
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultinuclear iron–phenyl species in reactions of simple iron salts with PhMgBr: identification of Fe4(μ-Ph)6(THF)4 as a key reactive species for cross-coupling catalysis†
Stephanie H.
Carpenter
 ,
Tessa M.
Baker
,
Salvador B.
Muñoz
III
,
William W.
Brennessel
and
Michael L.
Neidig
*
,
Tessa M.
Baker
,
Salvador B.
Muñoz
III
,
William W.
Brennessel
and
Michael L.
Neidig
*
Department of Chemistry, University of Rochester, Rochester, New York 14627, USA. E-mail: neidig@chem.rochester.edu
First published on 24th August 2018
Abstract
The first direct syntheses, structural characterizations, and reactivity studies of iron–phenyl species formed upon reaction of Fe(acac)3 and PhMgBr in THF are presented. Reaction of Fe(acac)3 with 4 equiv. PhMgBr in THF leads to the formation of [FePh2(μ-Ph)]22− at −80 °C, which can be stabilized through the addition of N-methylpyrrolidone. Alternatively, at −30 °C this reaction leads to the formation of the tetranuclear iron–phenyl cluster, Fe4(μ-Ph)6(THF)4. Further synthetic studies demonstrate that analogous tetranuclear iron clusters can be formed with both 4-F-PhMgBr and p-tolylMgBr, illustrating the generality of this structural motif for reactions of simple ferric salts and aryl Grignard reagents in THF. Additional studies isolate and define key iron species involved in the synthetic pathway leading to the formation of the tetranuclear iron–aryl species. While reaction studies demonstrate that [FePh2(μ-Ph)]22− is unreactive towards electrophile, Fe4(μ-Ph)6(THF)4 is found to rapidly react with bromocyclohexane to selectively form phenylcyclohexane. Based on this reactivity, a new catalytic reaction protocol has been developed that enables efficient cross-couplings using Fe4(μ-Ph)6(THF)4, circumventing the current need for additives such as TMEDA or supporting ligands to achieve effective cross-coupling of PhMgBr and a secondary alkyl halide.
Introduction
Iron-catalyzed organic transformations continue to attract significant interest due to the low cost, improved sustainability and potential for novel reactivity of iron compared to more traditional precious metal catalytic systems.1–17 Catalytic reactions involving simple ferric salts and phenyl nucleophiles (most extensively phenylmagnesium bromide, PhMgBr) are of particular interest as this combination has shown to be effective in catalysis in both iron-catalyzed cross-coupling and iron-catalyzed C–H functionalization systems.1–17 Unfortunately, the nature of the in situ formed and reactive iron species generated from simple ferric salts and PhMgBr in catalysis remains ambiguous. Such structural insight is essential for defining the role of various iron–phenyl species for both productive and off-cycle reactivity, and for the development of more efficient catalytic methodologies using these reagents.Motivated by the critical need to define the iron–phenyl species involved in catalysis, several recent studies have employed NMR and electron paramagnetic resonance (EPR) spectroscopies, as well as density functional theory (DFT) calculations to investigate iron speciation in reactions of simple ferric salts and PhMgBr.18–25 Such studies have hypothesized the formation of mononuclear FeI and Fe0 species in situ.18–25 Alternatively, extended X-ray absorption fine structure (EXAFS) studies have led to the proposed formation of FeII dimers in situ in these reactions, whereas mass spectrometry studies have suggested that iron–phenyl-ate species of even higher nuclearity can form.26,27 While these studies represent important contributions towards the identification of some of the in situ formed iron–phenyl species, the resulting lack of consensus on the nature of these species (e.g. oxidation state, nuclearity) reflects the limited availability of isolated and structurally defined iron–phenyl species. To date, the only structurally defined species reported have been a mononuclear iron(II)–phenyl-ate species formed from the reaction between simple iron salts and PhLi, as well as a reduced iron(I)–phenyl species formed using the same nucleophile (Scheme 1).18,24 The latter was found to be unreactive towards electrophile, likely representing an unproductive iron–phenyl species for cross-coupling.24 Notably, no structurally defined iron–phenyl species generated from the reaction of simple ferric salts with PhMgBr in THF, the reagents and solvents employed in both cross-coupling and C–H functionalization reactions, have been reported. This lack of structural insight greatly contrasts analogous reactions with the more sterically encumbered mesitylmagnesium bromide (MesMgBr), where both FeMes3− and Fe2Mes4 have been isolated.28
 | ||
| Scheme 1 Structurally defined iron–phenyl species formed using PhLi implicated in iron-cross coupling reactions. | ||
In this study, we report the isolation and characterization of the first multinuclear iron–phenyl species formed from the reaction of Fe(acac)3 and ArMgBr in THF. Insight into the reaction pathway of formation of both di- and tetranuclear iron-complexes is presented, as well as the generality of the tetranuclear structure across several aryl Grignard reagents. Stoichiometric reaction studies are utilized to evaluate the potential roles of the multinuclear iron–phenyl complexes in catalysis. These combined synthetic and reaction studies have led to the identification of a highly reactive iron–phenyl species for the selective formation of cross-coupled product, resulting in a new ligand- and additive-free reaction protocol for cross-coupling with Fe(acac)3 and PhMgBr.
Results and discussion
Isolation and characterization of [FePh2(μ-Ph)]22− from low temperature reactions of Fe(acac)3 and PhMgBr
Previous iron-catalyzed cross-coupling studies have indicated that a minimum of 4 equiv. of Grignard reagent is required to achieve effective catalysis using simple ferric salts and PhMgBr.29,30 Therefore, synthetic studies focused on the isolation of iron–phenyl species formed using this ratio of iron to Grignard reagent in THF. While viable single crystals could not be isolated directly from reactions at −80 °C, a modified synthetic procedure involving layering and warming of 4 equiv. of PhMgBr on top of Fe(acac)3 (see Experimental section for further details) produced orange crystalline material suitable for single crystal X-ray diffraction (SC-XRD). The orange crystals were found to have extreme air-, moisture-, and temperature-sensitivities; crystalline material could only be handled at or below −80 °C under a nitrogen atmosphere to prevent decomposition. Despite these handling limitations, the crystalline material was successfully characterized by SC-XRD which revealed the identity of the orange crystals as the dinuclear iron species, [Mg(acac)(THF)4]2[FePh2(μ-Ph)]2·4THF (1a) (Fig. 1). This complex exhibits an Fe–Fe distance of 2.5175(9) Å, and has average terminal and bridging Fe–Ph bond lengths of 2.083(4) Å and 2.203(4) Å, respectively. Notably, this Fe–Fe distance is significantly shorter than the corresponding distance in Fe2Mes4 and [Ar*Fe(μ-Ph)]2 (Ar* = C6H3-2,6-(C6H2-2,4,6-iPr3)2); Fe–Fe distances of 2.612(1) Å to 2.635(1) Å for Fe2Mes4 and 2.7207(14) Å for [Ar*Fe(μ-Ph)]2.31–35 Based upon the crystal structure, both iron sites are formally iron(II), where the presence of distorted tetrahedral iron sites suggest the presence of high-spin iron(II) centers due to the tetrahedral ligand fields. The 57Fe Mössbauer spectrum of crystalline 1a was characterized by a single doublet with an isomer shift (δ) of 0.34 mm s−1 and a quadrupole splitting (ΔEQ) of 2.28 mm s−1. The observed quadrupole splitting is consistent with the presence of high-spin iron(II) sites and the low isomer shift is in agreement with previously reported transmetalled iron(II) complexes corresponding to the highly covalent Fe–C bonds.36,37 Unfortunately, further characterization of pure 1a was not possible due to the high thermal instability of the complex. | ||
| Fig. 1 The crystal structure, selected bond distances and angles, and 80 K 57Fe Mössbauer spectrum of [Mg(acac)(THF)4]2[Fe(Ph)2(μ-Ph)]2·4THF (1a). The hydrogen atoms are omitted for clarity and the thermal ellipsoids are shown at 50% probability. | ||
N-Methylpyrrolidone (NMP) has been previously employed as an additive in cross-coupling reactions with simple ferric salts, and has been demonstrated in the case of MeMgBr to stabilize [FeMe3]−, favoring its formation over [Fe8Me12]−.38,39 Interestingly, NMP was found to bind to the Mg cation in this case38 as opposed to binding directly to the iron center as suggested in earlier studies by Holland and co-workers.40 It was hypothesized that the inclusion of NMP as an additive might enable the stabilization of [FePh2(μ-Ph)]22−. Therefore, an analogous reaction of Fe(acac)3 with 4 equiv. PhMgBr in THF in the presence of excess NMP (36 equiv.) was performed at −30 °C. This reaction led to the formation of an analogous dimer [Mg(NMP)6][FePh2(μ-Ph)]2·3.5THF (1b) as confirmed by SC-XRD (Fig. 2). A slight decrease in the Fe–Fe distance in 1b to 2.4969(8) Å is present compared to 1a, with average terminal and bridging Fe–Ph bond lengths of 2.075(4) Å to 2.205(4) Å, respectively. The 57Fe Mössbauer spectrum of 1b was found to be identical to that of 1a, with parameters of δ = 0.34 mm s−1 and ΔEQ = 2.28 mm s−1. Consistent with previous studies of [FeMe3]−,39 the change of counterion in the complex significantly improved the thermal stability of the iron–phenyl dimer at −30 °C, enabling additional characterization of 1b. While Evans method NMR coupled with atomic absorption spectroscopy (AAS; to quantify the amount of 1b dissolved in solution as the material is too thermally unstable to weigh) would be an ideal way to calculate the spin state of 1b, the complex was found to be too unstable to obtain meaningful NMR data. Instead, magnetic circular dichroism (MCD) was employed to determine the spin state of 1b. No MCD signal is observed in both mull and solution samples at 5 K which, combined with the absence of an EPR signal at 10 K, is consistent with the assignment of a S = 0 ground state due to antiferromagnetic coupling of the iron(II) sites in the dimer. Lastly, it was important to evaluate whether 1b could undergo additional transformations upon reaction with additional PhMgBr. 57Fe Mössbauer spectroscopy confirmed that addition of excess PhMgBr (>20 equiv.) directly to a solution of 1b at −80 °C resulted in no reaction within 5 min (a catalytically relevant time frame, vide infra). The formation of both 1a and 1b differs drastically from the formation of Fe2Mes4; Fe2Mes4 is accessible at RT, illustrating how the sterically bulkyl mesityl ligands modulate the formation and stability of the resulting iron dimer.28
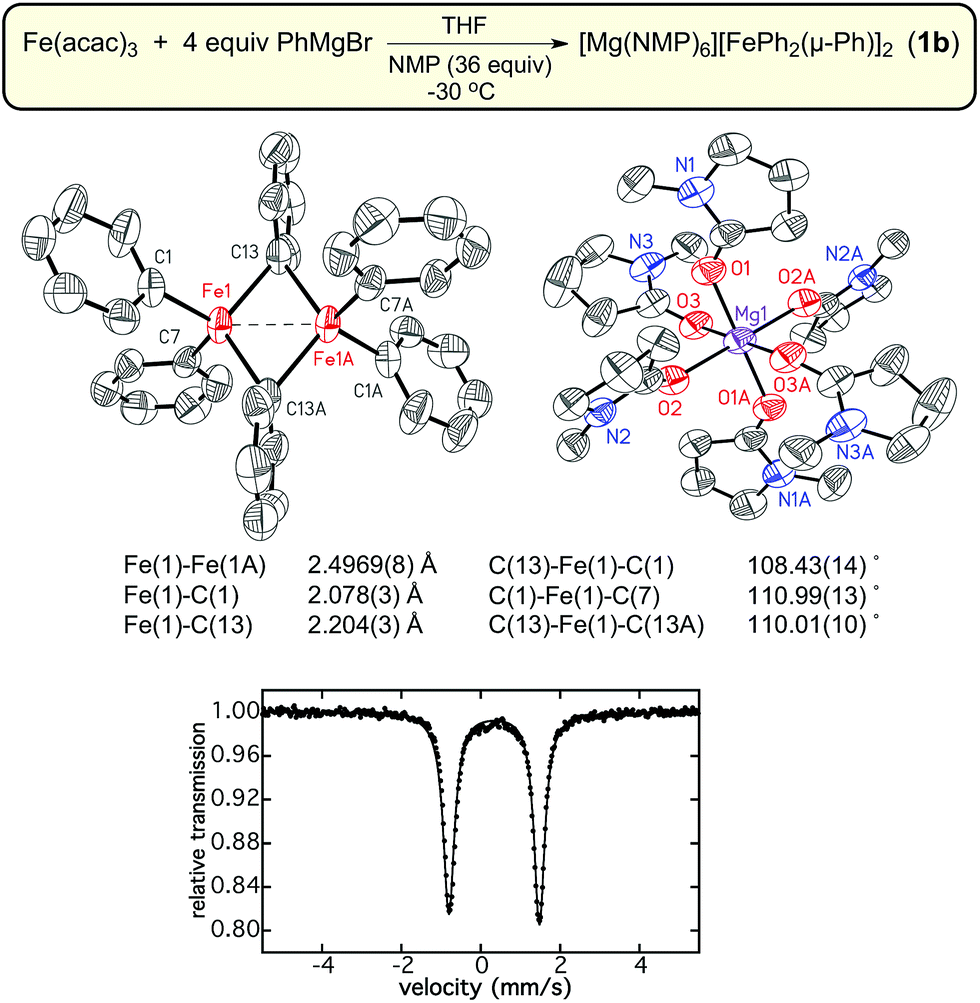 | ||
| Fig. 2 The crystal structure, selected bond distances and angles, and 80 K 57Fe Mössbauer spectrum of [Mg(NMP)6][Fe(Ph)2(μ-Ph)]2·3.5THF (1b). The hydrogen atoms are omitted for clarity and the thermal ellipsoids are shown at 50% probability. | ||
Isolation and characterization of tetranuclear iron–aryl species
Due to the thermal instability of 1a, it was critical to determine the nature of the iron species formed at elevated temperatures as a more reduced iron–phenyl species might be obtainable. Reaction of Fe(acac)3 with 4 equiv. of PhMgBr in THF at −30 °C, in the absence of NMP, yielded a brown solution from which brown crystalline material could be obtained. The overall connectivity, geometry, and chemical formulation could be unambiguously assigned by SC-XRD as the tetranuclear iron cluster Fe4(μ-Ph)6(THF)4·2THF (2a) (Fig. 3). This cluster is more reduced than 1a or 1b, formally containing two iron(I) and two iron(II) ions. Unfortunately, these crystals diffracted very weakly and, hence, a more detailed discussion of the structural parameters of 2a is not possible. It was hypothesized that changing the aryl group of the Grignard reagent might enable access to higher quality crystalline material for SC-XRD analysis. An analogous reaction utilizing p-tolylMgBr yielded high quality crystals identified by SC-XRD as Fe4(μ-p-tolyl)6(THF)4·2THF·C5H12 (2b) upon crystallization at −80 °C. Of note, an analogous crystallization at −30 °C yielded the formation of a slightly perturbed tetranuclear cluster, Fe4(μ-p-tolyl)6(THF)3·THF (2c), which lacks one THF ligand to iron (note that the co-crystallized THF solvent molecule is >4.6 Å from the open iron center). Thus slight structural variation demonstrates the potential lability of coordinated THF in these clusters in order to generate an open coordination site for reaction with electrophile (vide infra). The Fe–Fe bond distances in 2b and 2c range from 2.3751(9) Å to 2.5532(10) Å, which are notably shorter compared to reported tetranuclear iron species such as [Fe4(CO)12(CCH3)]−, [(η5-C5H4Me)4Fe4(μ3-CH)2(μ3-CNPh)2](PF6)2, and [Fe4(CO)12(μ3-CO)]2−, where the Fe–Fe bond distances typically range from 2.469 Å to 2.618 Å.41–44 Interestingly, a [Fe8Me12]− cluster has been previously isolated and characterized as a key intermediate in reactions involving simple ferric salts and MeMgBr.45 The Fe–Fe distances in [Fe8Me12]− are closer to the Fe–Fe distances in 2b and 2c, ranging from 2.4188(15) to 2.4514(15) Å.45 The bridging Fe–Ph bonds range from 2.139(3) Å to 2.266(3) Å for 2b and from 2.078(5) Å to 2.412(5) Å for 2c. Lastly, an analogous tetranuclear iron complex could also be synthesized using 4-fluorophenylmagnesium bromide (4-F-PhMgBr), identified by SC-XRD as Fe4(μ-4-F-Ph)6(THF)4 (2d). As with 2a, the isolated crystalline material was weakly diffracting, and hence, the SC-XRD data was solely used for the unambiguous assignment of connectivity. Overall, the ability to access analogous tetranuclear iron–aryl clusters across both electron withdrawing and electron donating substituents on the aryl ligands demonstrates the generality of this structural motif in reactions of simple ferric salts and aryl Grignard reagents.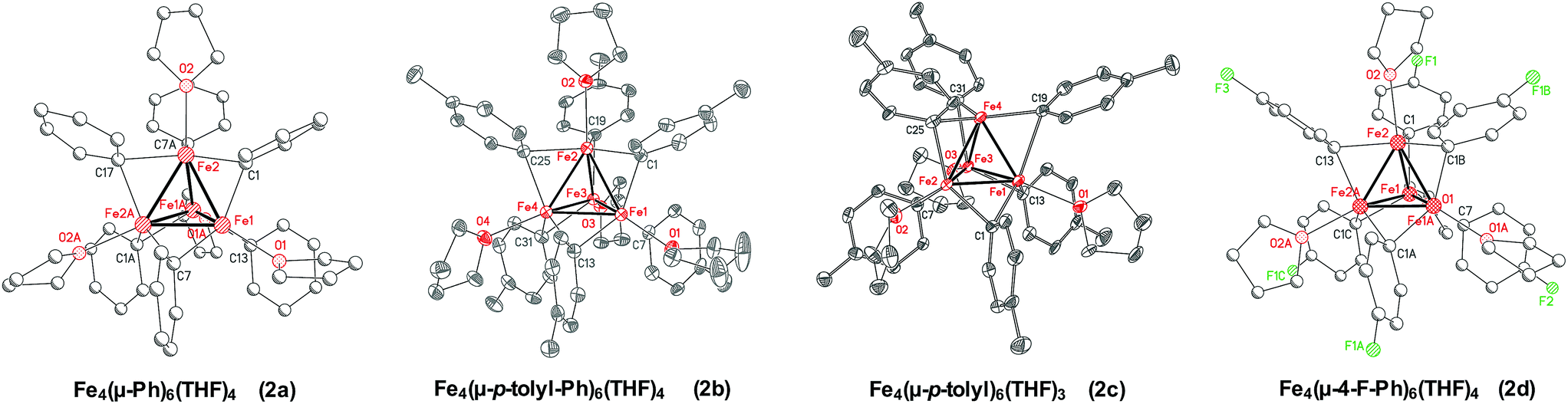 | ||
| Fig. 3 Crystal structures of the tetranuclear iron–aryl clusters, with hydrogen atoms omitted for clarity. Due to weakly diffractly crystals, the structures of Fe4(μ-Ph)6(THF)4 (2a) and Fe4(μ-4-F-Ph)6(THF)4 (2d) are used solely for connectivity and chemical formulation as shown. For structures with p-tolyl groups, Fe4(μ-p-tolyl)6(THF)3 (2b) and Fe4(μ-p-tolyl)6(THF)4 (2c), adequate intensity data could be obtained and the ellipsoids are shown at 50% probability. | ||
Complex 2a was the most synthetically robust tetranuclear complex for larger scale isolation and was utilized for further characterization and reactivity (vide infra) studies. The 57Fe Mössbauer spectrum of 2a is a broad quadrupole doublet with δ = 0.60 mm s−1 and ΔEQ = 0.84 mm s−1 (see ESI†). Analogous broad features were also previously observed for [Fe8Me12]−.45 The tetranuclear complex is not EPR active, but exhibits an intense C-term MCD spectra in both the near-infrared and UV-vis regions (see ESI†) consistent with the presence of an integer spin paramagnetic complex. Unfortunately, variable-temperature, variable-field (VTVH) MCD studies on the ground state of 2a did not enable the assignment of the spin state of this complex, likely due to complications from decay to additional paramagnetic iron species which complicates both these measurements and attempted Evans studies.
Investigation of the iron species involved in the synthetic pathway for formation of di- and tetranuclear iron–aryl species
Stoichiometric reactions of Fe(acac)3 with varying equivalents of 4-F-PhMgBr enabled the further investigation into the underlying synthetic pathway leading to formation of the dinuclear and tetranuclear iron species. When 1 equiv. of 4-F-PhMgBr is slowly added to Fe(acac)3 in THF at −30 °C, a red color persists. From this reaction, crystalline material of [trans-Fe(acac)2(THF)2]0.58·[trans-Mg(acac)2(THF)2]0.42 (3) was isolated and characterized by SC-XRD (Fig. 4). Similar crystals were independently isolated from reactions of Fe(acac)3 with 1 equiv. of three different aryl Grignard reagents (PhMgBr, 4-F-PhMgBr, and p-tolylMgBr), consistent with the first equivalent of ArMgBr reducing the iron(III) starting material to iron(II). Addition of a second equivalent of aryl Grignard reagent to the iron solution at −30 °C results in a color change from red to yellow. Although no usable crystals of the yellow material could be obtained, one could envision a complex analogous to the structurally characterized [(di-tBu-acac)Fe(μ-Mes)]2.46 The yellow iron solution turns orange upon the slow addition of a third equivalent of aryl Grignard reagent at −30 °C. From this solution, orange crystals were obtained at −80 °C suitable for SC-XRD and determined to be [Mg(acac)(THF)4]2[FeBr(4-F-Ph)(μ-4-F-Ph)]2·2.5THF (4). The Fe–Fe distance was found to be 2.5903(11) Å, which is longer than previously identified homoleptic iron(II)–phenyl dimers 1a and 1b. The unique terminal and two bridging Fe–Ph bond lengths, based on the independent dianion without disorder, are 2.072(4), 2.166(4) and 2.212(4) Å, respectively. The 80 K 57Fe Mössbauer spectrum of 4 has parameters of δ = 0.56 mm s−1 and ΔEQ = 1.81 mm s−1 (see ESI†). At low reaction temperatures, the addition of a fourth equiv. of 4-F-PhMgBr changes the orange solution to red, resulting in the formation of [Mg(acac)(THF)4]2[Fe(4-F-Ph)2(μ-4-F-Ph)]2·2THF (1c). Here, the additional equivalent of Grignard reagent is used to provide the last aryl group to each iron center, showing that dimers 1a and 1c can be constructed as each equivalent of Grignard reagent is slowly added to the iron solution. The Fe–Fe distance of 2.5847(5) Å in 1c is longer than those in 1a and 1b by ∼0.08 Å. The average Fe–Ph terminal and bridging bond lengths in 1c of 2.093(2) and 2.208(2) Å, respectively, are similar to those in 1a and 1b, although there exists a slight asymmetry in the core as bridging Fe–Ph bond lengths differ by ∼0.035 Å. The 80 K 57Fe Mössbauer spectrum of 1c is characterized by parameters of δ = 0.40 mm s−1 and ΔEQ = 2.21 mm s−1 (see ESI†). Much like 1a, complex 1c exhibits extreme temperature and moisture sensitivity, preventing further characterization. Lastly, the reduced tetranuclear iron species, Fe4(μ-4-F-Ph)6(THF)4 (2d) can be formed from the 4 equiv. reaction simply by warming to −30 °C.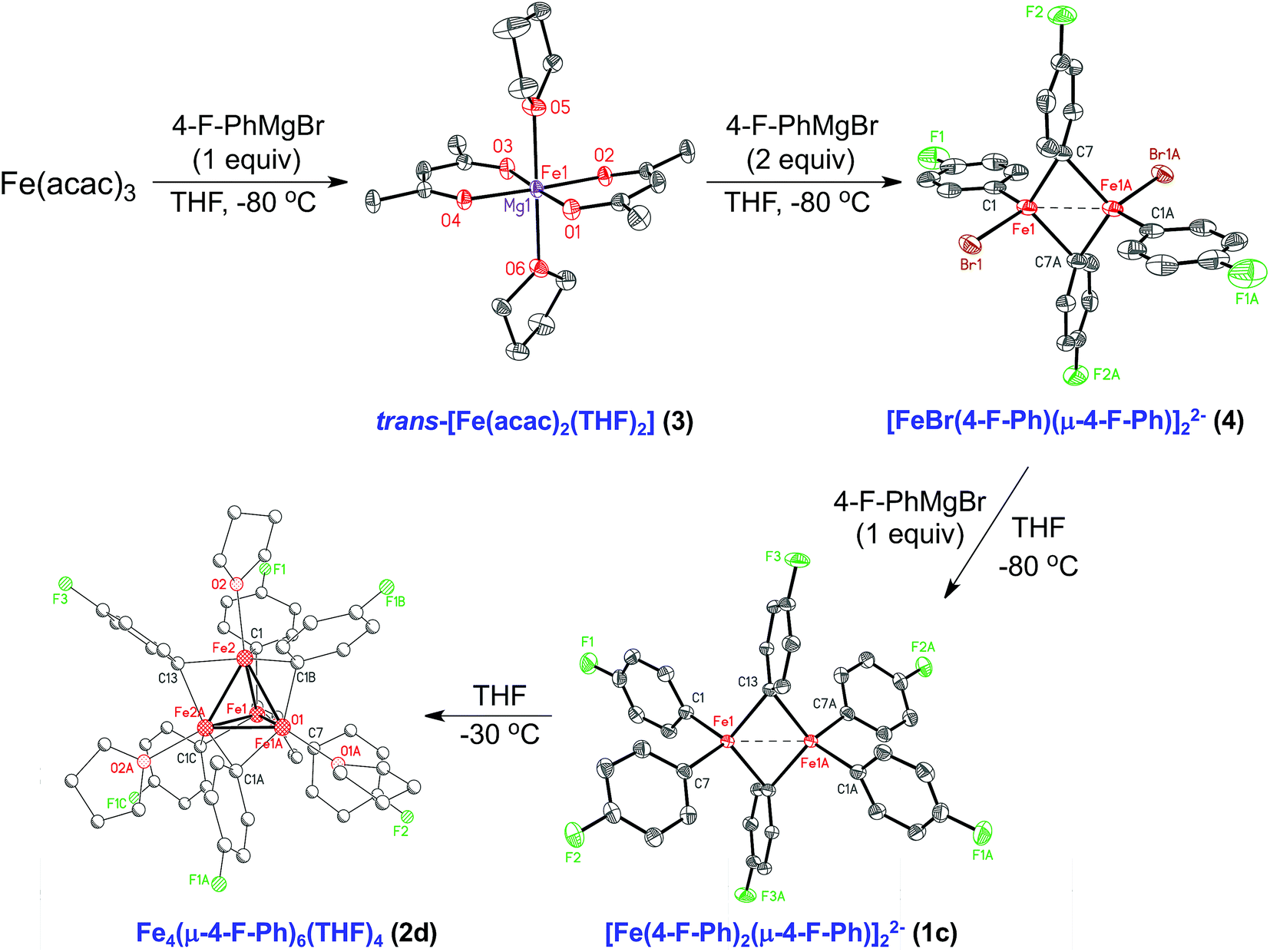 | ||
| Fig. 4 Iron species in the synthetic pathway for the formation of tetranuclear iron–aryl species utilizing Fe(acac)3 and 4-F-PhMgBr. Hydrogen atoms omitted for clarity and thermal ellipsoids are shown at 50% probability for 1c, 3 and 4. | ||
Reactivity studies of [FePh2(μ-Ph)]22− and Fe4(μ-Ph)6(THF)4 with bromocyclohexane
Beyond the unprecedented insight into iron speciation and structure for reactions of simple iron salts and aryl Grignard reagents described above, evaluation of the potential reactivity of these di- and tetranuclear complexes with electrophile is critical to understanding the potential roles of isolated species in cross-coupling catalysis. Due to the extreme thermal instability of 1a, only complex 1b could be utilized to evaluate the reactivity of the [FePh2(μ-Ph)]22− dimer (see ESI†). Employing time-resolved, freeze-quenched 57Fe Mössbauer spectroscopy, the decomposition of 57Fe-enriched 1b in solution was monitored at temperatures ranging from −20 °C to 0 °C in order to define its stability as a function of temperature and time. After re-dissolution of 1b in THF at −20 °C, only 70% of 1b was found to remain in solution after 45 s. As expected, repeating this experiment at 0 °C shows more rapid decay of the dimer with only ∼50% of 1b remaining in solution after 45 s. Therefore, reaction studies with electrophile were performed at −20 °C, focusing on a 45 s reaction window in order to minimize contributions from the decomposition of 1b. Bromocyclohexane was selected as an example electrophile for the reaction studies due to its common use in ferric salt catalyzed cross-coupling reactions with PhMgBr.47,48 GC-MS reaction studies showed no consumption of electrophile or generation of phenylcyclohexane within 45 s of reaction at −20 °C. Thus, the [FePh2(μ-Ph)]22− dimer exhibits no reactivity towards electrophile prior to its thermal decomposition in THF. Furthermore, the observation that NMP stabilizes the formation of this unreactive dimer is consistent with previous studies by Nakamura and co-workers, where NMP was shown to be an unfavorable co-solvent for iron-catalyzed cross-coupling reactions involving simple ferric salts and aryl Grignard reagents.47 In contrast to the lack of reactivity of 1b, formation of cross-coupled product was observed to be generated, albeit in low yield (24%), from the reaction of Fe2Mes4 and electrophile.28In order to evaluate the potential reactivity of the more reduced tetranuclear iron complex 2a with bromocyclohexane, it was again critical to first establish the thermal stability of 2a in solution at catalytically relevant temperatures. Fortunately, 2a was found to be stable at RT for up to 5 min in THF, enabling stoichiometric reactions to be performed within this time frame. Reactions of 2a with 15 equiv. bromocyclohexane at RT resulted in the rapid and selective formation of phenylcyclohexane (0.95 equiv. with respect to 2a within 5 s). Thus, 2a is a highly reactive species for the selective formation of cross-coupled product (kobs ∼ 12 min−1 for the initial turnover). Prolonged reaction times led to the generation of additional cross-coupled product, indicating that the iron products of each cross-coupling are capable of further reaction with electrophile (∼4 equiv. phenylcycohexane after 1 min of reaction (see ESI†); note that the reaction rate decreases for subsequent turnovers). It is noteworthy that 2a can directly react with electrophile to form cross-coupled product, whereas [Fe8Me12]− requires the addition of MeMgBr following initial reaction with electrophile to form product,45 indicating the presence of different underlying reaction mechanisms for iron–phenyl and iron–methyl clusters.
Cross-coupling catalysis using Fe4(μ-Ph)6(THF)4
Simple ferric salts were previously found by Nakamura and co-workers to perform poorly for catalytic cross-couplings of PhMgBr with secondary alkyl halides in the absence of TMEDA.47 Interestingly, Bedford and co-workers observed similar reactivity in the presence and absence of TMEDA when MesMgBr was employed, though this system was low yielding (∼35%).28 Hence, we were motivated by the observed reactivity of 2a to explore its potential effectiveness for catalytic cross-coupling in the absence of TMEDA. The utilization of the same reaction protocol as described for stoichiometric reactions of 2a but with the addition of PhMgBr (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with respect to electrophile) resulted in the formation of >95% cross-coupled product (Scheme 2a). Because 2a is challenging to synthesize and handle, a modified catalytic method targeting the formation of 2ain situ at −30 °C (a temperature where 2a is stable for days) was also evaluated as a potentially more convenient protocol that utilizes the selective reactivity of 2a without the need to isolate it (Scheme 2b). With this method, >95% of cross-coupled product could be obtained. Interestingly, removal of magnesium salts by filtration at −30 °C following in situ formation of 2a was found to be critical to achieve high yields of product, likely indicating an important role of cations on iron speciation during the initial synthesis of 2a or during catalysis. While a proof of concept, this initial evaluation of in situ generated 2a for catalysis will hopefully inspire future studies in the area of ligandless iron cross-coupling catalysis.
:1 with respect to electrophile) resulted in the formation of >95% cross-coupled product (Scheme 2a). Because 2a is challenging to synthesize and handle, a modified catalytic method targeting the formation of 2ain situ at −30 °C (a temperature where 2a is stable for days) was also evaluated as a potentially more convenient protocol that utilizes the selective reactivity of 2a without the need to isolate it (Scheme 2b). With this method, >95% of cross-coupled product could be obtained. Interestingly, removal of magnesium salts by filtration at −30 °C following in situ formation of 2a was found to be critical to achieve high yields of product, likely indicating an important role of cations on iron speciation during the initial synthesis of 2a or during catalysis. While a proof of concept, this initial evaluation of in situ generated 2a for catalysis will hopefully inspire future studies in the area of ligandless iron cross-coupling catalysis.
 | ||
| Scheme 2 Catalytic cross-couplings of PhMgBr and bromocyclohexane using (a) isolated 2a and (b) in situ generated 2a. | ||
While the current study has demonstrated the importance of dinuclear and tetranuclear iron–phenyl species in reactions of simple ferric salts and aryl Grignard reagents, additional iron–phenyl species beyond those isolated herein might also be accessible in such reactions. For example, previous EPR studies by Bedford indicated the in situ formation of a S = 1/2 species in reactions of FeCl3 and p-tolylMgBr at −30 °C though, unfortunately, this species was never spin quantified.21,28 We have observed the formation of the same S = 1/2 species in reaction of FeCl2 and 4 equiv. PhMgBr at −30 °C, though spin quantitated EPR indicated that it is a very minor species in solution (<5% of all iron) (see ESI†).21,28 Additionally, Bedford and co-workers also suggested that a monomeric iron(II) species, [Fe(p-tolyl)3]− can also form based on 1H NMR studies of reactions of FeCl2 with p-tolylMgBr,28 where the resonances assigned to this mononuclear species are significantly downfield shifted compared to those observed for 2b (see ESI†). Again, however, the amount of the mononuclear species in solution was not quantified and it remains unclear whether it is formed significantly in solution compared to other iron–p-tolyl species. Future studies should continue to define the diverse iron–aryl species accessible in such reactions as a function of concentration, solvent, aryl nucleophile (e.g. ArMgBr, ArLi, Ar2Zn, etc.), temperature, and reaction time.
Lastly, it is interesting to consider the origin of the reactivity differences observed for [FePh2(μ-Ph)]22− and Fe4(μ-Ph)6(THF)4. Since previous studies have proposed an Fe(I) active species for cross-coupling with PhMgBr and simple ferric salts (such as [PhFeI(acac)(THF)]−),18–25 the presence of two formally iron(I) sites in the mixed valent tetranuclear iron complex 2a might suggest iron reduced below iron(II) is important for reactivity in the isolated multimetallic complexes. Specifically, the THF ligation differences between complexes 2b and 2c demonstrate the ability of the tetranuclear complexes to lose a THF ligand to generate an open coordination position for reaction with electrophile. This ability to readily form an open coordination site might be equally significant in facilitating reactivity, whereas the dinuclear complexes would require a more significant geometric distortion in order to react with electrophile.
Conclusions
In this study, the first direct syntheses, structural characterizations, and reactivity studies of iron–phenyl species formed upon reaction of Fe(acac)3 and PhMgBr in THF have been presented. At −80 °C, this reaction leads to formation of [FePh2(μ-Ph)]22−, which was found to be unreactive towards electrophile. Alternatively, at −30 °C the formation of a more reduced, tetranuclear iron–phenyl cluster, Fe4(μ-Ph)6(THF)4, is observed, where this species is found to rapidly react with bromocyclohexane to selectively form cross-coupled product. Further synthetic studies demonstrate that analogous tetranuclear iron clusters can be formed with both 4-F-PhMgBr and p-tolylMgBr, illustrating the generality of this structural motif for reactions of simple ferric salts and aryl Grignard reagents in THF. Lastly, Fe4(μ-Ph)6(THF)4 can be utilized for efficient catalytic cross-coupling of PhMgBr and bromocyclohexane, circumventing the current need for additives such as TMEDA or supporting ligands to achieve high yields of cross-coupled product in this reaction.Experimental
General considerations
All reagents were purchased from commercial sources. All air- and moisture-sensitive manipulations were carried out in an MBraun inert-atmosphere (N2) glovebox equipped with a direct liquid nitrogen feed through inlet line. All anhydrous solvents were freshly dried using activated alumina/4 Å molecular sieves and stored under an inert atmosphere. Gas chromatography mass spectrometry was performed using a Shimadzu GCMS QP 2010. Atomic absorption spectroscopy (AAS) analysis was performed using a Shimadzu AAS 7000. Details on low temperature crystal manipulations, sample preparations for spectroscopy and MCD and EPR spectroscopy are given in the ESI.†57Fe Mössbauer spectroscopy
Solution samples for 57Fe Mössbauer spectroscopy were prepared from 57Fe(acac)3 to enable data collection from dilute, freeze-trapped solution samples; solid samples were made from non-enriched Fe(acac)3. All samples were prepared in an inert atmosphere glovebox equipped with a liquid nitrogen fill port to enable sample freezing to 77 K within the glovebox. Each sample was loaded into a Delrin Mössbauer cup for measurements and loaded under liquid nitrogen. Low temperature 57Fe Mössbauer measurements were performed using a See Co. MS4 Mössbauer spectrometer integrated with a Janis SVT-400T He/N2 cryostat for measurements at 5 K, 80 K, and 150 K. Isomer shifts were determined relative to an α-Fe at 298 K. All Mössbauer spectra were fit using the program WMoss (SeeCo). Errors of the analyses are δ ± 0.02 mm s−1 and ΔEQ ± 3%.Magnetic circular dichroism spectroscopy
All samples were prepared in an inert atmosphere glovebox equipped with a liquid nitrogen filling port to enable sample freezing to 77 K. Low temperature near-infrared (NIR) MCD experiments were conducted using a JASCO J-730 spectropolarimeter and a shielded S-20 photomultiplier tube. Both instruments have a modified sample compartment, which incorporates focusing optics and an Oxford Instruments SM4000-7T superconducting magnetic/cryostat. This set-up allows for measurements from 1.6 K to 290 K, with magnetic fields up to 7 T. A calibrated Cernox sensor directly inserted in the copper sample holder is used to measure the temperature at the sample to 0.001 K. All MCD spectra were baseline-corrected against zero-field scans.Electron paramagnetic resonance spectroscopy
A cold spatula was used to transfer material to a vial containing a known amount of THF at −80 °C. A cold pipette was then used to transfer the redissolved crystalline material to a precooled (in liquid nitrogen) 4 mm OD suprasil quartz EPR tube from Wilmad Labglass. The solution in the EPR tube was immediately frozen in liquid nitrogen. The remaining solution not used for the EPR sample was saved for AAS, so that spin integration of any EPR signal could be completed. All X-band EPR spectra were collected on a Bruker EMXplus spectrometer containing a 4119HS cavity and an Oxford ESR-900 helium flow cryostat. All EPR spectra were collected at 10 K, 9.38 GHz.Preparation of [Mg(acac)(THF)4]2[FePh2(μ-Ph)]2·4THF (1a)
Solid Fe(acac)3 (70.5 mg, 0.2 mmol) was added to a frozen solution of THF (2 mL) and PhMgBr in THF (1.0 M, 800 μL, 0.8 mmol) in a 20 mL scintillation vial. The frozen solution containing the solid Fe(acac)3 was allowed to stir at −30 °C for 5 minutes before quickly transferring the vial to −80 °C. At −80 °C, the solution was allowed to sit for 30 min at prior to filtering the solution through cold Celite. Following the filtration, cold toluene (1 mL) was added to the solution at −80 °C. The red solution was allowed to sit at −80 °C for an additional 15 minutes before filtering through cold Celite. The vial was then sealed with Apiezon N-grease and stored in a −80 °C freezer. Red crystals of 1a formed within a few days. Note that1ais extremely air and temperature sensitive, precluding further analyses. Analogous complications exist for the other di- and tetrameric iron species whose synthesis are described below.Preparation of [Mg(NMP)6][FePh2(μ-Ph)]2·3.5THF (1b)
Solid Fe(acac)3 (100 mg, 0.28 mmol) was dissolved in a solution of THF (10 mL) and 1-methyl-2-pyrrolidone (NMP) (0.98 mL, 10.1 mmol). The solution was then cooled to 0 °C, where PhMgBr in THF (1.0 M, 1.128 mL, 1.128 mmol) was added at 0.33 mmol min−1. The orange solution was allowed to stir at 0 °C for 5 minutes at 780 rpm. The orange solution was then filtered through cold Celite and cold pentane (3 mL) was layered on top of the THF solution and stored at −30 °C. Red crystals of 1b were observed the next day.Preparation of Fe4(μ-Ph)6(THF)4·2THF (2a)
Solid Fe(acac)3 (100 mg, 0.28 mmol) was dissolved in THF (2 mL). The solution was then cooled to −30 °C, where PhMgBr in THF (1.0 M, 1.133 mL, 1.133 mmol) was added dropwise at 0.33 mmol min−1. The brown solution was allowed to react at −30 °C for 5 minutes at 620 rpm prior to filtering through cold Celite. The brown solution was allowed to sit at −30 °C an additional 20 min. The solution was filtered through cold Celite once more in order to remove Mg salts. The solution was stored at −30 °C. Crystalline material of 2a formed over the course of several weeks.Preparation of Fe4(μ-p-tolyl)6(THF)4·2THF·C5H12 (2b)
Solid Fe(acac)3 (75 mg, 0.21 mmol) was dissolved in THF (10 mL). The solution was then cooled to −30 °C, where p-tolylMgBr in THF (1.0 M, 849 μL, 0.849 mmol) was added dropwise at 0.33 mmol min−1. The brown solution was allowed to react at −30 °C for 5 minutes at 620 rpm. The brown solution was then filtered through cold Celite. Cold pentane (3 mL) was then layered on top of the THF solution, and was then stored at −80 °C until crystalline material was observed.Preparation of Fe4(μ-p-tolyl)6(THF)3·THF (2c)
Solid Fe(acac)3 (76 mg, 0.22 mmol) was dissolved in THF (2 mL). The solution was then cooled to −30 °C, where p-tolylmagnesium bromide (p-tolylMgBr) in THF (1.0 M, 861 μL, 0.861 mmol) was added dropwise at 0.33 mmol min−1. The solution was allowed to react at −30 °C for 5 minutes at 620 rpm. The brown solution was then filtered through cold Celite. Cold pentane (2 mL) was then layered on top of the THF solution, and the solution was stored at −30 °C until crystalline material was observed.Preparation of Fe4(μ-4-F-Ph)6(THF)4 (2d)
Solid Fe(acac)3 (70 mg, 0.2 mmol) was dissolved in THF (2 mL). The solution was then cooled to 0 °C, where 4-F-PhMgBr in THF (1.0 M, 794 μL, 0.794 mmol) was added dropwise at 0.33 mmol min−1. The brown solution was then filtered through cold Celite. Cold toluene (1 mL) was then layered on top of the THF solution, and the sample was stored at −30 °C until crystalline material formed.Preparation of [trans-Fe(acac)2(THF)2]0.58·[trans-Mg(acac)2(THF)2]0.42 (3)
Solid Fe(acac)3 (66 mg, 0.19 mmol) was dissolved in THF (5 mL). The solution was then cooled to −30 °C, where 4-F-PhMgBr in THF (1.0 M, 186 μL, 0.186 mmol) was added dropwise at 0.33 mmol min−1. The orange solution was allowed to stir at −30 °C for 5 minutes at 620 rpm prior to filtering through cold Celite. Cold pentane (10 mL) was then layered on top of the solution, and the sample was stored at −80 °C until crystalline material was formed.Preparation of [Mg(acac)(THF)4]2[FeBr(4-F-Ph)(μ-4-F-Ph)]2·2.5THF (4)
Solid Fe(acac)3 (63 mg, 0.18 mmol) was dissolved in THF (5 mL). The solution was then cooled to −30 °C, where 4-F-PhMgBr in THF (1.0 M, 535 μL, 0.535 mmol) was added dropwise at 0.33 mmol min−1. The dark orange solution was allowed to stir at −30 °C for 5 minutes at 620 rpm. The dark orange was then filtered through cold Celite. Cold pentane (2 mL) was then layered on top of the solution, and the sample was stored at −80 °C until crystalline material was formed.Preparation of [Mg(acac)(THF)4]2[Fe(4-F-Ph)2(μ-4-F-Ph)]2·2THF (1c)
Solid Fe(acac)3 (75 mg, 0.20 mmol) was dissolved in THF (5 mL). The solution was then cooled to −30 °C, where 4-fluorophenylmagnesium bromide (4-F-PhMgBr) in THF (1.0 M, 850 μL, 0.85 mmol) was added dropwise at 0.33 mmol min−1. The dark red solution was allowed to stir at −30 °C for 5 minutes at 780 rpm. The dark red solution was then filtered through cold Celite. Cold toluene (3 mL) was then layered on top of the THF solution, and the solution was stored at −80 °C until crystalline material was formed.Thermal stability of 1b and 2a in solution
Crystalline material was collected as described in the ESI.† Crystalline material was transferred to a vial containing a known amount of THF at −80 °C for 1b and at −30 °C for 2a using a cold spatula. Once the crystalline material was completely redissolved, the solution was transferred to a vial containing a known amount of THF and stir bar at a warmer temperature using a cold pipette. Aliquots of the decaying solution were taken at various points over 20 minutes. Aliquots were taken using a cold pipette, and were transferred into a Delrin Mössbauer sample cup. Samples were immediately frozen in liquid nitrogen. This process was repeated for various temperatures, including, −20 °C and 0 °C. AAS was used to determined to the concentration of the respective complexes in solution.Reaction of 1b with bromocyclohexane
Dark red blocks of 1b were collected as described in the ESI.† Crystalline material was transferred to a vial containing a known amount of THF at −80 °C using a cold spatula. Once the crystalline material was completely redissolved, the solution was transferred to a vial containing THF, bromocyclohexane, PhMgBr, and stir bar at a warmer temperature using a cold pipette. The crystalline material was allowed to react, and aliquots were taken at various points over 20 minutes. The aliquots were quenched in 1:1 (v/v) THF:MeOH solution. A known amount of dodecane was then added to the quenched samples, and the samples were diluted to 1 mM prior to filtering through silica. AAS was used to determine the concentration of 1b in solution.
Reaction of 2a with bromocyclohexane
Crystalline material was collected as described in the ESI.† Crystalline material was transferred to a vial containing a known amount of THF at −30 °C using a cold spatula. Once the crystalline material was completely dissolved, the solution was transferred to a vial containing THF, bromocyclohexane (15 equiv. wrt 2a), and a stir bar at a warmer temperature using a cold pipette. The crystalline material was allowed to react, and aliquots were taken at various points over 20 minutes. The aliquots were quenched in MeOH (50 μL). A known amount of dodecane was then added to the quenched samples, and the samples were diluted to 1 mM prior to filtering through silica. AAS was used to determine the concentration of 2a in solution.Catalytic reaction protocol using isolated 2a as catalyst
Crystalline material was collected as described in the ESI.† Crystalline material was transferred to a vial containing a known amount of THF at −30 °C using a cold spatula. Once the crystallined was completely dissolved, the solution was quickly transferred to a vial containing THF, bromocyclohexane (13 equiv. wrt 2a), PhMgBr (1.0 M in THF, 13 equiv. wrt 2a), and a stir bar at RT. The crystalline material was allowed to react with electrophile and excess nucleophile, and aliquots were taken over the course of 20 minutes. The aliquots were quenching in MeOH (50 μL). A known amount of dodecane was then added to the quenched samples, and the samples were diluted to 1 mM prior to filtering though silica. AAS was used to determine the concentration of 2a in solution.Catalytic reaction protocol targeting 2a formation in situ
Fe(acac)3 (4 mg, 0.011 mmol) was dissolved in 2 mL THF and cooled to −30 °C. PhMgBr in THF (1.0 M, 45 μL, 4 equiv.) was added dropwise at 0.33 mmol min−1. The resulting brown solution was allowed to mix for 5 min at 620 rpm prior to filtering through cold Celite. The solution was allowed to sit at −30 °C for an additional 20 min, and filtered through cold Celite once more in order to remove any Mg salts. The solution was immediately transferred to a vial at RT containing THF, bromocyclohexane, PhMgBr, and a stir bar. Aliquots were taken at various time points over a course of 20 min, and quenched with MeOH (50 μL). A known amount of dodecane was then added to the quenched samples, and the samples were diluted to 1 mM prior to filtering through silica.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the National Institutes of Health (R01GM111480 to M. L. N.), funding from the National Science Foundation (CHE-1454370 to M. L. N. for initial dinuclear iron–phenyl synthetic work), an NSF Graduate Research Fellowship (T. M. B.) and a Ruth L. Kirchstein National Research Service award (F32GM120823 to S. B. M. III). The NSF is gratefully acknowledged for support for the acquisition of an X-ray diffractometer (CHE-1725028).Notes and references
- M. Tamura and J. Kochi, J. Am. Chem. Soc., 1971, 93, 1487–1489 CrossRef.
- A. Fürstner, A. Leitner, M. Méndez and H. Krause, J. Am. Chem. Soc., 2002, 124, 13856–13863 CrossRef.
- A. Fürstner and R. Martin, Chem. Lett., 2005, 34, 624–629 CrossRef.
- B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef PubMed.
- T. L. Mako and J. A. Byers, Inorg. Chem. Front., 2016, 3, 766–790 RSC.
- S. H. Carpenter and M. L. Neidig, Isr. J. Chem., 2017, 57, 1106–1116 CrossRef PubMed.
- I. Bauer and H.-J. Knolker, Chem. Rev., 2015, 115, 3170–3387 CrossRef PubMed.
- R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485–1493 CrossRef PubMed.
- J. Norinder, A. Matsumoto, N. Yoshikai and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 5858–5859 CrossRef PubMed.
- N. Yoshikai, A. Matsumoto, J. Norinder and E. Nakamura, Angew. Chem., Int. Ed., 2009, 48, 2925–2928 CrossRef PubMed.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef PubMed.
- G. Cera and L. Ackermann, Top. Curr. Chem., 2016, 374, 191–224 CrossRef PubMed.
- Q. Gu, H. H. Al Mamari, K. Gracyzk, E. Diers and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 3868–3871 CrossRef PubMed.
- J. Quintin, X. Franck, R. Hocquemiller and B. Figadère, Tetrahedron Lett., 2002, 43, 3547–3549 CrossRef.
- H. Felkin and B. Meunier, J. Organomet. Chem., 1978, 146, 169–178 CrossRef.
- A. Fürstner, ACS Cent. Sci., 2017, 2, 778–789 CrossRef PubMed.
- C. Bolm, J. Legros, J. L. Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef PubMed.
- A. Fürstner, R. Martin, H. Krause, G. Seidel, R. Goddard and C. W. Lehmann, J. Am. Chem. Soc., 2008, 130, 8773–8787 CrossRef PubMed.
- M. Clémancey, T. Cantat, G. Blondin, J.-M. Latour, P. Dorlet and G. Lefèvre, Inorg. Chem., 2017, 56, 3834–3848 CrossRef PubMed.
- A. Hedström, E. Lindstedt and P.-O. Norrby, J. Organomet. Chem., 2013, 748, 51–55 CrossRef.
- G. Lefèvre and A. Jutand, Chem.–Eur. J., 2014, 20, 4796–4805 CrossRef PubMed.
- A. Hedström, U. Bollmann, J. Bravidor and P.-O. Norrby, Chem.–Eur. J., 2011, 17, 11991–11993 CrossRef PubMed.
- K. Adams, A. K. Bell, J. Birkett, L. Brown, B. Chappell, D. M. Gill, P. K. T. Lo, N. J. Patmore, C. R. Rice, J. Ryan, P. Raubo and J. B. Sweeney, Nat. Chem., 2017, 9, 396–401 CrossRef PubMed.
- F. E. Zhurkin, M. D. Wodrich and X. Hu, Organometallics, 2017, 36, 499–501 CrossRef.
- A. Hedström, E. Lindstedt and P.-O. Norrby, J. Organomet. Chem., 2011, 748, 51–55 CrossRef.
- T. Parchomyk and K. Koszinowski, Chem.–Eur. J., 2016, 22, 15609–15613 CrossRef PubMed.
- R. Schoch, W. Desens, T. Werner and M. Bauer, Chem.–Eur. J., 2013, 19, 15816–15821 CrossRef PubMed.
- R. B. Bedford, P. B. Brenner, E. Carter, P. M. Cogswell, M. F. Haddow, J. N. Harvey, D. M. Murphy, J. Nunn and C. H. Woodall, Angew. Chem., Int. Ed., 2014, 53, 1804–1808 CrossRef PubMed.
- B. Bogdanovic and M. Schwickardi, Angew. Chem., Int. Ed., 2000, 39, 4610–4612 CrossRef PubMed.
- L. E. Aleandri, B. Bogdanovic, P. Bons, C. Duerr, A. Gaidies, T. Hartwig, S. C. Huckett, M. Lagarden, U. Wilczok and R. A. Brand, Chem. Mater., 1995, 7, 1153–1170 CrossRef.
- A. Klose, E. Solari, C. Floriani, A. Chiesi-Villa, C. Rizzoli and N. Re, J. Am. Chem. Soc., 1994, 116, 9123–9135 CrossRef.
- H. Müller, W. Seidel and H. Görls, J. Organomet. Chem., 1993, 445, 133–136 CrossRef.
- C.-L. Sun, H. Krause and A. Fürstner, Adv. Synth. Catal., 2014, 356, 1281–1291 CrossRef.
- J. Sundberg, M. S. Vad, J. E. McGrady, P. M. Bjoremark, M. Hakansson and C. J. McKenzie, J. Organomet. Chem., 2015, 786, 40–47 CrossRef.
- C. Ni and P. P. Power, Organometallics, 2009, 28, 6541 CrossRef.
- S. L. Daifuku, M. H. Al-Afyouni, B. E. R. Synder, J. L. Kneebone and M. L. Neidig, J. Am. Chem. Soc., 2014, 136, 9132–9143 CrossRef PubMed.
- S. L. Daifuku, J. L. Kneebone, B. E. R. Synder and M. L. Neidig, J. Am. Chem. Soc., 2015, 137, 11432–11444 CrossRef PubMed.
- G. Cahiez and H. Avedissian, Synthesis, 1998, 1998, 1199–1205 CrossRef.
- S. B. Muñoz III, S. L. Daifuku, J. D. Sears, T. M. Baker, S. H. Carpenter, W. W. Brennessel and M. L. Neidig, Angew. Chem., Int. Ed., 2018, 57, 6496–6500 CrossRef PubMed.
- K. Ding, F. Zannat, J. C. Morris, W. W. Brennessel and P. L. Holland, J. Organomet. Chem., 2009, 694, 4204–4208 CrossRef.
- E. M. Holt, K. H. Whitmire and D. F. Shriver, J. Am. Chem. Soc., 1982, 104, 5621–5626 CrossRef.
- M. Okazaki, K. Suto, N. Kudo, M. Takano and F. Ozawa, Organometallics, 2012, 31, 4110–4113 CrossRef.
- C. Femoni, M. C. Iapalucci, G. Longoni, S. Zacchini and E. Zazzaroni, Dalton Trans., 2007, 2644–2651 RSC.
- C. Femoni, M. C. Iapalucci, G. Longoni and S. Zacchini, Dalton Trans., 2011, 40, 8685–8694 RSC.
- S. B. Muñoz III, S. L. Daifuku, W. W. Brennessel and M. L. Neidig, J. Am. Chem. Soc., 2016, 138, 7492–7495 CrossRef PubMed.
- H. Müller, W. Seidel and H. Görls, Z. Anorg. Allg. Chem., 1996, 622, 1968–1974 CrossRef.
- M. Nakamura, K. Matsuo, S. Ito and E. Nakamura, J. Am. Chem. Soc., 2004, 126, 3686–3687 CrossRef PubMed.
- R. Martin and A. Fürstner, Angew. Chem., Int. Ed., 2004, 43, 3955–3957 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1851551–1851559. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02915f |
| This journal is © The Royal Society of Chemistry 2018 |