
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenetically encoded tags for real time dissection of protein assembly in living cells†
Guolin
Ma‡
 *a,
Qian
Zhang‡
ab,
Lian
He
a,
Nhung T.
Nguyen
a,
Shuzhong
Liu
a,
Zuojiong
Gong
b,
Yun
Huang
*cd and
Yubin
Zhou
*ae
*a,
Qian
Zhang‡
ab,
Lian
He
a,
Nhung T.
Nguyen
a,
Shuzhong
Liu
a,
Zuojiong
Gong
b,
Yun
Huang
*cd and
Yubin
Zhou
*ae
aCenter for Translational Cancer Research, Institute of Biosciences and Technology, College of Medicine, Texas A&M University, 2121 W Holcombe Blvd, Houston, TX 77030, USA. E-mail: gma@ibt.tamhsc.edu; yzhou@ibt.tamhsc.edu
bDepartment of Infectious Diseases, Renmin Hospital of Wuhan University, Wuhan 430060, China
cCenter for Epigenetics and Disease Prevention, Institute of Biosciences and Technology, College of Medicine, Texas A&M University, 2121 W Holcombe Blvd, Houston, TX 77030, USA. E-mail: yun.huang@ibt.tamhsc.edu
dDepartment of Molecular and Cellular Medicine, College of Medicine, Texas A&M University, College Station, TX 77843, USA
eDepartment of Medical Physiology, College of Medicine, Texas A&M University, Temple, TX 76504, USA
First published on 23rd May 2018
Abstract
Simple methods with straightforward readouts that enable real-time interrogation of protein quaternary structure are much needed to facilitate the physicochemical characterization of proteins at the single-cell level. After screening over a series of microtubule (MT) binders, we report herein the development of two genetically encoded tags (designated as “MoTags” for the monomer/oligomer detection tag) that can be conveniently fused to a given protein to probe its oligomeric state in cellulo when combined with routine fluorescence microscopy. In their monomeric form, MoTags are evenly distributed in the cytosol; whereas oligomerization enables MoTags to label MT or track MT tips in an oligomeric state-dependent manner. We demonstrate here the broad utility of engineered MoTags to aid the determination of protein oligomeric states, dissection of protein structure and function, and monitoring of protein–target interactions under physiological conditions in living cells.
Introduction
Proteins with dimeric and high-order oligomeric states comprise one third or more of cellular proteins.1 Maintaining appropriate oligomeric states is essential for proteins to execute their biological functions.2,3 In cell signalling, domain multimerization is often utilized to trigger the initiation of a myriad of cellular events.4 To date, several techniques have been applied to identify the status of protein self-association in living cells, including Förster resonance energy transfer (FRET),5 bimolecular fluorescence complementation (BiFC),6 fluorescence correlation spectroscopy (FCS)7,8 and fluorescence fluctuation spectroscopy (FFS).9 However, FRET and BiFC are more suitable for discriminating proteins from monomeric to oligomeric assembly without providing concrete information on their exact oligomeric state.5,6 In addition, the application of FCS and FFS is currently limited due to their low sensitivity, the requirement of sophisticated instrumentation and complicated post-processing of acquired images.7–9 Simple methods with straightforward readouts, which can be routinely used in laboratories while circumventing laborious protein expression and purification procedures, are needed to aid rapid quantitative dissection of protein behaviours under physiological conditions in living cells.Microtubules (MTs) are cytoskeletal filaments not only serving as tracks for intracellular transport but also playing indispensable roles in critical cellular processes, such as cell movement and division.10,11 The dynamics and network of microtubules are spatially and temporally controlled by microtubule-associated proteins (MAPs).10–12 Most MAPs share common features in their structurally conserved regions, including the MT association/binding domains (MADs) and coil–coil domains (CC).10–12 Dimerization or oligomerization mediated by CC domains has been known to be essential for maintaining a dynamic yet stable MAD-MT association.10,13 This feature prompted us to hypothesize that, when fused with a fluorescent probe, MADs can be engineered to determine the oligomeric states of proteins in single cells using a fluorescence microscope (Scheme 1).
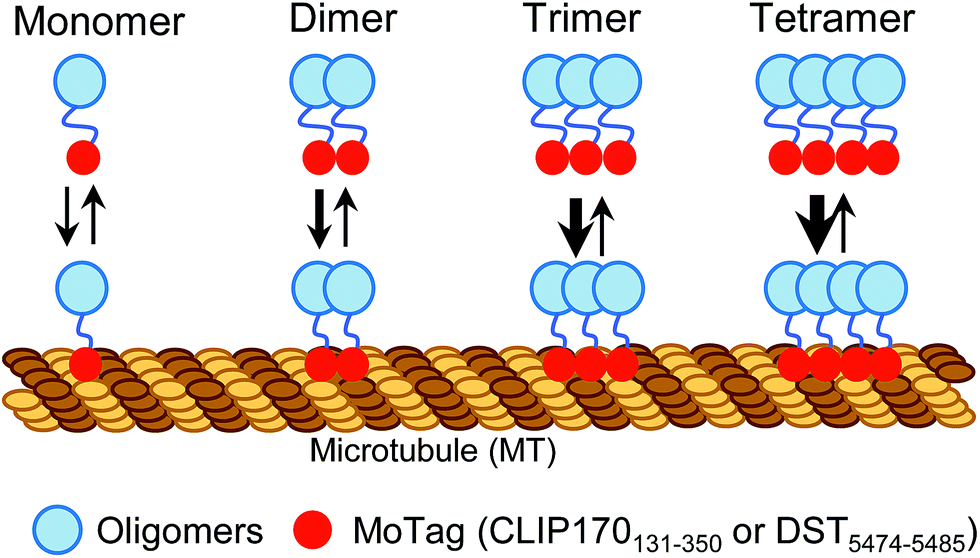 | ||
| Scheme 1 Schematic illustrating the design of MoTags to probe protein oligomeric states in living cells. | ||
In this study, we identified two classes of genetically encoded mini-tags derived from MADs that could serve this purpose (Fig. 1, S1†). We illustrate herein the broad applications of these engineered tags (designated as MoTags for the monomer/oligomer detection Tag) for assessing protein oligomeric states, the structure–function relationship, protein–protein interactions, and protein–DNA interactions (e.g., p53 or HSF1 (heat shock factor 1) complexes with their target DNAs), as well as protein–ligand interactions (e.g., Ca2+ binding to the ER luminal domain of stromal interaction molecule 1, or STIM1). Notably, this simple method can be easily applied in any laboratory equipped with a standard fluorescence microscope.
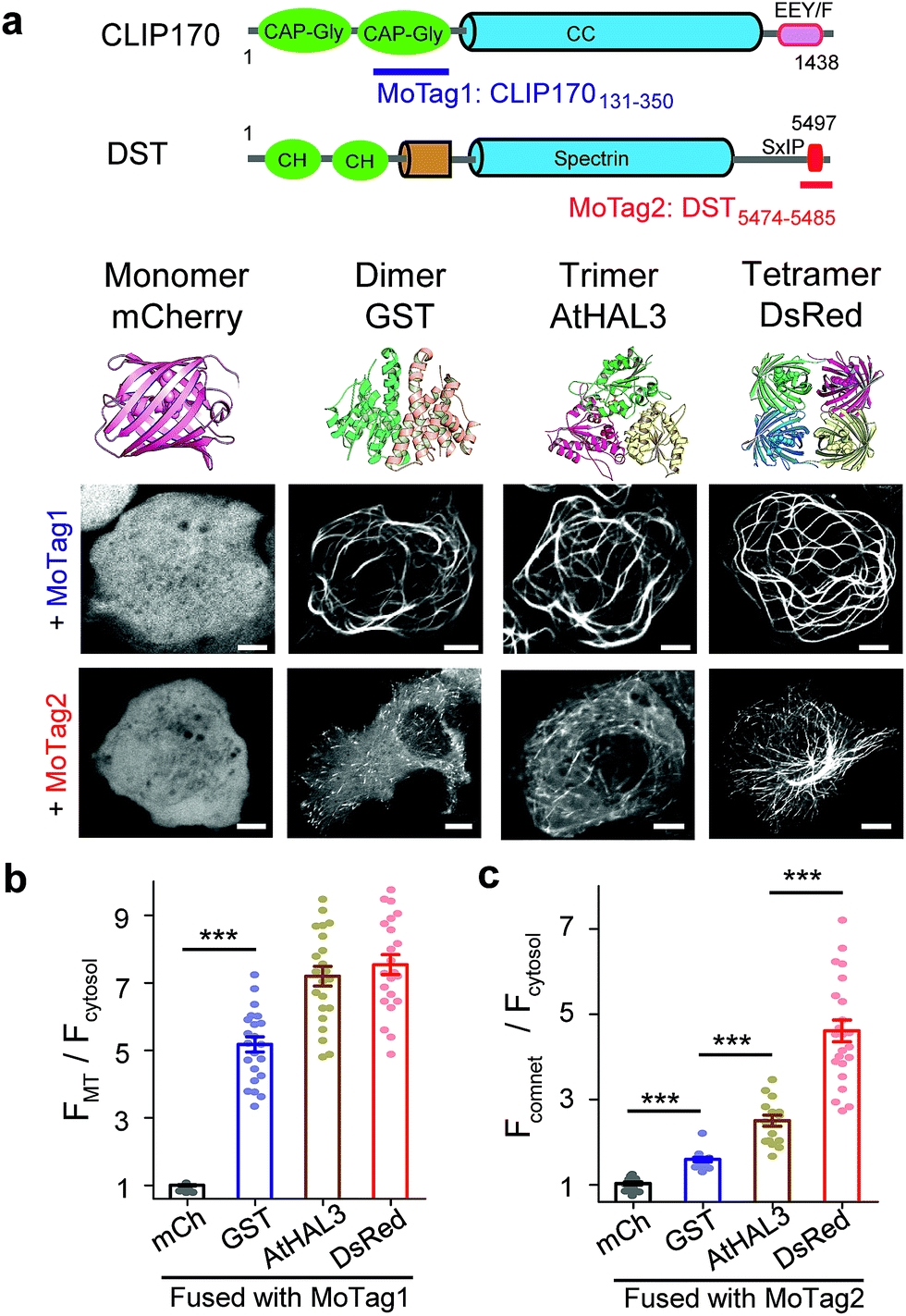 | ||
| Fig. 1 MoTags can be used to discriminate well-known oligomeric proteins. (a) Representative confocal images of mCherry (mCh) or the indicated oligomeric proteins (GST as dimer, AtHAL3 as trimer and DsRed as tetramer) fused to either MoTag1 or MoTag2. The domain architectures of MoTag1 and MoTag2 are shown on the top. See Fig. S1† for the design and screening procedures. Scale bar, 5 μm. (b and c) Quantification of the MT-to-cytosol (FMT/Fcytosol; b) or comet-to-cytosol fluorescence intensity ratio (Fcomet/Fcytosol; c) in HeLa cells expressing monomeric or oligomeric MoTag1 (b) or MoTag2 (c). See Fig. S4–9† for more images and statistical analyses. n= 18 cells from three independent experiments. All data are shown as mean ± s.e.m. ***P < 0.001 (two-tailed Student's t-test). | ||
Results
To quickly identify MADs that are suitable for discriminating monomers from oligomers, we performed an initial screening by fusing two red fluorescent proteins (FP),14 a monomeric mCherry (mCh) or a tetrameric DsRed, with a set of MT association/binding domains derived from various MT binders (Fig. 1a and b, S1a–c†). We reasoned that monomeric mCh-tagged MADs would have no or a relatively weak binding toward MT compared to their tetrameric DsRed-fusion counterparts, which would be reflected in their strengths for MT labelling in living cells. We tested this idea with several MAD fragments derived from the calponin homology (CH) domain of MT plus end binding protein EB1 (Fig. S2†),15 the CKK domain of MT minus end binding proteins16 (CAMSAP1/2; Fig. S3†), and the Gly-rich domain (CAP-Gly) from the cytoplasmic linker region of 170 kDa (ref. 15) (CLIP170; Fig. S4†).Among all the constructs, FP-tagged EB1-CH (EB11–191) or CLIP170131–350 showed an even distribution in the cytosol without noticeable background MT labelling (Fig. S2b and c, S4b and c†). By contrast, the DsRed-fusion constructs displayed clear labelling of MT (Fig. S2c and S4c†). The CAMSAP-based constructs, nonetheless, showed background labelling of MT even in its monomeric mCh-tagged form (Fig. S3b†), making them less ideal for further optimization. Overall, CLIP170131–350 stood out as the best performing candidate (designated as MoTag1). Upon fusion with GST (dimer), AtHAL3 (trimer) or DsRed (tetramer), MoTag1 invariably led to strong MT labelling (Fig. 1a, S4c†). To gain a more quantitative view on MT labelling, we calculated the MT-over-cytosol fluorescence intensity ratio (FMT/Fcytosol) in images acquired from HeLa cells expressing monomeric versus oligomeric MoTag1 (exemplified in Fig. S5a and b†). Compared to its basal value of 1 as a monomeric unit, oligomeric CLIP170131–350 constructs showed FMT/Fcytosol values of ∼4 to 7 (Fig. 1b). The expression levels of the MoTag1 fusion proteins did not seem to significantly affect FMT/Fcytosol values as readouts (Fig. S6a and c†). However, it failed to further detect the difference between trimers and tetramers. Regardless, the >4-fold change in the dynamic range of MT labelling makes MoTag1 a very sensitive tag to detect oligomeric assembly of proteins in single cells.
To more rigorously validate this finding within the same cell, we fused MoTag1 with a chemically inducible heteromerization system built upon the FK506 binding protein (FKBP) and FKBP12-rapamycin binding domain of mTOR (FRB).17 When covalently connected as a single-chain polypeptide, the FKBP-FRB fusion protein undergoes a monomer-to-dimer transition, whereas the FRB-FKBP chimera exhibits a monomer-to-tetramer transition upon the addition of rapamycin.18 Following fusion to either of the two chimeras, MoTag1 showed rapamycin-inducible MT labelling (Fig. S7, Movies S1 and 2†). Taken together, we have firmly established CLIP170131–350 (MoTag1) as an optimized tag that could easily discriminate monomers from dimers or higher-order oligomers.
Next, we moved on to explore the feasibility of further discriminating proteins assembled as dimers, trimers or tetramers. Given that our above engineering efforts with direct MT binders failed to achieve this goal, we shifted our focus on indirect MT binders, i.e., the SxIP (S/T-x-I-P) motif-containing protein family that can be recognized by EB1 (Fig. S1b, S5c, d, 6b, d, S8 and S9†). The SxIP motif is conserved in many microtubule end-tracking proteins that physically interact with EB1, thereby enabling an indirect docking toward the MT cytoskeleton.19,20 We selected SxIP motifs from three MT end tracking proteins,20,21 including adenomatous polyposis coli (APC), the stromal interaction molecule 1 (STIM1) and dystonin (DST, Fig. S1b and S8a†). We observed that the monomeric mCh-tagged SxIP motifs could barely track the MT tips (Fig. S8b†). Dimerization (fusion with GST) or tetramerization (fusion with DsRed) of SxIP motifs efficiently enhanced MT plus end tracking, as reflected by comet-like movements throughout the cytosol (Fig. S8b†). Among the three tested SxIP motifs, DST5474–5485 displayed the most striking comet tracking capability (Fig. 1a, c and S8b†) and was named as MoTag2. We further tested its performance by fusion with a set of well-characterized monomeric, dimeric, trimeric or tetrameric proteins (Fig. 1a and S9†). We monitored the degree of comet formation (quantified as Fcomet/Fcytosol; Fig S5c and d†) at varied expression levels (Fig. S6b and d†), and observed a positive correlation between comet formation and the protein oligomeric states (Fig. 1c, S9†).
In parallel, we tested the behaviour of MoTag2 in the same cells by utilizing the afore-mentioned rapamycin-inducible multimerization system18 (Fig. 2a). mCherry-tagged MoTag2 showed rapamycin-triggered MT plus end tracking (Fig. 2b, Movies S3 and 4†), with the comet intensities positively correlated with the oligomeric states of proteins (Fig. 2c and g). A similar trend was independently confirmed with light-inducible multimerization systems, including the optical dimerizer composed of iLID and sspB22 and the Arabidopsis cryptochrome 2 (CRY2)-based photoactivatable oligomerization system23,24 (Fig. 2d–g, Movie S5†). By plotting the comet intensities of a dozen tested proteins (in a logarithmic scale) against their known oligomeric states, we observed a linear relationship between the two variables (Fig. 2g, S9; see ESI†). The oligomeric states of representative proteins (monomeric mCherry, dimeric GST or tetrameric DsRed fused to MoTag2) were further validated by size exclusion chromatography in solution (Fig. S10†). Together, these findings unambiguously established the feasibility of using the degree of MoTag2 comet formation to discriminate proteins that are assembled in monomeric, dimeric, trimeric or tetrameric forms, as well as to monitor real time protein–protein interactions in living cells.
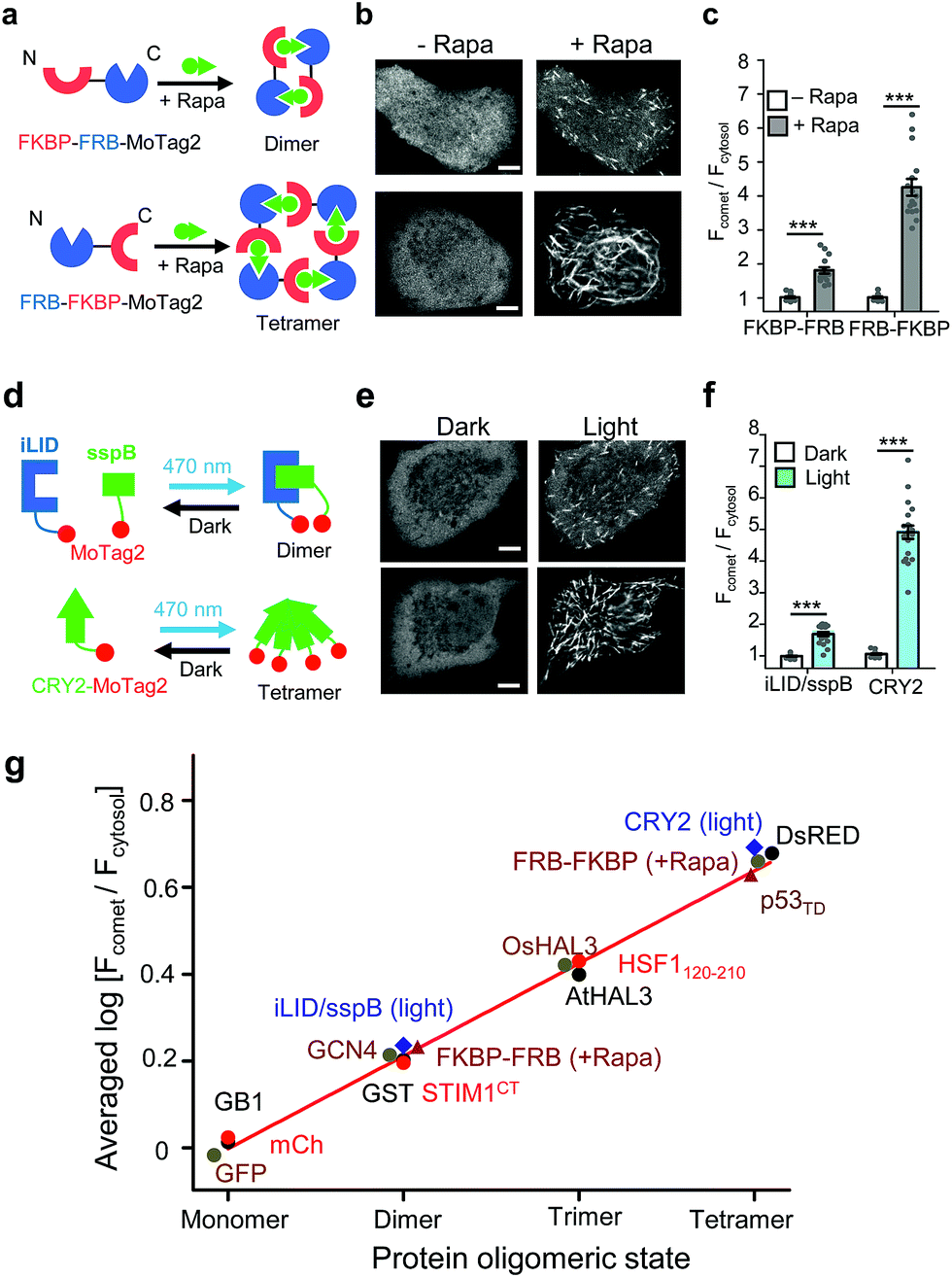 | ||
| Fig. 2 The 12-mer MoTag2 as a mini-tag for quantitative probing of protein oligomeric states in single cells. (a–f) MoTag2 was used to monitor chemical (a–c) or light-inducible (d–f) protein oligomerization. Schematic depicting the use of rapamycin (a) or light (d) to induce MoTag2 dimerization (FKBP-FRB; or iLID/sspB combinations) or tetramerization (FRB-FKBP or CYR2 tetramerization). The representative confocal images of transfected HeLa cells before and after treatment with 1 μM rapamycin (b; Movies S3 and 4) or blue light stimulation (d; 60 s, 470 nm, 40 μW mm−2; Movie S5†). Shown on the right (panels c and f) is the quantification of the comet-to-cytosol ratio of fluorescent signals from representative images. (g) The comet-to-cytosol ratio of fluorescent signals (in log10 scale) plotted against the oligomeric states of indicated proteins. A positive correlation was noted between the two variables. n = 15 cells from three independent experiments (mean ± s.e.m.; ***P < 0.001; two-tailed Student's t-test). Scale bar, 5 μM. | ||
We next sought to apply MoTag2 to assess how mutations and truncations in human p53 (ref. 25) (Fig. 3a–f, S11†) or heat-shock transcription factor 1 (ref. 26) (HSF1; Fig. S12†) would affect their quaternary structures, which has thus far not been systematically dissected in living cells owing to the lack of appropriate tools. Among all the p53-MoTag2 constructs we generated, the N-terminal transcription-activation domain (TAD) or the DNA-binding domain (DBD) failed to induce comet formation and thus existed as a monomer (T2, T3, T7 in Fig. 3b and S11†). In contrast, the tetramerization domain (TD) fused with MoTag2 elicited marked comet formation, indicating a tetrameric configuration25 (p53324–356, T6 in Fig. 3b). Other truncated p53 variants harbouring TD, along with wild-type (WT) p53, exhibited varying degrees of comet formation, likely because of their dimeric or mixed oligomeric assembly as reported by in vitro studies.27 When a TD-disruptive mutation,28 L330P, was introduced into p53, we observed the disappearance of comet formation due to compromised tetrameric assembly of TD. Conversely, a phosphorylation mimic mutation, S392E,29 in the p53 regulatory domain (REG) seemed to stabilize p53 in a tetrameric configuration (Fig. 3c and d). Most interestingly, when we transfected DBD-expressing HeLa cells with liposomes containing its cognate DNA duplexes, p53-DBD changed its oligomeric state from monomer to tetramer, as judged by the comet intensities under the corresponding conditions (Fig. 3e and f). A similar scenario was visualized in monomeric HSF1-DBD when complexed with its cognate DNA, except that it underwent a monomer-to-trimer transition (Fig. S12†). In aggregate, we demonstrated that MoTag2 could be exploited to not only identify domains that are essential for protein self-association but also report changes in protein quaternary structure upon formation of protein–DNA complexes.
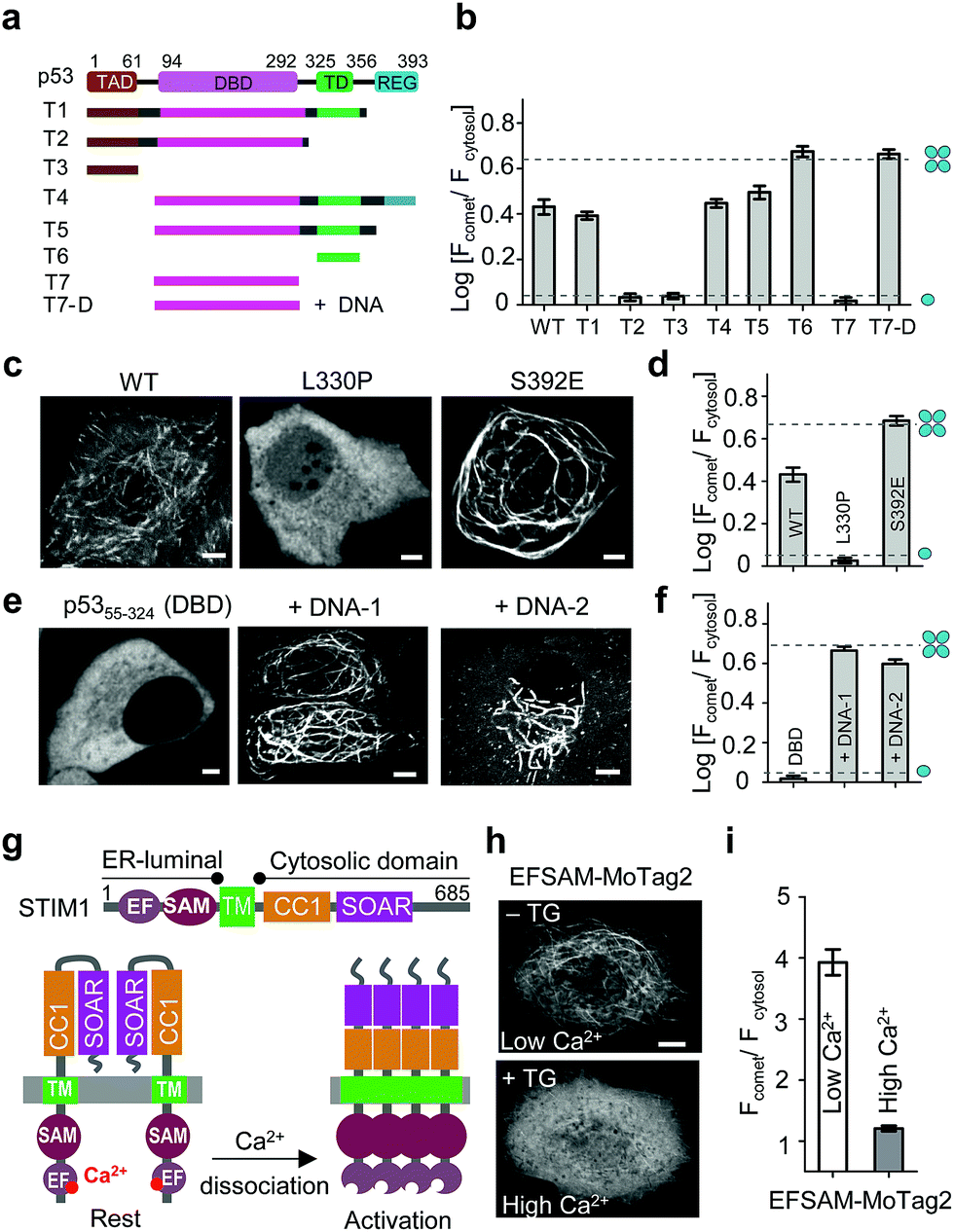 | ||
| Fig. 3 MoTag2 for real-time assessment of protein activities. (a–d) Characterization of the oligomeric states of p53 truncations (a and b) and mutations (c and d) fused with mCh-MoTag2. (a) Domain architecture of p53 and truncated variants used in the study. (b and d) Quantification of Fcomet/Fcytosol in HeLa cells transfected with the indicated p53 variants. (c) Representative confocal images of HeLa cells expressing WT or mutant p53. See Fig. S11† for more representative images and statistical analyses. (e and f) MoTag2 used to monitor p53–DNA interactions. (e) Representative imaging of mCh-DBD-MoTag2 transfected HeLa cells in the absence or presence of 10 μg cognate DNA duplexes. (f) Quantification of Fcomet/Fcytosol for free p53 DBD and DNA-bound DBD. (g–i) MoTag2 applied to report a ligand (Ca2+)-induced oligomer-to-monomer transition of the luminal domain of STIM1 (EFSAM). (g) Schematic depicting the domain architecture of STIM1 and Ca2+ dissociation induced STIM1 activation and oligomerization. (h) Representative confocal images of HeLa cells expressing mCh-EFSAM-MoTag2 before (low Ca2+) and after (high Ca2+) addition of 1 μM thapsigargin (TG). (i) Quantification of Fcomet/Fcytosol for the STIM1 luminal EFSAM domain under low and high Ca2+ conditions (n = 21 cells from three independent experiments; mean ± s.e.m.). Scale bar, 5 μM. | ||
We further explored the application of MoTag2 to dissect the activation and oligomerization of an endoplasmic reticulum (ER)-resident Ca2+-sensing protein STIM1 (Fig. 3g–i).30,31 STIM1 senses fluctuation of ER Ca2+ concentrations by its ER-luminal EFSAM domain. Ca2+ dissociation from the EF-hand arising from ER luminal Ca2+ store depletion could cause EFSAM oligomerization, which in turn activates the cytoplasmic domain (STIM1CT) by exposing the SOAR domain to engage and open ORAI Ca2+ channels on the plasma membrane32–37 (Fig. 3g). We used this system to test whether the MoTag can be used for reporting ligand (Ca2+ in this case)-induced structural changes in protein. The isolated EFSAM domain was fused with mCh-MoTag2 and expressed in the cytoplasm of HeLa cells. Live cell fluorescence imaging showed that EFSAM-MoTag2 clearly tracked MT with a comet-to-cytosol ratio of 4 (Fig. 3h and i), indicating a tetrameric or high-order assembly due to the low cytoplasmic Ca2+ concentration mimicking ER store depletion. After inducing cytosolic Ca2+ elevation by using thapsigargin (TG), we observed that the EFSAM-MoTag2 gradually stopped tracking MT plus ends and ultimately exhibited a more even cytosolic distribution (Fig. 3h and i). This finding suggested that the binding of Ca2+ to EFSAM could switch EFSAM from oligomer to monomer. Hence, the MoTag2 fusing strategy can be broadly extended to screen key structural determinants that govern the quaternary structure of a given protein, and to report molecular events that trigger monomer-to-oligomer interconversion.
Discussions
In the current study, we have developed a convenient method to quantitatively examine protein oligomeric states in single cells. By using the MT/comet-to-cytosol fluorescence signal ratio as a simple and straightforward readout, our method offers an easy solution to quantitatively dissect the quaternary structure of proteins in their native environment within intact cells by either confocal or epifluorescence microscopy (Fig. S13†). Based on our extensive analyses on a dozen proteins with varying oligomerization tendencies, it is feasible to apply this method to assay proteins with self-association dissociation constants ranging from nM to μM in living cells (Table S1†).Notably, the expression of MoTags did not seem to exert overt adverse effects on the host cell division or viability (Fig. S14a–c†). However, our method has its inherent limitations. First, because the readout of this approach relies on the binding of MoTag fusion proteins to the MT cytoskeleton, membrane-embedded proteins and cytosolic proteins that tend to damage microtubules are not suitable for this assay. Nonetheless, signalling proteins residing within subcellular compartments, such as the DNA binding domain of p53 in the nucleus or the EFSAM domain of STIM1 in the ER lumen, can be ectopically expressed in the cytosol to faithfully recapitulate ligand (DNA for p53 or Ca2+ for EFSAM; Fig. 3)-induced changes in oligomerization. Thus, under certain circumstances, it is possible to use our method to study the dynamics of protein assembly while maintaining the in situ function of the assayed protein. Second, the binding of MoTag fusion proteins might perturb the host cell MT dynamics since DsRed-MoTag fusion proteins tend to slow down the comet velocity of microtubule plus end binding protein EB1 (Fig. S14d†). However, these changes do not seem to adversely affect the readouts (MT or comet-to-cytosol fluorescence intensity ratio, rather than comet velocity) for oligomer discrimination in our assay. Congruently, regardless of these potential caveats, we anticipate that our MoTags will likely have many applications in the study of cytosolic protein structure and function, protein–protein or protein–ligand interactions.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge the financial support from the National Institutes of Health (R01GM112003, R21GM126532, and R01HL134780), the Welch Foundation (BE-1913), the American Cancer Society (RSG-16-215-01-TBE and RSG-18-043-01-LIB), the Cancer Prevention and Research Institute of Texas (RR140053 and RP170660), the John S. Dunn Foundation, and by an allocation from the Texas A&M University Health Science Centre Start-up Fund and the Texas A&M Triad for Transformation Program.Notes and references
- D. S. Goodsell and A. J. Olson, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 105–153 CrossRef PubMed.
- M. D. W. Griffin and J. A. Gerrard, in Protein Dimerization and Oligomerization in Biology, ed. J. M. Matthews, Springer New York, New York, NY, 2012, pp. 74–90, DOI:10.1007/978-1-4614-3229-6_5.
- M. H. Ali and B. Imperiali, Bioorg. Med. Chem., 2005, 13, 5013–5020 CrossRef PubMed.
- A. P. W. Funnell and M. Crossley, in Protein Dimerization and Oligomerization in Biology, ed. J. M. Matthews, Springer New York, New York, NY, 2012, pp. 105–121, DOI:10.1007/978-1-4614-3229-6_7.
- D. W. Piston and G. J. Kremers, Trends Biochem. Sci., 2007, 32, 407–414 CrossRef PubMed.
- T. K. Kerppola, Annu. Rev. Biophys., 2008, 37, 465–487 CrossRef PubMed.
- Y. Guan, M. Meurer, S. Raghavan, A. Rebane, J. R. Lindquist, S. Santos, I. Kats, M. W. Davidson, R. Mazitschek, T. E. Hughes, M. Drobizhev, M. Knop and J. V. Shah, Mol. Biol. Cell, 2015, 26, 2054–2066 CrossRef PubMed.
- M. Sergeev, S. Costantino and P. W. Wiseman, Biophys. J., 2006, 91, 3884–3896 CrossRef PubMed.
- Y. Chen, L. N. Wei and J. D. Muller, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15492–15497 CrossRef PubMed.
- A. Akhmanova and M. O. Steinmetz, Nat. Rev. Mol. Cell Biol., 2015, 16, 711–726 CrossRef PubMed.
- A. Desai and T. J. Mitchison, Annu. Rev. Cell Dev. Biol., 1997, 13, 83–117 CrossRef PubMed.
- A. Akhmanova and M. O. Steinmetz, J. Cell Sci., 2010, 123, 3415–3419 CrossRef PubMed.
- L. Truebestein and T. A. Leonard, BioEssays, 2016, 38, 903–916 CrossRef PubMed.
- R. N. Day and M. W. Davidson, Chem. Soc. Rev., 2009, 38, 2887–2921 RSC.
- K. C. Slep and R. D. Vale, Mol. Cell, 2007, 27, 976–991 CrossRef PubMed.
- J. Atherton, K. Jiang, M. M. Stangier, Y. Luo, S. Hua, K. Houben, J. J. E. van Hooff, A. P. Joseph, G. Scarabelli, B. J. Grant, A. J. Roberts, M. Topf, M. O. Steinmetz, M. Baldus, C. A. Moores and A. Akhmanova, Nat. Struct. Mol. Biol., 2017, 24, 931–943 Search PubMed.
- C. J. Sabers, M. M. Martin, G. J. Brunn, J. M. Williams, F. J. Dumont, G. Wiederrecht and R. T. Abraham, J. Biol. Chem., 1995, 270, 815–822 CrossRef PubMed.
- T. Inobe and N. Nukina, J. Biosci. Bioeng., 2016, 122, 40–46 CrossRef PubMed.
- P. Kumar and T. Wittmann, Trends Cell Biol., 2012, 22, 418–428 CrossRef PubMed.
- S. Honnappa, S. M. Gouveia, A. Weisbrich, F. F. Damberger, N. S. Bhavesh, H. Jawhari, I. Grigoriev, F. J. van Rijssel, R. M. Buey, A. Lawera, I. Jelesarov, F. K. Winkler, K. Wuthrich, A. Akhmanova and M. O. Steinmetz, Cell, 2009, 138, 366–376 CrossRef PubMed.
- A. Akhmanova and M. O. Steinmetz, Nat. Rev. Mol. Cell Biol., 2008, 9, 309–322 CrossRef PubMed.
- G. Guntas, R. A. Hallett, S. P. Zimmerman, T. Williams, H. Yumerefendi, J. E. Bear and B. Kuhlman, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 112–117 CrossRef PubMed.
- R. A. Hallett, S. P. Zimmerman, H. Yumerefendi, J. E. Bear and B. Kuhlman, ACS Synth. Biol., 2016, 5, 53–64 CrossRef PubMed.
- L. J. Bugaj, A. T. Choksi, C. K. Mesuda, R. S. Kane and D. V. Schaffer, Nat. Methods, 2013, 10, 249–252 CrossRef PubMed.
- A. C. Joerger and A. R. Fersht, Annu. Rev. Biochem., 2008, 77, 557–582 CrossRef PubMed.
- D. W. Neef, A. M. Jaeger and D. J. Thiele, Nat. Rev. Drug Discovery, 2011, 10, 930–944 CrossRef PubMed.
- C. D. Nicholls, K. G. McLure, M. A. Shields and P. W. Lee, J. Biol. Chem., 2002, 277, 12937–12945 CrossRef PubMed.
- T. Kawaguchi, S. Kato, K. Otsuka, G. Watanabe, T. Kumabe, T. Tominaga, T. Yoshimoto and C. Ishioka, Oncogene, 2005, 24, 6976–6981 CrossRef PubMed.
- N. M. Nichols and K. S. Matthews, Biochemistry, 2002, 41, 170–178 CrossRef PubMed.
- I. Derler, I. Jardin and C. Romanin, Am. J. Physiol., 2016, 310, C643–C662 CrossRef PubMed.
- P. B. Stathopulos, L. Zheng, G. Y. Li, M. J. Plevin and M. Ikura, Cell, 2008, 135, 110–122 CrossRef PubMed.
- G. Ma, S. Zheng, Y. Ke, L. Zhou, L. He, Y. Huang, Y. Wang and Y. Zhou, Curr. Mol. Med., 2017, 17, 60–69 CrossRef PubMed.
- G. Ma, M. Wei, L. He, C. Liu, B. Wu, S. L. Zhang, J. Jing, X. Liang, A. Senes, P. Tan, S. Li, A. Sun, Y. Bi, L. Zhong, H. Si, Y. Shen, M. Li, M. S. Lee, W. Zhou, J. Wang, Y. Wang and Y. Zhou, Nat. Commun., 2015, 6, 7826 CrossRef PubMed.
- J. Soboloff, B. S. Rothberg, M. Madesh and D. L. Gill, Nat. Rev. Mol. Cell Biol., 2012, 13, 549–565 CrossRef PubMed.
- Y. Zhou, P. Srinivasan, S. Razavi, S. Seymour, P. Meraner, A. Gudlur, P. B. Stathopulos, M. Ikura, A. Rao and P. G. Hogan, Nat. Struct. Mol. Biol., 2013, 20, 973–981 Search PubMed.
- Y. Zhou, P. Meraner, H. T. Kwon, D. Machnes, M. Oh-hora, J. Zimmer, Y. Huang, A. Stura, A. Rao and P. G. Hogan, Nat. Struct. Mol. Biol., 2010, 17, 112–116 Search PubMed.
- A. Gudlur, Y. Zhou and P. G. Hogan, Curr. Top. Membr., 2013, 71, 33–58 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00839f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |