
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLet there be light: stability of palmitic acid monolayers at the air/salt water interface in the presence and absence of simulated solar light and a photosensitizer†
Mona
Shrestha‡
a,
Man
Luo‡
a,
Yingmin
Li
b,
Bo
Xiang
b,
Wei
Xiong
 ab and
Vicki H.
Grassian
*acd
ab and
Vicki H.
Grassian
*acd
aDepartment of Chemistry & Biochemistry, University of California, La Jolla, San Diego, CA 92093, USA. E-mail: vhgrassian@ucsd.edu
bMaterials Science and Engineering Program, University of California, La Jolla, San Diego, CA 92093, USA
cScripps Institution of Oceanography, University of California, La Jolla, San Diego, CA 92093, USA
dDepartment of Nanoengineering, University of California, La Jolla, San Diego, CA 92093, USA
First published on 5th June 2018
Abstract
Long-chain fatty acid monolayers are known surfactants present at air/water interfaces. However, little is known about the stability of these long-chain fatty acid monolayers in the presence of solar radiation. Here we have investigated, for the first time, the stability of palmitic acid monolayers on salt water interfaces in the presence and absence of simulated solar light with and without a photosensitizer in the underlying salt subphase. Using surface sensitive probes to measure changes in the properties of these monolayers upon irradiation, we found that the monolayers become less stable in the presence of light and a photosensitizer, in this case humic acid, in the salt solution. The presence of the photosensitizer is essential in significantly reducing the stability of the monolayer upon irradiation. The mechanism for this loss of stability is due to interfacial photochemistry involving electronically excited humic acid and molecular oxygen reacting with palmitic acid at the interface to form more oxygenated and less surface-active species. These oxygenated species can then more readily partition into the underlying solution.
Introduction
Oceans cover ∼70% of the Earth's surface and contribute to a majority of primary aerosol emissions known as sea spray aerosol (SSA).1 SSA can affect the Earth's radiative balance by scattering or absorbing solar radiation.2 It can also act as cloud condensation nuclei (CCN).3 SSA is comprised of a variety of organic compounds, salts and biological species, depending on the specific aerosol size range.1,4 In the supermicron size range, a large number of the particles that make up SSA are mostly comprised of salts although their surfaces are often coated by surfactants, such as long-chain fatty acid.5 Long-chain fatty acids are ubiquitous in nature and are highly surface-active molecules due to their hydrophobic tails and hydrophilic headgroups. These molecules are present at the ocean surface and they can be incorporated into SSA upon bubble bursting.6–8 The most prevalent long-chain fatty acid in SSA are palmitic and stearic acid, comprising more than two-thirds of the fatty acids present in SSA.5 Furthermore, there is a selective enrichment of Ca2+ compared to other cations present that can transfer along with these surface-active components due to binding interactions with the carboxylate headgroups.9Fatty acid organic coatings on aerosol surfaces can affect the chemical, physical and optical properties of the aerosol particles.10–12 Therefore, the stability of surfactant organic thin films can also impact the surface properties of aerosols. For example, the ordering of organic coated interfaces can affect the transfer between the gas and the aqueous phase as in the case of water evaporation and solubilization of trace gases.11 In particular, highly ordered organics on the surface can prevent the transport of volatile organics as well as decrease the evaporation of water through the surface leading to an increased aerosol lifetime.13,14 If the organic film is destabilized, leading to a loss of order, the aerosol can be more permeable to water, which can impact the overall size of the particle as a function of relative humidity.15
Very recent studies, based mostly on mass spectrometry techniques, have shown the importance of the photochemistry of shorter chain fatty acids, namely nonanoic acid, in the presence and absence of a photosensitizer in generating more oxidized, functionalized and unsaturated compounds in the gas and aqueous phases.16–19 It has been suggested that this photochemistry, both direct excitation of the fatty acid and excitation through photosensitized reactions, can lead to an abiotic source of precursors for secondary organic aerosol formation in the atmosphere.10,16,17 However, in a more recent study, Rapf et al. showed that shorter chain fatty acid and fatty alcohols such as nonanoic acid, hexanoic acid, nonanol and hexanol can undergo photochemical reactions only in the presence of α-keto acids that act as radical initiators and that direct photochemical reactions of these acids and alcohols did not occur.20 Thus, although it is clear that photochemistry can be important in reactions within atmospheric aerosols,21 some questions remain about the photochemistry of fatty acids and the direct excitation mechanism.
Given the importance of both photo-initiated processes in atmospheric chemistry and the stability of organic films at aerosol surfaces, we aim here to better understand the impact of solar radiation on the stability of palmitic acid, one of the dominant long-chain fatty acids present in SSA, at the air/salt water interface in the presence and absence of a photosensitizer using surface sensitive techniques including surface pressure–area isotherms along with infrared reflection absorption spectroscopy (IRRAS). Langmuir monolayers have been used as a proxy for aerosols coated with fatty acids in various studies and surface isotherms can provide structural information, as well as the packing density, of the monolayer present at the air/water interface.13,22–25 Using a Langmuir trough, the stability of these palmitic acid monolayers with and without a photosensitizer in the presence of salts such as NaCl and CaCl2 found in the ocean is investigated. Humic acid was chosen as the photosensitizer for this work as it represents chromophoric dissolved organic matter (cDOM), natural photosensitizers found in seawater, that have been found to concentrate on the sea surface.17,26 These photosensitizers are also expected to get into the aerosol phase. To our knowledge, this is the first study of the stability of palmitic acid monolayers, or any fatty acid monolayer, at the air/salt water interface with a photosensitizer and simulated solar radiation using surface sensitive probes.
Experimental methods
Palmitic acid (>99%) and humic acid were purchased from Sigma Aldrich and were used without further purification. Chloroform (>99.9%) was purchased from Fisher Scientific. The chloroform was used to prepare 1 mg mL−1 palmitic acid solution. Milli-Q water with an electric resistance of 18.2 MΩ was used. CaCl2 and NaCl salts were purchased from Fisher Scientific and were purified by baking at 500 °C overnight and filtering twice with a Whatman Carbon-Cap activated carbon filter (Whatman Carbon Cap 75, Fisher Scientific) to remove organic contaminants that are known to be surface-active. The final concentrations of the salt solutions were obtained by Mohr titration.A computer-controlled film balance with a Langmuir trough (KSV NIMA LB, S/N AAA100505) enclosed in a dry air purged plexiglass chamber was used for these experiments. Irradiation experiments were performed using a solar simulator (Newport Corporation, Oriel LCS-100) equipped with a 100 W Xe arc lamp. A water filter was placed after the lamp to cut off infrared radiation. The spectral output of the lamp is comparable to that of the solar spectrum and provides irradiation in an area of approximately 4 cm by 7 cm with a power density of 160 to 170 W m−2. The spectral output of the simulator is shown in Fig. S1.† The temperature of the solution in the trough was controlled using a water circulator (Beckman Geneline Cooler). The pH of the subphase ranged from 5 to 6. These pH values lie in the range of aerosol acidity.27,28 We used six different subphases for our experiments: CaCl2 solution (0.005 M), NaCl solution (0.5 M), humic acid (10 mg L−1) with CaCl2 (0.005 M) solution, humic acid (10 mg L−1) with NaCl (0.5 M) solution, a mixture of salt solutions (0.005 M CaCl2 and 0.5 M NaCl) and humic acid (10 mg L−1) with a mixture of salt solutions. The concentrations were chosen to be close to sea water concentrations.
Palmitic acid in chloroform was injected onto the subphase in the trough using a microsyringe (10 μL). After the injection of palmitic acid, the monolayer was left for 20 minutes to allow the evaporation of the solvent. Monolayer stability was investigated in different ways including area and pressure relaxation measurements. For area relaxation measurements, the palmitic acid monolayer is held at a constant surface pressure and the change in molecular area is recorded over time. In particular, for these experiments, the monolayer was held at 25 mN m−1 for ∼3 hours. This surface pressure was chosen since the monolayer is in a condensed phase with fatty acids concentrated at the interface, and it allows full utilization of the irradiation area. Irradiation was started 10 minutes after reaching the surface pressure of 25 mN m−1. For pressure relaxation experiments, the monolayer was held at a particular molecular area/barrier position corresponding to a certain surface pressure and the change in surface pressure with time was measured. For these experiments, an isotherm was collected up to 25 mN m−1 and the monolayer was then expanded after obtaining the isotherm. After 10 minutes, the second isotherm was held at a mean molecular area corresponding to an initial surface pressure of 25 mN m−1 for ∼3 hours. Irradiation was started after holding the monolayer for 10 minutes. All of these experiments were done both in the dark and under simulated irradiation with replicate measurements performed. Errors reported are from these replicate measurements.
Fig. 1 shows a schematic of the IRRAS setup. The IR beam exits from an external port of an infrared spectrometer (Bruker Tensor 37). The incident beam is directed onto the aqueous solution surface in the Langmuir trough at a 30° angle from the surface normal using a gold mirror. The reflected beam is then collected by another gold mirror and is sent to the MCT detector (Infrared Associates Inc., mid-band with a ZnSe window). Each IRRAS spectrum is a collection of 300 scans taken at a spectral resolution of 8 cm−1. IRRAS spectra were collected at two time points: as soon as irradiation started (after 10 minutes of holding for non-irradiation experiments), and after 3 hours of irradiation (after 3 hours and 10 minutes of holding for non-irradiation experiments). Due to low signals and water interference in other regions, here we will focus on the results obtained in the C–H stretching region. For these IRRAS spectra, reflectance–absorbance (RA) is plotted as a function of wavenumber, where
| RA = −log(R/R0) | (1) |
 | ||
| Fig. 1 Experimental setup showing the IRRAS as well as the Langmuir trough. | ||
The same sample preparation protocol was used for sum frequency generation (SFG) measurements. A salt solution was loaded in a Petri dish, and the palmitic acid solution was injected onto the subphase using a microsyringe. Before beginning the SFG measurements, the sample was equilibrated for 20 min for the solvent to evaporate. The powers of the 800 nm and mid-IR beams used are 0.5 μJ and 1 μJ, respectively, with an exposure time of 5 min.
Results and discussion
Palmitic acid monolayers on salt water surfaces
Fig. 2a shows a surface pressure–area isotherm of the palmitic acid monolayer on a NaCl subphase. This isotherm has been discussed in detail previously.29 Briefly, the phase transitions in the palmitic acid isotherm from higher to lower mean molecular area (MMA) are gas and tilted condensed (G-TC) coexistence phase, tilted condensed (TC) phase, and untilted condensed (UC) phase, respectively, as indicated in Fig. 2a. After the UC phase, the monolayer collapses with decreasing MMA. Fig. 2b shows the IRRAS spectra at different surface pressures (0, 10 and 25 mN m−1) representing various phases of the palmitic acid monolayer at the air/salt water interface. The IRRAS results for the palmitic acid monolayer reveal 3 peaks at 2854 cm−1, 2919 cm−1 and 2960 cm−1 that correspond to the methylene symmetric, methylene asymmetric and methyl asymmetric stretches, respectively. Fig. 2b shows that increasing the surface pressure, which leads to a more ordered monolayer and a higher surface coverage in the infrared beam path, can increase the IRRAS spectral intensity. In addition, Fig. 2c displays the SFG spectrum obtained for the palmitic acid monolayer on the NaCl subphase in a condensed aqueous phase. The SFG setup used for this experiment has been described in Xiang, Li et al.30 SFG spectroscopy is interface sensitive and has been previously employed to study atmospherically relevant systems at air/water interfaces.31–35 The ssp-polarized SFG spectrum in Fig. 2c contains two peaks at 2870 cm−1 and 2940 cm−1 and can be assigned to the methyl symmetric stretch and methyl fermi resonance, respectively, based on the assignment of Tang et al.29 The SFG spectrum further confirms the presence of palmitic acid molecules at the air/salt water interface. The difference in IRRAS and SFG spectra in the C–H stretching region could be due to differences in the symmetry selection rules between the two spectroscopic techniques. | ||
| Fig. 2 (a) Surface pressure–area isotherm of a palmitic acid monolayer on NaCl (aq) showing various phases: gas and tilted condensed (G-TC); tilted condensed (TC); and untilted condensed (UC). (b) IRRAS spectra of palmitic acid on NaCl (aq) at specified surface pressures. (c) SFG spectrum of palmitic acid on NaCl (aq). (d) Normalized area (MMA/MMA0) relaxation curve at a constant surface pressure of 25 mN m−1 and (e) normalized pressure (π/π0) relaxation curve at a MMA corresponding to a pressure of 25 mN m−1 of the palmitic acid monolayer on NaCl. In the relaxation curves, time = 0 when the holding of the monolayer begins. | ||
The curves of the palmitic acid monolayer on the NaCl subphase for area and surface pressure relaxation experiments are also shown in Fig. 2d and e, respectively. When the underlying solution contains salts such as NaCl or CaCl2, the cation can bind to the headgroup of palmitic acid as Allen and co-workers have described in detail.29,36 Even with large differences in the concentrations of Ca2+ and Na+, the decay rates of the relaxation curves of the palmitic acid monolayer on NaCl only and CaCl2 only (not shown here) were comparable. Although the concentrations of these salts are very different, 0.5 M and 0.005 M for NaCl and CaCl2, respectively, the similarity in the observed phenomenon may be due to the stronger binding affinity of Ca2+ to COO− compared to Na+ as Ca2+ can form ionic complexes in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 bridging configuration even at lower concentrations while Na+ favors a solvent-separated Na+:COO− that has a weaker binding affinity to the COO− headgroup.29,35,36 As shown in Fig. 2, over time, these monolayers are not stable as there is a gradual decrease in both the area and pressure relaxation curves. This loss of stability has been attributed to several factors including leakage of palmitic acid molecules through the barriers, rearrangement of molecules, three-dimensional nuclei formation and dissolution into the underlying phase.37,38
:1 bridging configuration even at lower concentrations while Na+ favors a solvent-separated Na+:COO− that has a weaker binding affinity to the COO− headgroup.29,35,36 As shown in Fig. 2, over time, these monolayers are not stable as there is a gradual decrease in both the area and pressure relaxation curves. This loss of stability has been attributed to several factors including leakage of palmitic acid molecules through the barriers, rearrangement of molecules, three-dimensional nuclei formation and dissolution into the underlying phase.37,38
Palmitic acid monolayers on salt water in the presence and absence of light and a photosensitizer
Changes in the stability of palmitic acid monolayers on salt solutions were monitored using area and surface pressure relaxation curves under different conditions. In particular, the area relaxation curves of palmitic acid monolayers on NaCl and CaCl2 with and without humic acid for dark versus irradiation experiments are shown in Fig. 3a and b. Irradiating the palmitic acid monolayers led to an increase in the decay rates of the relaxation curves irrespective of the underlying solutions (Fig. 3a–c). However, it is evident that a much greater change in the decay rate is observed upon the addition of humic acid to the salt subphase. The relative molecular area decrease (as shown in the bar graph in Fig. 3c) at 180 minutes in irradiation experiments with respect to dark experiments for humic acid with salts is at least 20% while for salt subphases, the decrease is less than ∼5%. | ||
| Fig. 3 Normalized area relaxation curves of the palmitic acid monolayer for both irradiation and no irradiation experiments on the salt mixture (NaCl and CaCl2) (a) without and (b) with humic acid. (c) Bar graph showing the decrease in relative area at time = 180 min in irradiation experiments with respect to no irradiation experiments for three different salt subphases in the presence (solid bars) and absence (crosshatched bars) of humic acid. | ||
The IRRAS spectra in the C–H stretching region appear similar for palmitic acid monolayers on different underlying solutions in both dark and irradiation experiments. ESI Fig. S2a and S2b† display the IRRAS spectra of the monolayer on humic acid and a mixture of salts (NaCl and CaCl2) with and without irradiation as examples. The difference spectra shown in Fig. S2c† were obtained by subtracting the intensity of the IRRAS spectra at the beginning of the irradiation (or after holding the monolayer for 10 minutes) from the intensity of the IRRAS spectra after 3 h of irradiation or no irradiation. The difference spectra obtained for dark and irradiation experiments are comparable in terms of intensity. There are only small changes in the intensity in the C–H stretching region for both experiments and this result is expected since the monolayer is held at the same surface pressure throughout both experiments and as shown in Fig. 2b, the spectral intensity changes only for different surface pressures.
Like the area relaxation curves, the pressure relaxation curves showed similar results for the various underlying solutions in the presence and absence of solar radiation (Fig. 4a & b). Irradiation experiments display a higher decay rate of the relaxation curves than those in the dark. The biggest change in the decay rate was observed for palmitic acid monolayers when humic acid is mixed with the salts (NaCl, CaCl2 or NaCl and CaCl2) as seen for the area relaxation experiments (Fig. 4b). There is also a distinct shift in the slope of the stability curve when the solar simulator is turned on and this trend is only seen for the salt solutions containing humic acid (Fig. 4b inset).
 | ||
| Fig. 4 Normalized pressure relaxation curves for NaCl and CaCl2 (a) without and (b) with humic acid. The inset shows the distinct shift in the slope of the curve upon the start of irradiation. (c) Difference IRRAS spectra obtained for the palmitic acid monolayer on NaCl and CaCl2 in humic acid. The difference spectrum (Δ reflectance–absorbance) is the spectrum collected after 3 h of either irradiation or no irradiation minus the spectrum collected initially. Because of the way in which these reflectance spectra are plotted, positive peaks in the difference spectrum reflect a loss of light absorbing molecular species. | ||
As seen for the area relaxation experiments, the IRRAS spectra for pressure relaxation experiments show three C–H stretches for the palmitic acid monolayer. The difference spectra in Fig. 4c clearly show that palmitic acid monolayers on salt solutions containing humic acid have a large difference in spectral intensity between light and dark experiments. In contrast, the difference spectra obtained from the experiments containing only the salt do not show much difference in the spectral intensity for monolayers that were irradiated and not irradiated (not shown here). These results in the C–H stretching region indicate that there is a higher loss of palmitic acid from the surface in the presence of a photosensitizer and salts upon irradiation in comparison to the dark experiment.
Models to describe the loss of palmitic acid at the interface
It is thus concluded from both area and pressure relaxation experiments conducted using surface sensitive techniques that irradiated palmitic acid monolayers on salt solutions containing humic acid are significantly less stable than those without. These results suggest that there could be a loss of palmitic acid from the surface at a higher rate upon irradiation and in the presence of humic acid. There are two theoretical models that can be applied to understand the expulsion of fatty acids from the surface: (1) nucleation and (2) dissolution. Nucleation theory by Vollhardt et al. is based on the formation of a 3-dimensional solid phase at the air/water interface.37 Dissolution of interfacial molecules into the underlying solution can be driven by differences in chemical potential.38 These two models yield different-shaped area relaxation curves, monotonic versus S-shaped for dissolution and nucleation, respectively.38 In the current study, the area relaxation curves obtained for our experiments are monotonic and not S-shaped, which means that the dissolution model is the more relevant one. Thus, the dissolution model is used to fit these curves to get a better understanding of the stability behavior of the palmitic acid monolayers with and without irradiation.Accordingly as discussed in Patil et al., the desorption kinetics of fatty acids changing with time in area relaxation experiments can be fitted to a two stage model.39 In particular, ln(MMA/MMA0) decreases linearly with t1/2 and t at the initial and later stages, respectively. The desorption coefficients are as follows:
 | (2) |
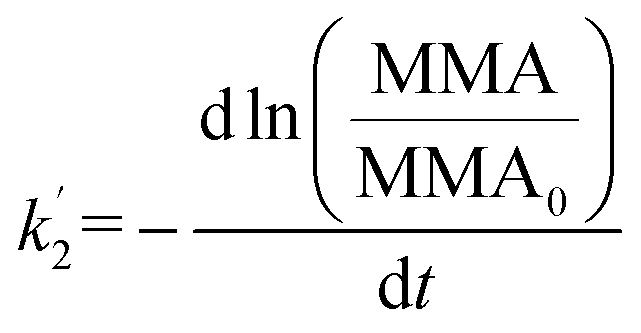 | (3) |
In order to understand and quantify the kinetics observed in these experiments, we used the area relaxation curves obtained for palmitic acid monolayers on various subphases for dark and irradiation experiments. It must be noted that the irradiation was started after holding the monolayer for 10 min at a constant surface pressure; so, only the kinetics for the later stages is measured from our results. We thus replotted the curves obtained for palmitic acid monolayers on different salt subphases with and without humic acid (for dark and irradiation experiments) as ln(MMA/MMA0) versus time to calculate the slope which is equal to −k′2. The desorption coefficients for the non-irradiation and irradiation experiments and their ratios for all the subphases are shown in Table 1. It can be observed from Table 1 that the ratios for irradiation over no irradiation rate constants (light compared to dark) are close to 1 for salt subphases while the ratios are at least 3 and above when humic acid is added to the salt subphases. These results indicate that compared to dark experiments, palmitic acid monolayers are considerably less stable upon irradiation with solar radiation only in the presence of a photosensitizer, and when humic acid is present, there is a significant increase in the dissolution of interfacial molecules into the underlying salt solution.
| Subphases | k ′2 (s−1) | Ratio | |
|---|---|---|---|
| Light | Dark | ||
| NaCl | 1.8 ± 0.1 × 10−5 | 1.5 ± 0.1 × 10−5 | 1.2 ± 0.1 |
| NaCl + humic acid | 3.6 ± 0.5 × 10−5 | 1.2 ± 0.1 × 10−5 | 3.0 ± 0.5 |
| CaCl2 | 1.4 ± 0.1 × 10−5 | 8.3 ± 0.1 × 10−6 | 1.6 ± 0.2 |
| CaCl2 + humic acid | 3.7 ± 0.1 × 10−5 | 1.2 ± 0.1 × 10−5 | 3.0 ± 0.1 |
| NaCl + CaCl2 | 1.6 ± 0.3 × 10−5 | 1.4 ± 0.2 × 10−5 | 1.1 ± 0.2 |
| NaCl + CaCl2 + humic acid | 2.9 ± 0.3 × 10−5 | 8.3 ± 0.1 × 10−6 | 3.5 ± 0.5 |
There can be various reasons for the increased dissolution rate. Temperature changes could lead to increased solubility of palmitic acid into the subphase. However, when we performed control experiments, neither increasing nor decreasing the temperature of the trough with or without irradiation had any effect on the decay rate of the area relaxation curve (Fig. S3†). Similarly, an increase of the subphase pH has also been suggested to contribute to higher dissolution of the fatty acid monolayer.38 In our case, the pH did not change upon irradiation, which rules out changes in pH as a possible reason for the increased dissolution. Therefore, another mechanism must be operative for the loss of stability that involves interfacial photochemistry which occurs upon initiation by the photosensitizer in the underlying salt solution. A mechanism for this interfacial photosensitized chemistry is discussed in more detail below.
Oxygenation of palmitic acid monolayers and product dissolution into the subphase
It is well known that light absorption by humic acid can produce a variety of reactive species such as OH, 3HA*, 1O2, and O2−.40,41 These reactive oxygen species along with excited humic acid can react with fatty acids, including palmitic acid molecules, at the interface resulting in various photochemical degradation pathways. It is proposed here that photosensitized reactions of palmitic acid at the interface form fewer surface-active species that dissolve into the bulk, which leads to higher rates of loss of molecules from the interface. Since the addition of humic acid to the salt subphase slightly shifts the isotherm of the palmitic acid monolayer to a higher mean molecular area by ten percent, as seen in Fig. S4,† humic acid is found to be in the near-interfacial region. In particular, hydrogen abstraction of palmitic acid by the excited humic acid at the interface can lead to condensed phase products that can partition into the bulk. Tinel et al. recently proposed a mechanism for short chain fatty acid photochemistry at interfaces.19 In that mechanism, there is the addition of molecular oxygen to the fatty acid radical which forms the peroxy radical that can either fragment to a saturated ketone or form compounds with hydroxyl and carbonyl functional groups.19 These compounds may continue to react with the excited photosensitizer and upon addition of molecular oxygen to the fatty acid radical, forming more oxidized products, like more highly oxygenated acids. Gas phase products were also reported by Tinel et al. for shorter chain fatty acids; however, we did not observe any such products using Chemical Ionization Mass Spectrometry (CIMS) upon irradiating palmitic acid with humic acid and salts. Although the absence of gas phase products can be due to the low concentration of palmitic acid used in the experiments and monolayer coverages, the photo-initiated reaction of palmitic acid forming gas phase products can only be a minor pathway due to the long carbon chain and the low volatility of the products formed. Thus, when palmitic acid is irradiated in the presence of a photosensitizer we propose the formation of more oxidized species that are less surface-active and can more readily dissolve and partition into the bulk salt water subphase.19 A summary of this proposed mechanism is shown in Scheme 1. | ||
| Scheme 1 Proposed photosensitized reaction scheme for palmitic acid in the presence of humic acid. The triplet excited state of humic acid, 3HA*, is shown to initiate the reaction. The scheme is a modified version of Tinel et al. for nonanoic acid in an oxygen rich environment.19 | ||
It is concluded that this photosensitized degradation pathway is the reason for the decreased stability of the monolayer and the higher loss of molecules at the interface upon exposure to solar radiation with a photosensitizer in the salt subphase as shown for the first time by these surface sensitive measurements.
Conclusions
The stability of palmitic acid monolayers on salt solutions with and without a photosensitizer in dark compared to light experiments has been investigated using surface sensitive probes in a Langmuir trough. It was observed that the irradiated monolayers in the presence of humic acid in the near-surface region were significantly less stable due to photosensitized reactions of palmitic acid producing highly oxidized products in the bulk that partition into the underlying subphase. One of the atmospheric implications of this study would be that the fatty acid coating on aerosols with photosensitizers may not be as impermeable to water and other trace gases upon irradiation, which could affect the size and lifetime of marine-derived and other aqueous atmospheric aerosols. The direct photochemistry of palmitic acid is minor and difficult to discern in some cases, and overall not as significant at air/sea water interfaces compared to the photosensitized reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation through the Centers of Chemical Innovation Program under Grant CHE1305427. We would like to thank Professor Heather Allen, Ellen Adams, Bethany Rudd and Kimberly Carter for their insights regarding monolayer preparation. We also want to acknowledge Professor Kimberly Prather and Jon Sauer for conducting CIMS experiments and Professor Michael Tauber for helpful discussions.References
- P. K. Quinn, D. B. Collins, V. H. Grassian, K. A. Prather and T. S. Bates, Chem. Rev., 2015, 115, 4383–4399 CrossRef PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere, Academic Press, San Diego, 2000 Search PubMed.
- D. K. Farmer, C. D. Cappa and S. M. Kreidenweis, Chem. Rev., 2015, 115, 4199–4217 CrossRef PubMed.
- R. E. Cochran, O. Laskina, J. V. Trueblood, A. D. Estillore, H. S. Morris, T. Jayarathne, C. M. Sultana, C. Lee, P. Lin, J. Laskin, A. Laskin, J. A. Dowling, Z. Qin, C. D. Cappa, T. H. Bertram, A. V. Tivanski, E. A. Stone, K. A. Prather and V. H. Grassian, Chem, 2017, 2, 655–667 Search PubMed.
- R. E. Cochran, O. Laskina, T. Jayarathne, A. Laskin, J. Laskin, P. Lin, C. Sultana, C. Lee, K. A. Moore, C. D. Cappa, T. H. Bertram, K. A. Prather, V. H. Grassian and E. A. Stone, Environ. Sci. Technol., 2016, 50, 2477–2486 CrossRef PubMed.
- F. Cavalli, M. C. Facchini, S. Decesari, M. Mircea, L. Emblico, S. Fuzzi, D. Ceburnis, Y. J. Yoon, C. D. O'Dowd, J. P. Putaud and A. Dell'Acqua, J. Geophys. Res., 2004, 109, D24215 CrossRef.
- M. Mochida, N. Umemoto, K. Kawamura, H.-J. Lim and B. J. Turpin, J. Geophys. Res., 2007, 112, D15209 CrossRef.
- H. Tervahattu, J. Juhanoja and K. Kupiainen, J. Geophys. Res., 2002, 107(D16), 4319 CrossRef.
- R. E. Cochran, O. S. Ryder, V. H. Grassian and K. A. Prather, Acc. Chem. Res., 2017, 50, 599–604 CrossRef PubMed.
- V. Vaida, Science, 2016, 353, 650 CrossRef PubMed.
- D. J. Donaldson and V. Vaida, Chem. Rev., 2006, 106, 1445–1461 CrossRef PubMed.
- C. R. Ruehl and K. R. Wilson, J. Phys. Chem. A, 2014, 118, 3952–3966 CrossRef PubMed.
- S. Li, L. Du, Z. Wei and W. Wang, Sci. Total Environ., 2017, 580, 1155–1161 CrossRef PubMed.
- J. F. Davies, R. E. H. Miles, A. E. Haddrell and J. P. Reid, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8807–8812 CrossRef PubMed.
- E. C. Griffith, T. R. C. Guizado, A. S. Pimentel, G. S. Tyndall and V. Vaida, J. Phys. Chem. C, 2013, 117, 22341–22350 CrossRef.
- R. Ciuraru, L. Fine, M. Pinxteren, B. D'Anna, H. Herrmann and C. George, Environ. Sci. Technol., 2015, 49, 13199–13205 CrossRef PubMed.
- R. Ciuraru, L. Fine, M. van Pinxteren, B. D'Anna, H. Herrmann and C. George, Sci. Rep., 2015, 5, 12741 CrossRef PubMed.
- S. Rossignol, L. Tinel, A. Bianco, M. Passananti, M. Brigante, D. J. Donaldson and C. George, Science, 2016, 353, 699–702 CrossRef PubMed.
- L. Tinel, S. Rossignol, A. Bianco, M. Passananti, S. Perrier, X. Wang, M. Brigante, D. J. Donaldson and C. George, Environ. Sci. Technol., 2016, 50, 11041–11048 CrossRef PubMed.
- R. J. Rapf, R. J. Perkins, M. R. Dooley, J. A. Kroll, B. K. Carpenter and V. Vaida, ACS Cent. Sci., 2018, 4(5), 624–630 CrossRef PubMed.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Chem. Rev., 2015, 115, 4218–4258 CrossRef PubMed.
- M. R. Sierra-Hernandez and H. C. Allen, Langmuir, 2010, 26, 18806–18816 CrossRef PubMed.
- R. Mendelsohn and C. R. Flach, in Handbook of Vibrational Spectroscopy, John Wiley & Sons, Ltd, 2006, DOI:10.1002/0470027320.s2205.
- B. T. Mmereki, D. J. Donaldson, J. B. Gilman, T. L. Eliason and V. Vaida, Atmos. Environ., 2004, 38, 6091–6103 CrossRef.
- L. F. Voss, M. F. Bazerbashi, C. P. Beekman, C. M. Hadad and H. C. Allen, J. Geophys. Res., 2007, 112, D06209 Search PubMed.
- S. Canonica, U. Jans, K. Stemmler and J. Hoigne, Environ. Sci. Technol., 1995, 29, 1822–1831 CrossRef PubMed.
- A. M. Fridlind and M. Z. Jacobson, J. Geophys. Res., 2000, 105, 17325–17340 CrossRef.
- T. Zhang, S. L. Brantley, D. Verreault, R. Dhankani, S. A. Corcelli and H. C. Allen, Langmuir, 2018, 34, 530–539 CrossRef PubMed.
- C. Y. Tang and H. C. Allen, J. Phys. Chem. A, 2009, 113, 7383–7393 CrossRef PubMed.
- B. Xiang, Y. Li, C. H. Pham, F. Paesani and W. Xiong, Sci. Adv., 2017, 3, e1701508 CrossRef PubMed.
- P. G. Blower, E. Shamay, L. Kringle, S. T. Ota and G. L. Richmond, J. Phys. Chem. A, 2013, 117, 2529–2542 CrossRef PubMed.
- S. N. Wren, B. P. Gordon, N. A. Valley, L. E. McWilliams and G. L. Richmond, J. Phys. Chem. A, 2015, 119, 6391–6403 CrossRef PubMed.
- S. M. Burrows, E. Gobrogge, L. Fu, K. Link, S. M. Elliott, H. Wang and R. Walker, Geophys. Res. Lett., 2016, 43(15), 8306–8313 CrossRef.
- E. M. Adams, D. Verreault, T. Jayarathne, R. E. Cochran, E. A. Stone and H. C. Allen, Phys. Chem. Chem. Phys., 2016, 18, 32345–32357 RSC.
- E. Adams and H. Allen, Atmosphere, 2013, 4, 315–336 CrossRef.
- C. Y. Tang, Z. Huang and H. C. Allen, J. Phys. Chem. B, 2010, 114, 17068–17076 CrossRef PubMed.
- D. Vollhardt, Adv. Colloid Interface Sci., 2006, 123, 173–188 CrossRef PubMed.
- A. M. Brzozowska, M. H. G. Duits and F. Mugele, Colloids Surf., A, 2012, 407, 38–48 CrossRef.
- G. S. Patil, R. H. Mathhews and D. G. Cornwell, J. Lipid Res., 1973, 14, 26–31 Search PubMed.
- F. al Housari, D. Vione, S. Chiron and S. Barbati, Photochem. Photobiol. Sci., 2010, 9, 78–86 RSC.
- J. P. Aguer, C. Richard and F. Andreux, Analusis, 1999, 27, 387–389 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01957f |
| ‡ Denotes co-first authors. |
| This journal is © The Royal Society of Chemistry 2018 |