
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective radical process for synthesis of chiral indolines by metalloradical alkylation of diverse C(sp3)–H bonds†
Xin
Wen
 ,
Yong
Wang
and
X. Peter
Zhang
*
,
Yong
Wang
and
X. Peter
Zhang
*
Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, USA. E-mail: peter.zhang@bc.edu
First published on 14th May 2018
Abstract
A new C–C bond formation strategy based on enantioselective radical alkylation of C(sp3)–H bonds via Co(II)-based metalloradical catalysis has been demonstrated for stereoselective synthesis of chiral indolines. The Co(II)-based system enables activation of aryldiazomethanes as radical precursors at room temperature for enantioselective intramolecular radical alkylation of broad types of C–H bonds, constructing 2-substituted indolines in high yields with excellent enantioselectivities. In addition to chemoselectivity and regioselectivity, this Co(II)-catalyzed alkylation features tolerance to functional groups and compatibility with heteroaryl substrates. Detailed mechanistic studies provide insight into the underlying stepwise radical pathway.
Introduction
Recent years have witnessed intense research efforts in exploring the unique features of radical reactions for organic synthesis.1 Among the diverse types of radical reactions, hydrogen atom abstraction (HAA) has been recognized as a general pathway to activate C(sp3)–H bonds, offering a potential approach for C–C bond formation via direct radical C–H alkylation.2 In addition to the prerequisite for controlled generation of the incoming radicals, development of HAA-based radical C–H alkylation, however, faces formidable challenges associated with governing the reactivity and selectivity of the outgoing alkyl radicals for ensuing C–C bond formation. In particular, control of enantioselectivity of radical reactions is typically difficult.1,2 Among recent developments,3 metalloradical catalysis (MRC), which involves the use of metal-centered radicals for catalytic generation of metal-stabilized organic radicals while controlling their radical reactions, has emerged as a conceptually new approach for the development of stereoselective radical processes.4,5 As stable metalloradicals, Co(II) complexes of D2-symmetric chiral amidoporphyrins [Co(D2-Por*)] exhibit the capability of homolytically activating diazo compounds as radical precursors to generate α-Co(III)-alkyl radicals.6 These Co-stabilized C-centered radicals can serve as key catalytic intermediates for asymmetric radical transformations.7 Recently, Co(II)-based MRC was further extended to the use of donor-substituted diazo compounds such as aryldiazomethanes as radical precursors.8 Upon activation, the resulting α-Co(III)-benzyl radicals could undergo radical addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and radical substitution for stereoselective radical cyclopropanation.8a Besides radical addition, we were interested in exploring the potential ability of α-Co(III)-benzyl radicals for HAA that might lead to radical alkylation of C–H bonds. Particularly, we were attracted to aryldiazomethane 1′ with ortho-amino functionality and hypothesized that the corresponding α-Co(III)-benzyl radical intermediate A would favor intramolecular HAA from the C–H bonds at the distal 5-position to form ε-Co(III)-aminoalkyl radical B, where the C-centered radical would be stabilized by the lone pair of the adjacent nitrogen (Scheme 1). If the α-aminoalkyl radical in B could proceed 5-exo-tet radical cyclization at the α-carbon for C–C bond formation in an asymmetric fashion, it would lead to a new catalytic system for enantioselective radical C–H alkylation to construct chiral 2-substituted indolines, which exist ubiquitously in natural and synthetic compounds (Fig. S1 in ESI†).9
C bonds and radical substitution for stereoselective radical cyclopropanation.8a Besides radical addition, we were interested in exploring the potential ability of α-Co(III)-benzyl radicals for HAA that might lead to radical alkylation of C–H bonds. Particularly, we were attracted to aryldiazomethane 1′ with ortho-amino functionality and hypothesized that the corresponding α-Co(III)-benzyl radical intermediate A would favor intramolecular HAA from the C–H bonds at the distal 5-position to form ε-Co(III)-aminoalkyl radical B, where the C-centered radical would be stabilized by the lone pair of the adjacent nitrogen (Scheme 1). If the α-aminoalkyl radical in B could proceed 5-exo-tet radical cyclization at the α-carbon for C–C bond formation in an asymmetric fashion, it would lead to a new catalytic system for enantioselective radical C–H alkylation to construct chiral 2-substituted indolines, which exist ubiquitously in natural and synthetic compounds (Fig. S1 in ESI†).9
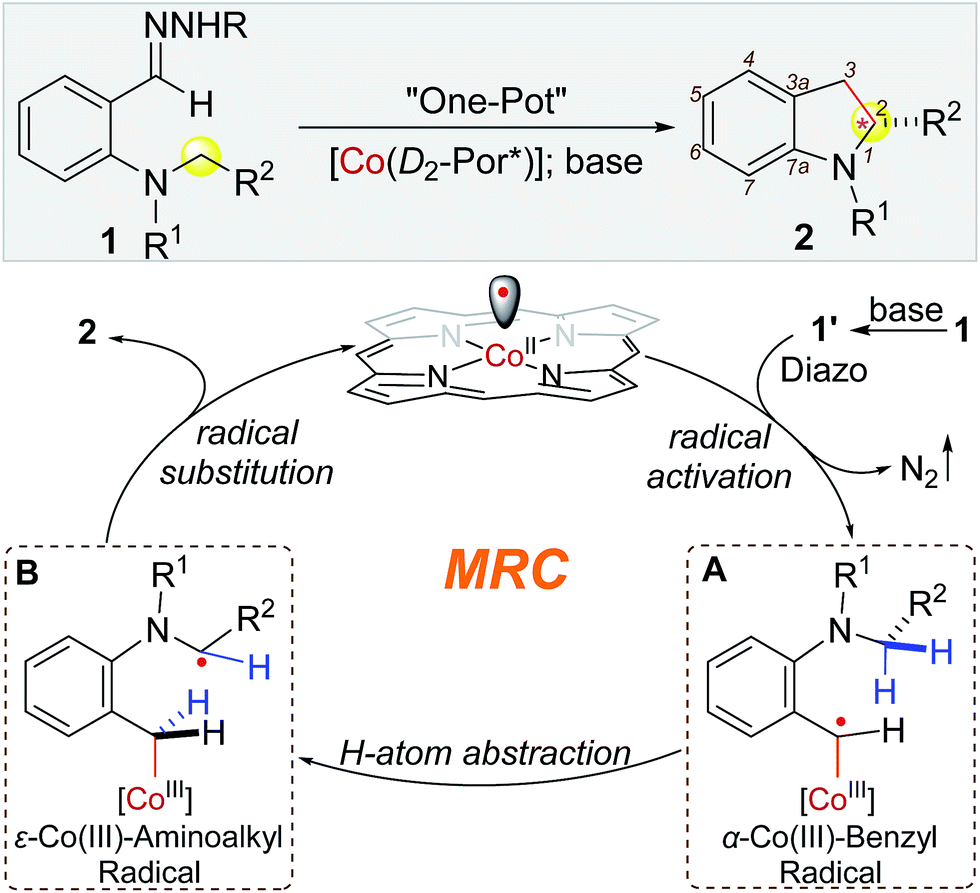 | ||
| Scheme 1 Working proposal for construction of 2-substituted indolines by radical C–H alkylation via Co(II)-MRC. | ||
Tremendous efforts have been devoted to asymmetric synthesis of 2-substituted indolines due to their biological importance.10 Among others,11 existing methods have explored strategies that are based on asymmetric hydrogenation of C2C3 bond,11a,b asymmetric alkylation at C2 position,11c asymmetric formation of C3–C3a bond,11d,e as well as asymmetric formation of N1–C7a11f–h and N1–C2 bonds.11i However, stereoselective construction of chiral 2-substituted indolines that is based on asymmetric formation of C2–C3 bond via C–H alkylation has been less developed.12 This underdevelopment may be attributed to the inherent challenge for enantioselective formation of C–C bonds between two sp3-carbons. To date, there is no previous report on asymmetric construction of 2-substituted indolines through C2(sp3)–C3(sp3) bond formation via stereoselective C(sp3)–H alkylation using donor-type diazo compounds. As a new synthetic application of Co(II)-MRC, we herein report the development of the first catalytic system for asymmetric synthesis of 2-substituted indolines via enantioselective radical C–H alkylation of aryldiazomethanes, which can be generated in situ from readily accessible aryl aldehyde-derived hydrazone precursors. Through the design of a new D2-symmetric chiral amidoporphyrin, the Co(II)-catalyzed process can alkylate diverse types of C–H bonds at room temperature to form chiral 2-substituted indolines.
Results and discussion
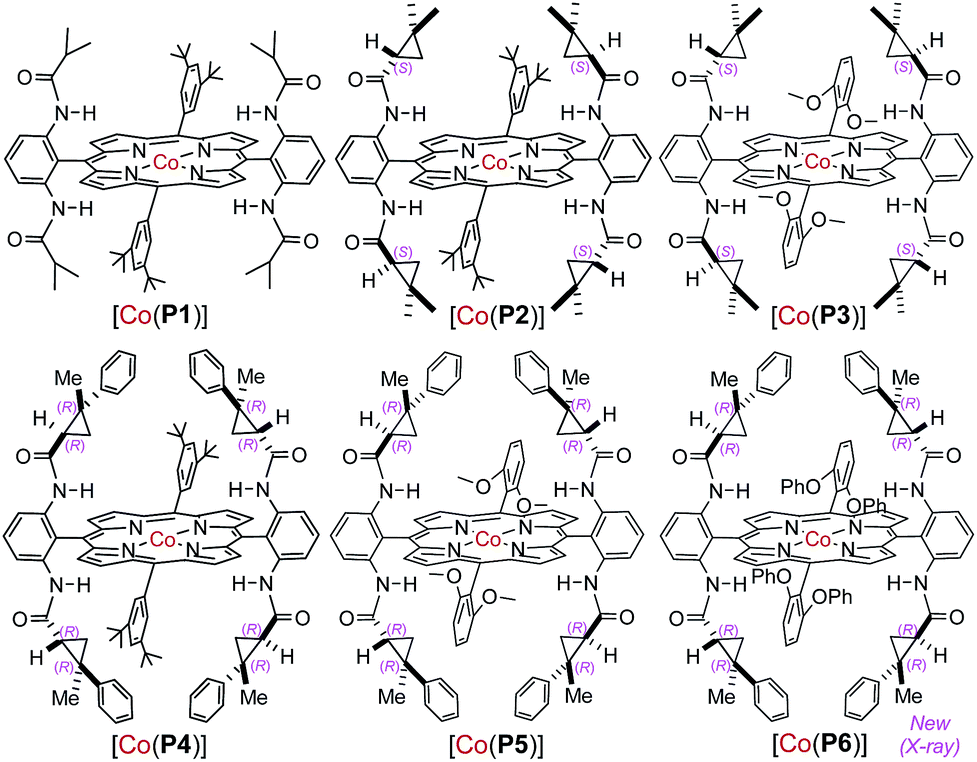
At the outset, o-aminobenzaldehyde-derived hydrazone 1a was selected to examine the feasibility of Co(II)-catalyzed radical C–H alkylation (Table 1). It was found that Co(II) complex of D2h-symmetric achiral amidoporphyrin [Co(P1)] (P1 = 3,5-DitBu-IbuPhyrin)13 was an effective metalloradical catalyst, delivering the desired 2-phenylindoline 2a in 81% yield even at room temperature (entry 1). The high yield implies that the in situ generation of the corresponding aryldiazomethane from 1a was facile and properly matched with the rate of its activation by the catalyst toward the C–H alkylation. To achieve enantioselectivity, the use of the first-generation chiral catalyst [Co(P2)] (P2 = 3,5-DitBu-ChenPhyrin)7e resulted in 2a in a similar yield with a low but significant enantioselectivity (entry 2). The asymmetric induction was improved without affecting the yield when the more sterically demanding catalyst [Co(P3)] (P3 = 2,6-DiMeO-ChenPhyrin) was used (entry 3). This ligand buttressing effect prompted us to evaluate the second-generation catalyst [Co(P4)] (P4 = 3,5-DitBu-QingPhyrin).7b Indeed, both the reactivity and enantioselectivity were significantly enhanced (entry 4). In a similar trend, when sterically more encumbered [Co(P5)] (P5 = 2,6-DiMeO-QingPhyrin) was used, improvement in enantioselectivity continued (entry 5). To amplify such effect, we synthesized the new catalyst [Co(P6)] (P6 = 2,6-DiPhO-QingPhyrin) by replacing the methoxy groups in P5 with phenoxy groups, which could catalyze 2a formation in 82% yield with 66% ee (entry 6). Using [Co(P6)], we then examined the effect of N-substituents in substrate 1 on the reaction. Change from t-butyl (1a) to ethyl (1b) to methyl (1c) carbamates led to a successive increase in both yield and ee, achieving almost quantitative yield and 86% ee in the case of 1c (entries 6–8). This outcome might be attributed to the potential hydrogen-bonding interaction between the carbonyl group of the carbamate and the amido group of the catalyst, which strengthens upon the decrease in sterics (Fig. S2†). Further investigation revealed that both polar and non-polar solvents were suitable (entries 8–13). The solvent of choice was methanol, affording 2-phenylindoline 2c in 92% yield with 94% ee (entry 12; see Table S1† for the effect of different sulfonyl groups).|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Catalyst | Solvent | Yieldb (%) | eec (%) |
| a Carried out with 1 (0.1 mmol) in the presence of Cs2CO3 (2.0 equiv.) by [Co(Por)] (2 mol%) in solvent (1.0 mL); TPS = 2,4,6-triisopropylphenyl sulfonyl. b Isolated yields. c Determined by chiral HPLC. | |||||
| 1 | t-Bu (1a) |

|
Toluene | 81 | – |
| 2 | t-Bu (1a) |

|
Toluene | 74 | 10 |
| 3 | t-Bu (1a) |

|
Toluene | 75 | 19 |
| 4 | t-Bu (1a) |

|
Toluene | 92 | 38 |
| 5 | t-Bu (1a) |

|
Toluene | 90 | 42 |
| 6 | t-Bu (1a) |

|
Toluene | 82 | 66 |
| 7 | Et (1b) |

|
Toluene | 92 | 86 |
| 8 | Me (1c) |

|
Toluene | 99 | 86 |
| 9 | Me (1c) |

|
Benzene | 99 | 86 |
| 10 | Me (1c) |

|
Tetrahydrofuran | 78 | 83 |
| 11 | Me (1c) |

|
Dimethoxyethane | 95 | 88 |
| 12 | Me (1c) |

|
Methanol | 92 | 94 |
| 13 | Me (1c) |

|
Diethyl ether | 98 | 82 |
|
|||||
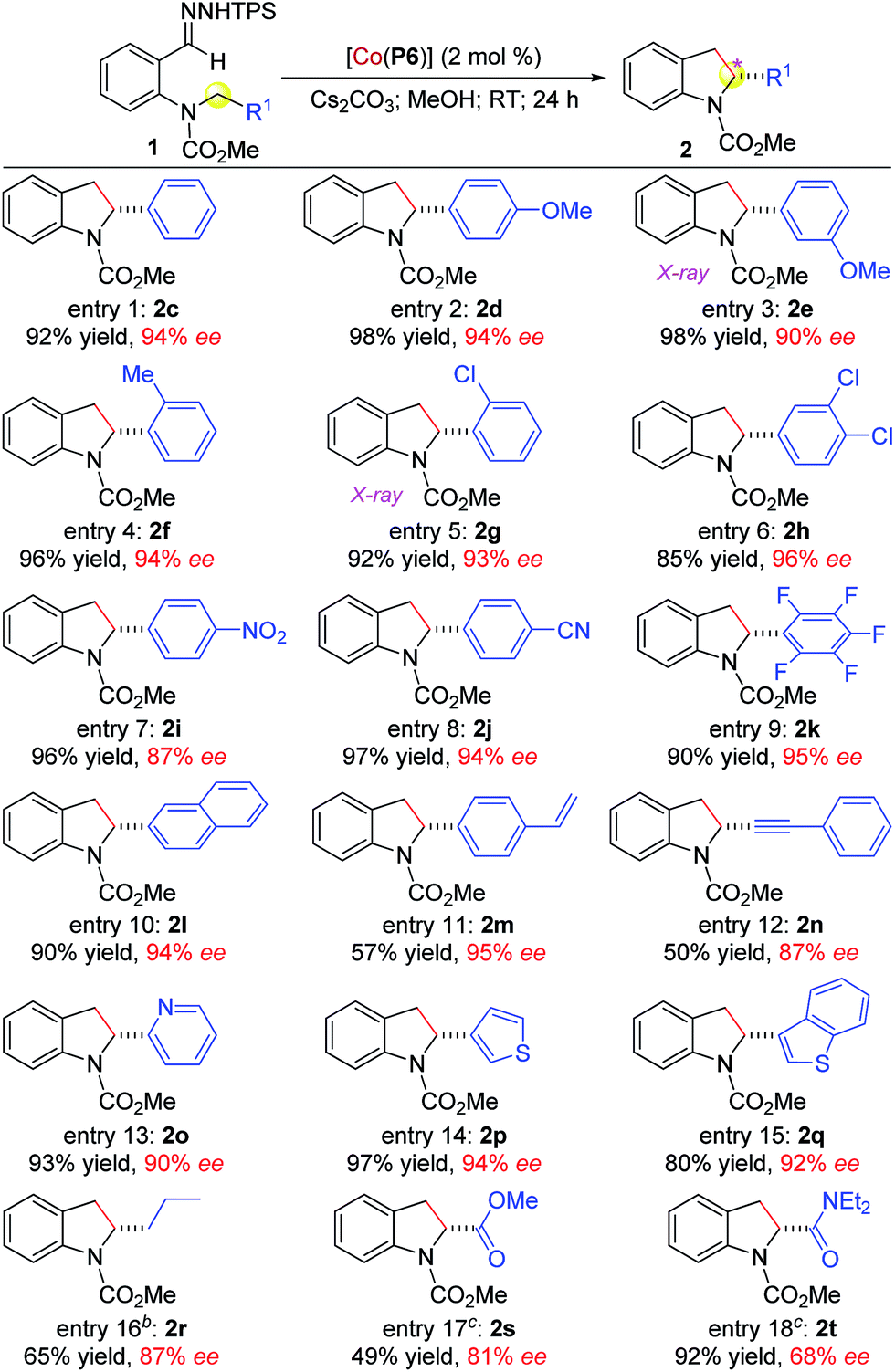
Under the optimized conditions, the scope of [Co(P6)]-catalyzed radical alkylation was evaluated by employing different C–H substrates (Table 2). As demonstrated with substrates 1c–1k, benzylic C–H bonds having varied electronic and steric properties could be effectively alkylated at room temperature in a highly enantioselective fashion, affording chiral 2-arylindolines 2c–2k in excellent yields (entries 1–9). The absolute configurations of 2e and 2g were established by X-ray crystal structural analysis as (R) (see ESI†). It is noteworthy to mention that even the highly electron-deficient pentafluorobenzylic C–H bond in 1k could successfully undergo radical alkylation, forming 2k in 90% yield with 95% ee (entry 9). The system could also alkylate C–H bonds adjacent to other arenes as shown with the 2-naphthyl-based substrate 1l (entry 10). Besides –NO2 and –CN functionalities (entries 7 and 8), the alkylation tolerated both alkenyl and alkynyl groups, as demonstrated by the stereoselective formation of 2m and 2n without complications from potential reactions with the CC and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds, respectively (entries 11 and 12). Notably, this system was equally effective for alkylation of C–H bonds next to heteroarenes, such as pyridine (1o), thiophene (1p), and benzothiophene (1q), providing 2-heteroarylindolines 2o–2q in high yields and enantioselectivities (entries 13–15). Given that both heteroarene and indoline are prevalent structural elements in bioactive natural and synthetic compounds, the access of these linked biheterocyclic compounds in high enantiopurity may find applications in pharmaceutical research and development. Furthermore, non-activated C–H bonds could also be alkylated, as exemplified by the regioselective 1,5-alkylation of 1r, forming 2-propylindoline (2r) in 65% yield with 87% ee although 60 °C was needed (entry 16). The alkylation was further highlighted by its applicability to even C–H bonds that are directly attached to electron-withdrawing groups. For example, electron-poor C–H bonds that are adjacent to ester (1s) and amide (1t) groups were smoothly alkylated at 40 °C to furnish the 2-ester (2s)- and 2-amido (2t)-indolines in varied yields and enantioselectivities (entries 17 and 18). These results manifested the low sensitivity of the Co(II)-based alkylation to the electronic properties of C–H bonds, which are consistent with its underlying radical mechanism.
C bonds, respectively (entries 11 and 12). Notably, this system was equally effective for alkylation of C–H bonds next to heteroarenes, such as pyridine (1o), thiophene (1p), and benzothiophene (1q), providing 2-heteroarylindolines 2o–2q in high yields and enantioselectivities (entries 13–15). Given that both heteroarene and indoline are prevalent structural elements in bioactive natural and synthetic compounds, the access of these linked biheterocyclic compounds in high enantiopurity may find applications in pharmaceutical research and development. Furthermore, non-activated C–H bonds could also be alkylated, as exemplified by the regioselective 1,5-alkylation of 1r, forming 2-propylindoline (2r) in 65% yield with 87% ee although 60 °C was needed (entry 16). The alkylation was further highlighted by its applicability to even C–H bonds that are directly attached to electron-withdrawing groups. For example, electron-poor C–H bonds that are adjacent to ester (1s) and amide (1t) groups were smoothly alkylated at 40 °C to furnish the 2-ester (2s)- and 2-amido (2t)-indolines in varied yields and enantioselectivities (entries 17 and 18). These results manifested the low sensitivity of the Co(II)-based alkylation to the electronic properties of C–H bonds, which are consistent with its underlying radical mechanism.
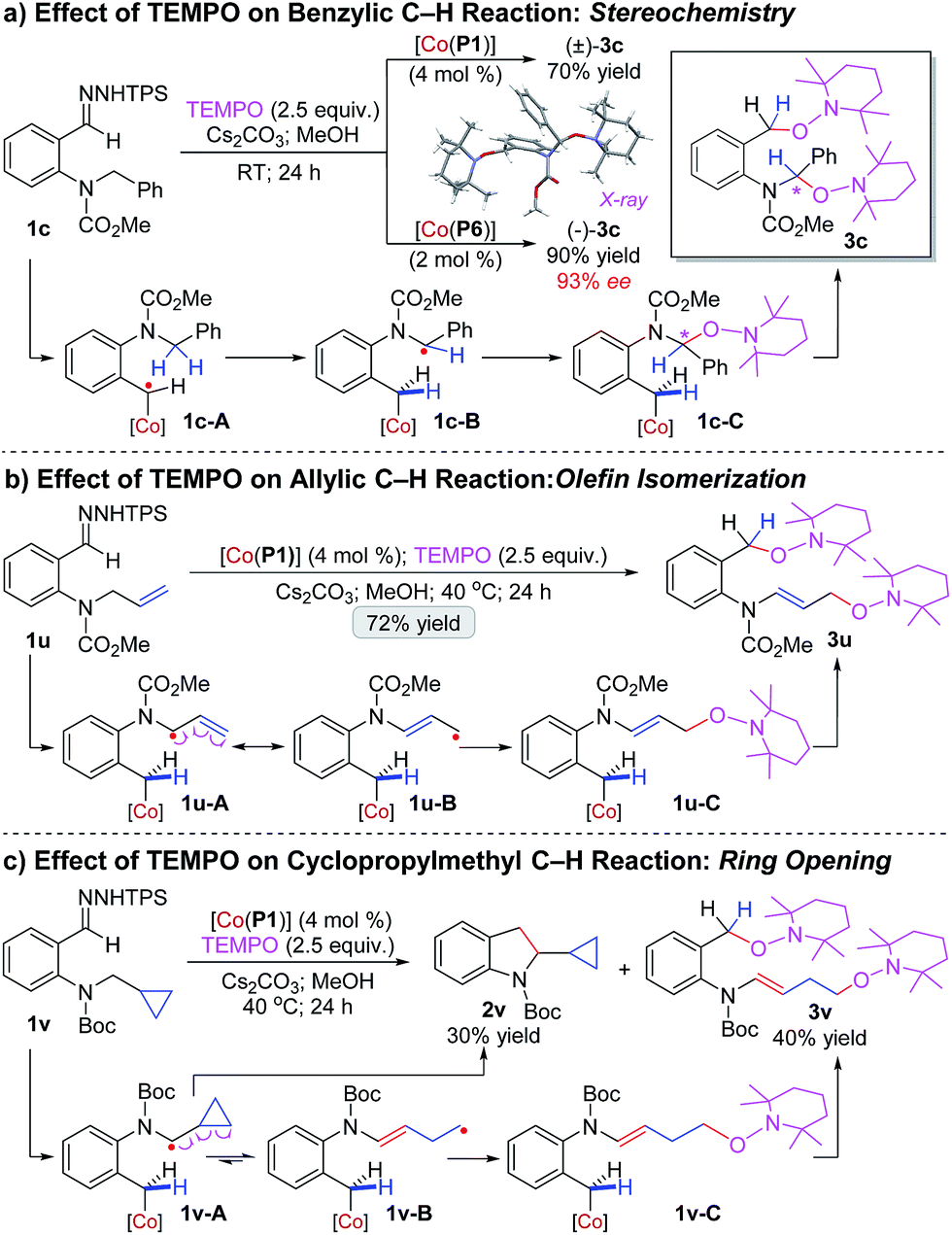
To gain insight into the underlying mechanism of this Co(II)-catalyzed C–H alkylation, a set of mechanistic experiments were conducted (Scheme 2). First, the effect of TEMPO was examined. Addition of TEMPO to the reaction of benzyl C–H substrate 1c by achiral catalyst [Co(P1)] resulted in no formation of C–H alkylation product 2c. Instead, compound (±)-3c was isolated in 70% yield, whose structure was confirmed by X-ray analysis to contain two TEMPO units at the 1- and 5-positions (Scheme 2a). The formation of (±)-3c is indicative of the existence of the initial α-Co(III)-benzyl radical (1c-A) as well as the resulting ε-Co(III)-aminoalkyl radical (1c-B) from 1,5-HAA, which was presumably capped subsequently by TEMPO at the ε-position through radical recombination to generate intermediate 1c-C and then followed by radical substitution with second TEMPO at the α-position to break the weak Co(III)–C bond for final production of (±)-3c. To gain information on stereochemistry, the same reaction was performed with chiral catalyst [Co(P6)] (Scheme 2a). The same bis-TEMPO-capped compound (−)-3c was generated, but in a much higher yield of 90% and, remarkably, with 93% ee. The fact that the enantioselectivity for the C–O bond formation (93% ee) of the TEMPO-capped product (−)-3c was almost identical to the one observed for the C–C bond formation (94% ee) of the C–H alkylation product 2c in the absence of TEMPO (Table 2: entry 1) implies that the prochiral α-aminoalkyl radical in 1c-B was confined inside the chiral pocket of [Co(P6)] to adapt a stable, well-defined configuration. In addition, the resulting Co(III)-supported alkyl radical intermediates from the reaction of 1c by [Co(P1)] in the absence of TEMPO could be directly detected by HRMS (Fig. S3†) and also spin-trapped by phenyl N-tert-butylnitrone (PBN) to exhibit the characteristic EPR signals (Fig. S4†).
 | ||
| Scheme 2 Mechanistic studies on Co(II)-catalyzed intramolecular radical C–H alkylation of o-aminoaryldiazomethanes. | ||
To gather further evidence for the stepwise radical mechanism, we designed specific substrates as radical probes to shed light on the nature of ε-Co(III)-aminoalkyl radical intermediates. First, allylic C–H substrate 1u was prepared as a radical resonance probe to evaluate potential olefin isomerization via the resulting allylic radical intermediate after 1,5-HAA (Scheme 2b). As observed for 1c (Scheme 2a), a similar bis-TEMPO-capped compound 3u was isolated in 72% yield without formation of the corresponding C–H alkylation product (Scheme 2b). Characterizations of 3u revealed that the CC double bond was isomerized from the terminal to internal position (Scheme 2b). Clearly, the resulting ε-Co(III)-aminoalkyl radical, which can be represented by its two resonance forms 1u-A and 1u-B as an allylic radical, was captured by TEMPO to give intermediate 1u-C and then underwent further radical substitution with second TEMPO to deliver 3u. The predominant production of 3u is presumably a result of the much faster capping rate of 1u-B (a primary radical) than 1u-A (a secondary radical) by TEMPO radical. Second, substrate 1v bearing a cyclopropyl ring was synthesized as a radical clock to examine ring-opening of the cyclopropylmethyl radical generated from 1,5-HAA (Scheme 2c). Interestingly, the reaction of 1v by [Co(P1)] in the presence of TEMPO resulted in the formation of bis-TEMPO-capped compound 3v in 40% yield as well as the C–H alkylation product 2v in 30% yield. Obviously, the corresponding ε-Co(III)-aminoalkyl radical intermediate 1v-A underwent two competitive pathways. While its radical substitution formed 2v, the cyclopropylcarbinyl radical in 1v-A also proceeded ring-opening competitively to generate homoallylic alkyl radical 1v-B, which was transformed to the enamine 3v upon two sequential captures by TEMPO via intermediate 1v-C. The fact that 2v and 3v were produced in similar yields indicated that the forming rate of C2–C3 bond via radical substitution to construct the indoline ring was fast.14
Conclusions
In summary, the new Co(II)-based metalloradical system for enantioselective radical alkylation of C(sp3)–H bonds has been developed for stereoselective synthesis of chiral indolines through asymmetric C2–C3 bond formation. Supported by the new ligand 2,6-DiPhO-QingPhyrin, this Co(II)-catalyzed system can activate in situ generated ortho-aminoaryldiazomethanes at room temperature for stereoselective radical alkylation of different types of C(sp3)–H bonds with varied electronic and steric properties. In addition to chemoselectivity and regioselectivity, this radical system features functional group tolerance as well as compatibility with heteroaryl units. It represents a new synthetic application of Co(II)-based MRC and offers a streamlined construction of chiral 2-substituted indolines from readily available starting materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support by NSF (CHE-1624216) and NIH (R01-GM102554).References
-
(a)
C. Chatgilialoglu and A. Studer, Encyclopedia of radicals in chemistry, biology, and materials, John Wiley & Sons, Chichester, West Sussex, Hoboken, N.J., 2012 Search PubMed
; (b) D. P. Curran, N. A. Porter and B. Giese, Stereochemistry of radical reactions: concepts, guidelines, and synthetic applications, John Wiley & Sons, 2008 Search PubMed
-
(a) Q. Lu and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 49–51 CrossRef PubMed
-
(a) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef PubMed
- For reviews and highlights on Co(II)-based MRC, see:
(a) H. Miyabe, A. Kawashima, E. Yoshioka and S. Kohtani, Chem.–Eur. J., 2017, 23, 6225–6236 CrossRef PubMed
- Select examples on Ti(III)-MRC, see:
(a) Y. Q. Zhang, E. Vogelsang, Z. W. Qu, S. Grimme and A. Gansäuer, Angew. Chem., Int. Ed., 2017, 56, 12654–12657 CrossRef PubMed
- For experimental and theoretical studies on the radical mechanism involving α-Co(III)-alkyl radical (also known as Co(III)-carbene radical) intermediates for olefin cyclopropanation, see:
(a) H. J. Lu, W. I. Dzik, X. Xu, L. Wojtas, B. de Bruin and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8518–8521 CrossRef PubMed
- For select examples on asymmetric radical transformations via Co(III)-alkyl radical intermediates, see:
(a) X. Xu, S. F. Zhu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2013, 52, 11857–11861 CrossRef PubMed
-
(a) Y. Wang, X. Wen, X. Cui, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2017, 139, 1049–1052 CrossRef PubMed
-
(a) D. Zhang, H. Song and Y. Qin, Acc. Chem. Res., 2011, 44, 447–457 CrossRef PubMed
- For a review on asymmetric synthesis of 2-substituted indolines, see: S. Anas and H. B. Kagan, Tetrahedron: Asymmetry, 2009, 20, 2193–2199 CrossRef
- For select examples on asymmetric synthesis of 2-substituted indolines, see:
(a) Z. Yang, F. Chen, Y. He, N. Yang and Q.-H. Fan, Angew. Chem., Int. Ed., 2016, 55, 13863–13866 CrossRef PubMed
-
(a) M. Santi, S. T. R. Müller, A. A. Folgueiras-Amador, A. Uttry, P. Hellier and T. Wirth, Eur. J. Org. Chem., 2017, 2017, 1889–1893 CrossRef
- J. V. Ruppel, J. E. Jones, C. A. Huff, R. M. Kamble, Y. Chen and X. P. Zhang, Org. Lett., 2008, 10, 1995–1998 CrossRef PubMed
- For the rate constant of ring-opening of the parent cyclopropylmethyl radical, see: V. W. Bowry, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1991, 113, 5687–5698 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1833864–1833865, 1833867–1833868. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01476k |
| This journal is © The Royal Society of Chemistry 2018 |