G. Centi, R. Passalacqua and S. Perathoner, Energy Environ. Sci., 2010, 3, 292–301 CAS.
- T. Yamamoto, H. Katsumata, T. Suzuki and S. Kaneco, ECS Trans., 2017, 75, 31–37 CrossRef.
- X. Zhou, R. Liu, K. Sun, Y. Chen, E. Verlage, S. A. Francis, N. S. Lewis and C. Xiang, ACS Energy Lett., 2016, 1, 764–770 CrossRef CAS.
- Y. Xu, Y. Jia, Y. Zhang, R. Nie, Z. Zhu, J. Wang and H. Jing, Appl. Catal., B, 2017, 205, 254–261 CrossRef CAS.
- Y. H. Yang, R. R. Xie, H. Li, C. J. Liu, W. H. Liu and F. Q. Zhan, Trans. Nonferrous Met. Soc. China, 2016, 26, 2390–2396 CrossRef CAS.
- I. Ganesh, P. P. Kumar, I. Annapoorna, J. M. Sumliner, M. Ramakrishna, N. Y. Hebalkar, G. Padmanabham and G. Sundararajan, Appl. Surf. Sci., 2014, 293, 229–247 CrossRef CAS.
- M. R. Hasan, S. B. Abd Hamid and W. J. Basirun, Appl. Surf. Sci., 2015, 339, 22–27 CrossRef CAS.
- L. Wang, Y. Jia, R. Nie, Y. Zhang, F. Chen, Z. Zhu, J. Wang and H. Jing, J. Catal., 2017, 349, 1–7 CrossRef CAS.
- J. W. Jang, S. Cho, G. Magesh, Y. J. Jang, J. Y. Kim, W. Y. Kim, J. K. Seo, S. Kim, K. H. Lee and J. S. Lee, Angew. Chem., Int. Ed., 2014, 53, 5852–5857 CrossRef CAS PubMed.
- Y. J. Jang, J. W. Jang, J. Lee, J. H. Kim, H. Kumagai, J. Lee, T. Minegishi, J. Kubota, K. Domen and J. S. Lee, Energy Environ. Sci., 2015, 8, 3597–3604 CAS.
- Y. J. Jang, I. Jeong, J. Lee, J. Lee, M. J. Ko and J. S. Lee, ACS Nano, 2016, 10, 6980–6987 CrossRef CAS PubMed.
- X. Xiong, M. Forster, J. D. Major, Y. Xu and A. J. Cowan, J. Phys. Chem. C, 2017, 121, 22073–22080 CAS.
- M. Li, P. Li, K. Chang, H. Liu, X. Hai, H. Zhang and J. Ye, Chem. Commun., 2016, 52, 8235–8238 RSC.
- G. Ghadimkhani, N. R. de Tacconi, W. Chanmanee, C. Janaky and K. Rajeshwar, Chem. Commun., 2013, 49, 1297–1299 RSC.
- K. Rajeshwar, N. R. de Tacconi, G. Ghadimkhani, W. Chanmanee and C. Janaky, ChemPhysChem, 2013, 14, 2251–2259 CrossRef CAS PubMed.
- H. Homayoni, W. Chanmanee, N. R. de Tacconi, B. H. Dennis and K. Rajeshwar, J. Electrochem. Soc., 2015, 162, E115–E122 CrossRef CAS.
- X. Ba, L. L. Yan, S. Huang, J. G. Yu, X. J. Xia and Y. Yu, J. Phys. Chem. C, 2014, 118, 24467–24478 CAS.
- J. F. de Brito, A. R. Araujo, K. Rajeshwar and M. V. B. Zanoni, Chem. Eng. J., 2015, 264, 302–309 CrossRef.
- J. F. De Brito, A. A. da Silva, A. J. Cavalheiro and M. V. B. Zanoni, Int. J. Electrochem. Sci., 2014, 9, 5961–5973 Search PubMed.
- C. Janaky, D. Hursan, B. Endrodi, W. Chanmanee, D. Roy, D. Liu, N. R. de Tacconi, B. H. Dennis and K. Rajeshwar, ACS Energy Lett., 2016, 1, 332–338 CrossRef CAS.
- M. Schreier, J. Luo, P. Gao, T. Moehl, M. T. Mayer and M. Gratzel, J. Am. Chem. Soc., 2016, 138, 1938–1946 CrossRef CAS PubMed.
- X. Chang, T. Wang, P. Zhang, Y. Wei, J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 8840–8845 CrossRef CAS PubMed.
- N. C. D. Nath, S. Y. Choi, H. W. Jeong, J. J. Lee and H. Park, Nano Energy, 2016, 25, 51–59 CrossRef.
- E. Kecsenovity, B. Endrődi, Z. Pápa, K. Hernádi, K. Rajeshwar and C. Janáky, J. Mater. Chem. A, 2016, 4, 3139–3147 CAS.
- B. Blackburn, I. Hassan, C. Zhang, C. Blackman, K. Holt and C. Carmalt, J. Nanosci. Nanotechnol., 2016, 16, 10112–10116 CrossRef CAS.
- X. Yang, E. A. Fugate, Y. Mueanngern and L. R. Baker, ACS Catal., 2017, 7, 177–180 CrossRef CAS.
- U. Kang, S. K. Choi, J. H. Dong, M. J. Sang, W. Choi, S. H. Dong, A. Abdel-Wahab and H. Park, Energy Environ. Sci., 2015, 8, 2638–2643 CAS.
- A. A. Tahir, H. Ullah, P. Sudhagar, M. A. M. Teridi, A. Devadoss and S. Sundaram, Chem. Rec., 2016, 16, 1591–1634 CrossRef PubMed.
- S. Zhou, Y. Liu, J. M. Li, Y. J. Wang, G. Y. Jiang, Z. Zhao, D. X. Wang, A. J. Duan, J. Liu and Y. C. Wei, Appl. Catal., B, 2014, 158, 20–29 CrossRef.
- Y. M. He, L. H. Zhang, M. H. Fan, X. X. Wang, M. L. Walbridge, Q. Y. Nong, Y. Wu and L. H. Zhao, Sol. Energy Mater. Sol. Cells, 2015, 137, 175–184 CrossRef CAS.
- Y. He, L. Zhang, B. Teng and M. Fan, Environ. Sci. Technol., 2015, 49, 649–656 CrossRef CAS PubMed.
- J. C. Wang, H. C. Yao, Z. Y. Fan, L. Zhang, J. S. Wang, S. Q. Zang and Z. J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 3765–3775 CAS.
- L. M. Azofra, D. R. MacFarlane and C. H. Sun, Phys. Chem. Chem. Phys., 2016, 18, 18507–18514 RSC.
- N. Sagara, S. Kamimura, T. Tsubota and T. Ohno, Appl. Catal., B, 2016, 192, 193–198 CrossRef CAS.
- D. Zhang, J. Shi, W. Zi, P. Wang and S. Liu, ChemSusChem, 2017, 10, 4324–4341 CrossRef CAS PubMed.
- Y. Zhang, W. Luc, G. S. Hutchings and F. Jiao, ACS Appl. Mater. Interfaces, 2016, 8, 24652–24658 CAS.
- K. D. Yang, Y. Ha, U. Sim, J. An, C. W. Lee, K. Jin, Y. Kim, J. Park, J. S. Hong, J. H. Lee, H. E. Lee, H. Y. Jeong, H. Kim and K. T. Nam, Adv. Funct. Mater., 2016, 26, 233–242 CrossRef CAS.
- J. T. Song, H. Ryoo, M. Cho, J. Kim, J. G. Kim, S. Y. Chung and J. Oh, Adv. Energy Mater., 2016, 7, 1601103–1601111 CrossRef.
- Y. Wang, S. Fan, B. AlOtaibi, Y. Wang, L. Li and Z. Mi, Chemistry, 2016, 22, 8809–8813 CrossRef CAS PubMed.
- F. Urbain, P. Tang, N. M. Carretero, T. Andreu, L. G. Gerling, C. Voz, J. Arbiol and J. R. Morante, Energy Environ. Sci., 2017, 10, 2256–2266 CAS.
- T. Sekimoto, H. Hashiba, S. Shinagawa, Y. Uetake, M. Deguchi, S. Yotsuhashi and K. Ohkawa, J. Phys. Chem. C, 2016, 120, 13970–13975 CAS.
- N. J. Firet and W. A. Smith, ACS Catal., 2016, 7, 606–612 CrossRef.
- S. Zhu, B. Jiang, W.-B. Cai and M. Shao, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS PubMed.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
Footnote |
| † These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |