
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElements of fractal geometry in the 1H NMR spectrum of a copolymer intercalation-complex: identification of the underlying Cantor set†
John S.
Shaw
,
Rajendran
Vaiyapuri
,
Matthew P.
Parker
 ,
Claire A.
Murray
,
Kate J. C.
Lim
,
Cong
Pan
,
Marcus
Knappert
,
Christine J.
Cardin
,
Barnaby W.
Greenland
,
Ricardo
Grau-Crespo
and
Howard M.
Colquhoun
*
,
Claire A.
Murray
,
Kate J. C.
Lim
,
Cong
Pan
,
Marcus
Knappert
,
Christine J.
Cardin
,
Barnaby W.
Greenland
,
Ricardo
Grau-Crespo
and
Howard M.
Colquhoun
*
Department of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, UK. E-mail: h.m.colquhoun@rdg.ac.uk
First published on 27th March 2018
Abstract
Sequence-selective intercalation of pyrene into the chain-folds of a random, binary copolyimide under fast-exchange conditions results in the development of self-similar structure in the diimide region of the 1H NMR spectrum. The resulting spectrum can be described by the mathematics of fractals, an approach that is rationalised in terms of a dynamic summation of ring-current shielding effects produced by pyrene molecules intercalating into the chain at progressively greater distances from each “observed” diimide residue. The underlying set of all such summations is found to be a defined mathematical fractal namely the fourth-quarter Cantor set, within which the observed spectrum is embedded. The pattern of resonances predicted by a geometric construction of the fourth-quarter Cantor set agrees well with the observed spectrum.
Introduction
The principle that digital information can be encoded at the molecular level, as a linear sequence of monomer residues in a high molecular weight copolymer chain, has provided a definitive basis for the whole of biology.1–4 Life, however, remains the sole example of an operational information technology working at the molecular level, despite the fact that even the simplest two-monomer linear copolymer is the logical equivalent of a binary string.5 Nevertheless, significant efforts are now being made to develop sequence-defined, synthetic copolymers and to explore their potential for information storage and processing.6–12Interestingly, even random copolymers have potential in this field, since it is well established that the information capacity of a linear sequence reaches a maximum when the sequence is fully random.13 This slightly counter-intuitive result makes sense when it is realised that “information” comes in two forms, (i) potential information (or information capacity), which is simply the number of digits present when information is expressed in the form of a binary string (ignoring any possible meaning), and (ii) contextual information, whereby sequences within such a string acquire meaning through the application of an external context.14 For example, the string of letters that comprise the title of this paper is, if not completely random, at least completely arbitrary – it only conveys information because short sequences within the string such as “fractal”, “spectrum”, and “intercalation” have previously been assigned meaning in the context of the English language. Indeed the letter-sequence “fractal” was given meaning only a few decades ago, by Mandelbrot;15 a good example of how a specific but meaningless sequence can become information through the application of context. A possible approach to generating information from an extended but random copolymer chain would therefore be to identify short, specific sequences within the copolymer, to which meaning may eventually be assigned, then to read out these sequences by selective interaction with some form of “reader-molecule”, and finally to give the identified sequences contextual meaning. Generating information by selecting sequences from a pre-existing copolymer chain is just as valid as synthesising the same sequences from scratch. Conceptually, it is the equivalent of selecting words from a dictionary rather than spelling them out letter by letter.
With regard to molecular-level reading of sequences in synthetic copolymers, some progress has been made using the ability of π-electron-rich tweezer-molecules16 to bind reversibly to specific triplet sequences in chain-folding aromatic copolyimides.17–20 This form of sequence-recognition involves sterically and electronically complementary π–π-stacking between the tweezer arms and the diimide residues of the copolymer, producing large ring-current shielding effects,21,22 and thereby resolving extended sequence-information in the 1H NMR spectrum.
Here we report the results of a different form of supramolecular sequence-reading, based on selective but dynamic intercalation of the simple aromatic probe-molecule pyrene into specific chain-folds of a novel copolyimide, 1 (Fig. 1). This type of assembly reveals progressively more extended sequence-information in the 1H NMR spectrum of the copolymer as the concentration of probe molecule increases. Wholly unexpectedly, the sequence-information being “read” is ultimately expressed in the form of a self-similar pattern in the diimide region of the spectrum. Self-similarity is perhaps the most general characteristic of objects showing fractal geometry,23–25 and mathematical or “true” fractals show perfect self-similarity over an infinite number of length scales. Real objects, however, are much less perfectly structured and are generally described as fractal “if they contain parts that at two or more smaller scales appear in some way similar to the whole”.26 There have been several reports of molecular and supramolecular structures that show self-similarity and approximate fractal geometry,27–29 most recently via the assembly, through halogen bonding, of brominated oligophenylenes on the Ag(111) surface to give some very realistic Sierpiński triangles as visualised by STM.30 In the present work however, self-similarity emerges not in the structure of a molecular assembly itself, but in the induced magnetic field (and consequent NMR spectrum) created by dynamic, sequence-specific intercalation of small aromatic molecules into a random copolymer chain.
 | ||
Fig. 1 Synthesis of binary copolymer 1 and the 1H NMR spectrum (diimide region) of this copolymer at 25 °C in the presence of increasing concentrations of pyrene-d10. (A) Pure copolymer 1 (in CDCl3/(CF3)2CHOH, 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at 4 mM concentration of NDI residues. (B, C, D) Copolymer 1 in the presence of 0.12, 0.50 and 1.0 molar equivalents (relative to NDI residues) respectively of pyrene-d10. :1 v/v) at 4 mM concentration of NDI residues. (B, C, D) Copolymer 1 in the presence of 0.12, 0.50 and 1.0 molar equivalents (relative to NDI residues) respectively of pyrene-d10. | ||
Results and discussion
In the absence of pyrene, copolymer 1 exhibits a three-line group of resonances (relative intensities 1:2:1) at lowest field (Fig. 1A). These arise from naphthalenediimide (NDI) residues flanked by either two aromatic diamine residues (Ar-NDI-Ar), two aliphatic diamine residues (Al-NDI-Al), or one of each (Ar-NDI-Al). The latter, unsymmetrical sequence is directionally degenerate, i.e. there also exists a reverse sequence (Al-NDI-Ar) which, when read backwards, is the same as the original. Thus, in a random 1:1 copolymer there are twice as many sequences of this type as there are of each of the other two sequences. The observed pattern of intensities (1:2:1) confirms that the monomer sequence, at least at the triplet level, is genuinely random.
Addition of progressively increasing amounts of pyrene (in the perdeuterated form, to avoid resonance-overlap) to a 4 mM solution of copolymer 1 produced marked splittings and large upfield shifts (Δδ up to 0.62 ppm) of some of the diimide resonances (Fig. 1A–D). These effects clearly result from the binding of pyrene-d10 molecules to the copolymer chain, and the absence of separate resonances corresponding to bound and unbound polymer shows that the system is dynamic, with fast exchange on the NMR timescale.
The most remarkable feature of the diimide region of the spectrum at high pyrene loadings (Fig. 1D) is its self-similarity, as illustrated in Fig. 2 where the original spectrum (a) is shown re-scaled by 1/4, 1/16, and 1/64. In each case, the re-scaled spectrum closely resembles the highest-field quarter of the previous-scale spectrum. Spectrum (b) is a geometric reconstruction in which these re-scaled copies successively replace the highest field 1/4 of the previous spectrum. Even allowing for the effects of scaling on spectroscopic linewidths, it is clear from the reconstruction shown in Fig. 2b that the experimental spectrum does indeed consist – to good approximation – of smaller copies of itself over at least three length scales, and can therefore be described as self-similar.26
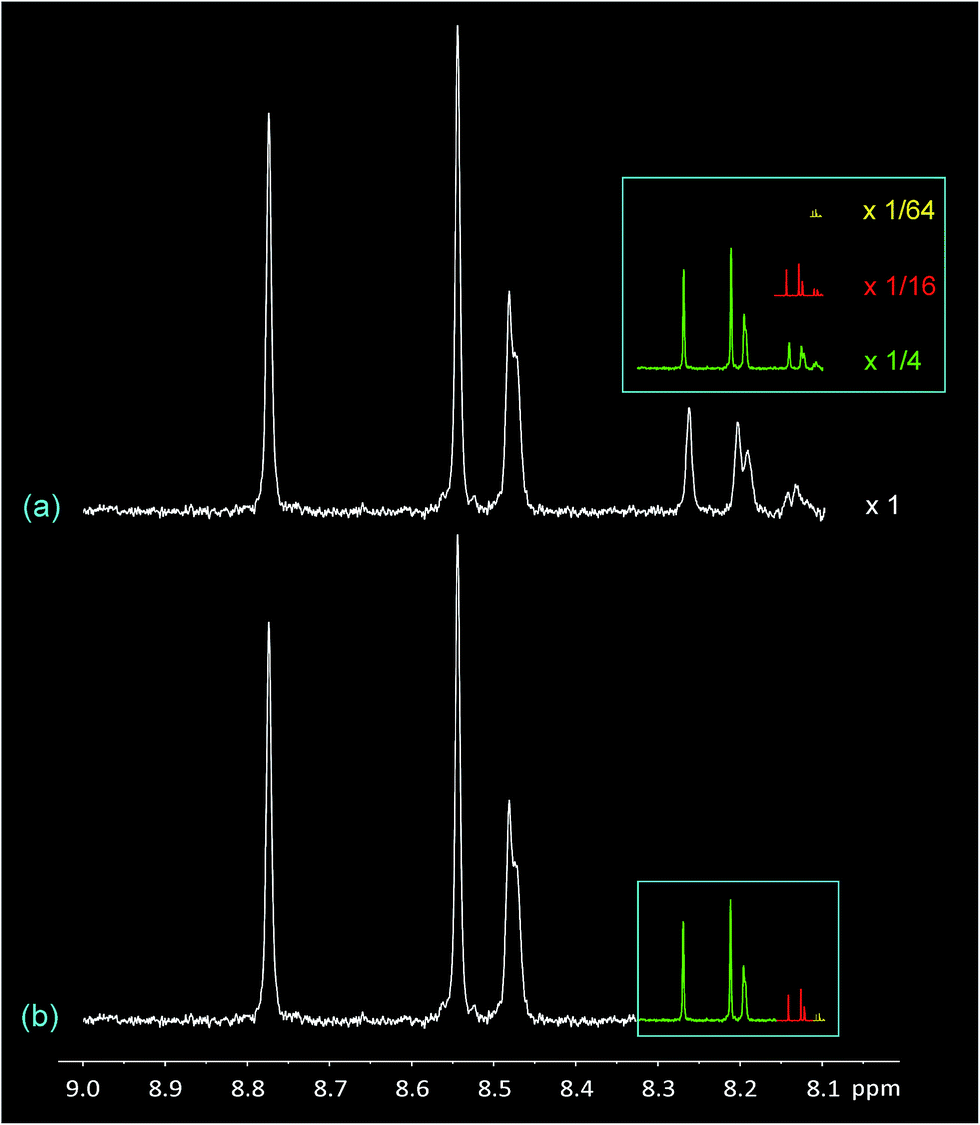 | ||
| Fig. 2 Self-similarity in the 1H NMR spectrum of copolymer 1 (diimide region) in the presence of one equivalent of pyrene-d10. (a) Spectrum “D” from Fig. 1, expanded vertically. The inset shows the entire spectrum re-scaled ×1/4 (green), ×1/16 (red) and ×1/64 (yellow) in both dimensions. In every case the rescaled spectrum closely resembles the highest field one-quarter of the spectrum at the previous length scale. (b) Reconstruction of spectrum “(a)” by deleting the highest field 1/4 of the spectrum and replacing it with a 1/4 scale copy of the original spectrum, and then repeating the process over two further length scales. Although this type of geometric reconstruction does not reproduce the original spectrum exactly, mainly owing to progressive narrowing of the spectroscopic linewidth, comparison of (a) and (b) brings out the self-similar character of spectrum (a) very clearly. | ||
We have developed an atomistically well-defined model that predicts the emergence of self-similar character in the 1H NMR spectrum of the pyrene-intercalated copolymer, and even allows the underlying mathematical fractal to be identified. This model builds on the idea that the total ring-current shielding experienced by a diimide proton is the sum of the diminishing shielding effects produced by intercalator-molecules that bind at progressively greater distances along the polymer chain from a central, “observed”, diimide residue (Fig. 3A).17–20 Defining a chain-fold comprising two NDI units linked by a 1,2-bis(2-aminoethoxy)ethane residue as “II” and one comprising an NDI unit adjacent to a 4,4′-biphenylenedisulfone group as “IS” (or “SI”), then our initial model is illustrated in Fig. 3A as an energy-minimised, chain-folded and intercalated septet sequence.
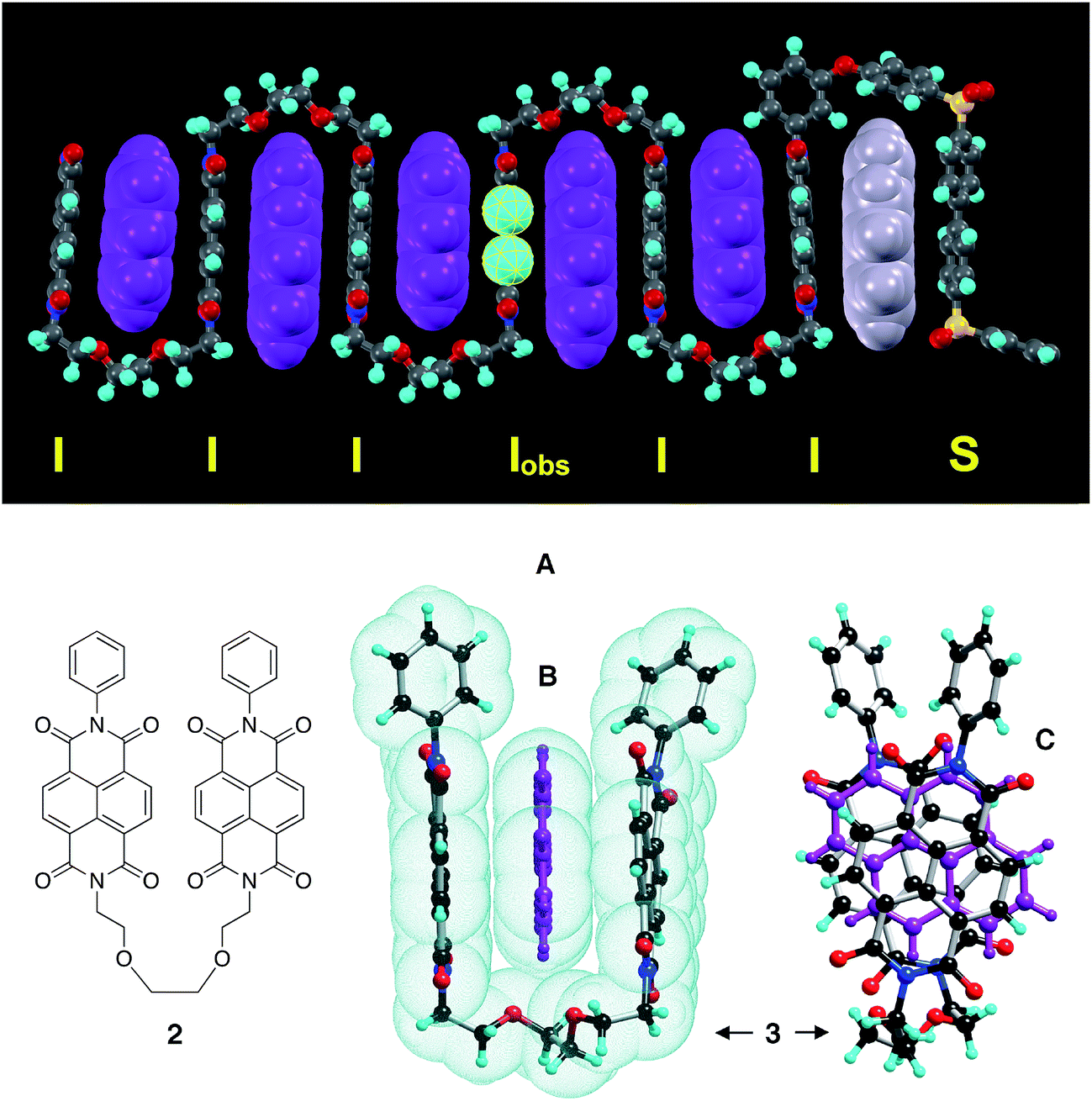 | ||
Fig. 3 (A) Energy-minimised model for a segment of copolymer 1 (septet sequence “ ”) intercalated by pyrene. Diimide residues are indicated as “I” and the biphenylenedisulfone residue as “S”. The central, “observed” diimide residue is indicated as “Iobs”. Pyrene molecules located in the strongly-binding “II” chain-fold are shown in purple and a pyrene molecule located in the very weakly-binding “IS” fold is shown in grey. (B) Single crystal X-ray structure of a chain-folded 1:1 complex (3) formed between dimer 2 and pyrene viewed along the plane of the intercalating pyrene molecule. The fold provides a binding site in which the measured perpendicular distance between the centroids of the two diimide residues is 6.82 Å. (C) Complex 3 viewed perpendicular to the plane of the pyrene molecule (shown in purple). ”) intercalated by pyrene. Diimide residues are indicated as “I” and the biphenylenedisulfone residue as “S”. The central, “observed” diimide residue is indicated as “Iobs”. Pyrene molecules located in the strongly-binding “II” chain-fold are shown in purple and a pyrene molecule located in the very weakly-binding “IS” fold is shown in grey. (B) Single crystal X-ray structure of a chain-folded 1:1 complex (3) formed between dimer 2 and pyrene viewed along the plane of the intercalating pyrene molecule. The fold provides a binding site in which the measured perpendicular distance between the centroids of the two diimide residues is 6.82 Å. (C) Complex 3 viewed perpendicular to the plane of the pyrene molecule (shown in purple). | ||
However, this initial model takes no account of differences in binding constant between pyrene and the different chain-folds present in copolymer 1. Fig. 1 shows clearly that there must be large differences of this type, because resonances representing different sequences are shifted upfield by anything from 0.07 ppm (for “SIS”-centred sequences) to 0.62 ppm (for “III”-centred sequences) [note that the sequence “SS” is forbidden by the chemistry of the copolyimide, since the diamine residue “S” must always be followed by (and preceded by) the diimide residue “I”].
Crystallographic evidence for strong complexation of pyrene at the “II” chain-fold was provided by single crystal X-ray analyses of the model complex 3 (Fig. 3B). Here, a pyrene molecule is bound by an oligomer (2) comprising two NDI residues linked by a single fold-unit and terminated by phenyl residues. The area of overlap between the pyrene and NDI units is very high (Fig. 3C), and their separations (atom-to-plane distances of 3.3–3.5 Å) correspond to tight van der Waals contact (Fig. 3B). Moreover, the recently-reported crystal structure of a different complex, now between oligomer 2 and a bis-pyrenyl tweezer molecule provides a very good model for the multiply-intercalated structure proposed in Fig. 3A.31 See ESI.†
Given the evidently large difference in binding constant between sequences “SI” and “II”, we next adopt the simplifying approximation that pyrene binds only to “II” sequences so that, in a predictive binding model, the positions of “II” pairs are the defining characteristics of any longer sequence. For example, reading outwards in both directions from the central “observed” diimide residue (highlighted here in red), the septet sequence  contains two “II” sites immediately adjacent to – and including – the central residue. In addition, there is one “II” site at a next-adjacent position, and one “II” site at an outermost position. In binding terms, the sequence ISIIIII can therefore be described by the 3-digit code [211]. See Fig. 4.
contains two “II” sites immediately adjacent to – and including – the central residue. In addition, there is one “II” site at a next-adjacent position, and one “II” site at an outermost position. In binding terms, the sequence ISIIIII can therefore be described by the 3-digit code [211]. See Fig. 4.
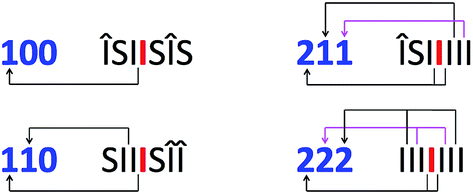 | ||
Fig. 4 Methodology for assigning 3-digit codes to different septet sequences in copolymer 1. For each septet sequence, the numbers of “II” pairs at the three types of position relative to the central, observed diimide residue  (adjacent, next-adjacent and next-next-adjacent) give the three digits of the corresponding code. Note that diimide residues outside an “S” residue (relative to the centre) are coded as Î. These do not contribute to the total ring current shielding of (adjacent, next-adjacent and next-next-adjacent) give the three digits of the corresponding code. Note that diimide residues outside an “S” residue (relative to the centre) are coded as Î. These do not contribute to the total ring current shielding of  for reasons discussed below. for reasons discussed below. | ||
Any imide-centred septet sequence can similarly be assigned a 3-digit code in which the highest possible value of any digit is 2 and the lowest value is zero since, in any linear copolymer the number of monomer residues of a given type at a specific distance from any other monomer residue can only be 0, 1 or 2. The code for  would then be [222] and, for
would then be [222] and, for  , [220]. The experimental data, however, reveal an additional requirement for translating copolymer sequences into digital codes based on “II” pairs. This is that pyrene intercalation into “II” binding sites outside an “S” residue (relative to the central “I” residue) must be ignored. This effect may result from the weakly binding “SI” sequence allowing a gross departure from the time-averaged, chain-folded conformation (Fig. 3A) that enables long range ring-current shielding of the central “I” by intercalating pyrene molecules. Examples of this methodology for assigning digital codes to copolymer sequences are illustrated in Fig. 4, where “I” residues outside an S residue are given the symbol “Δ.
, [220]. The experimental data, however, reveal an additional requirement for translating copolymer sequences into digital codes based on “II” pairs. This is that pyrene intercalation into “II” binding sites outside an “S” residue (relative to the central “I” residue) must be ignored. This effect may result from the weakly binding “SI” sequence allowing a gross departure from the time-averaged, chain-folded conformation (Fig. 3A) that enables long range ring-current shielding of the central “I” by intercalating pyrene molecules. Examples of this methodology for assigning digital codes to copolymer sequences are illustrated in Fig. 4, where “I” residues outside an S residue are given the symbol “Δ.
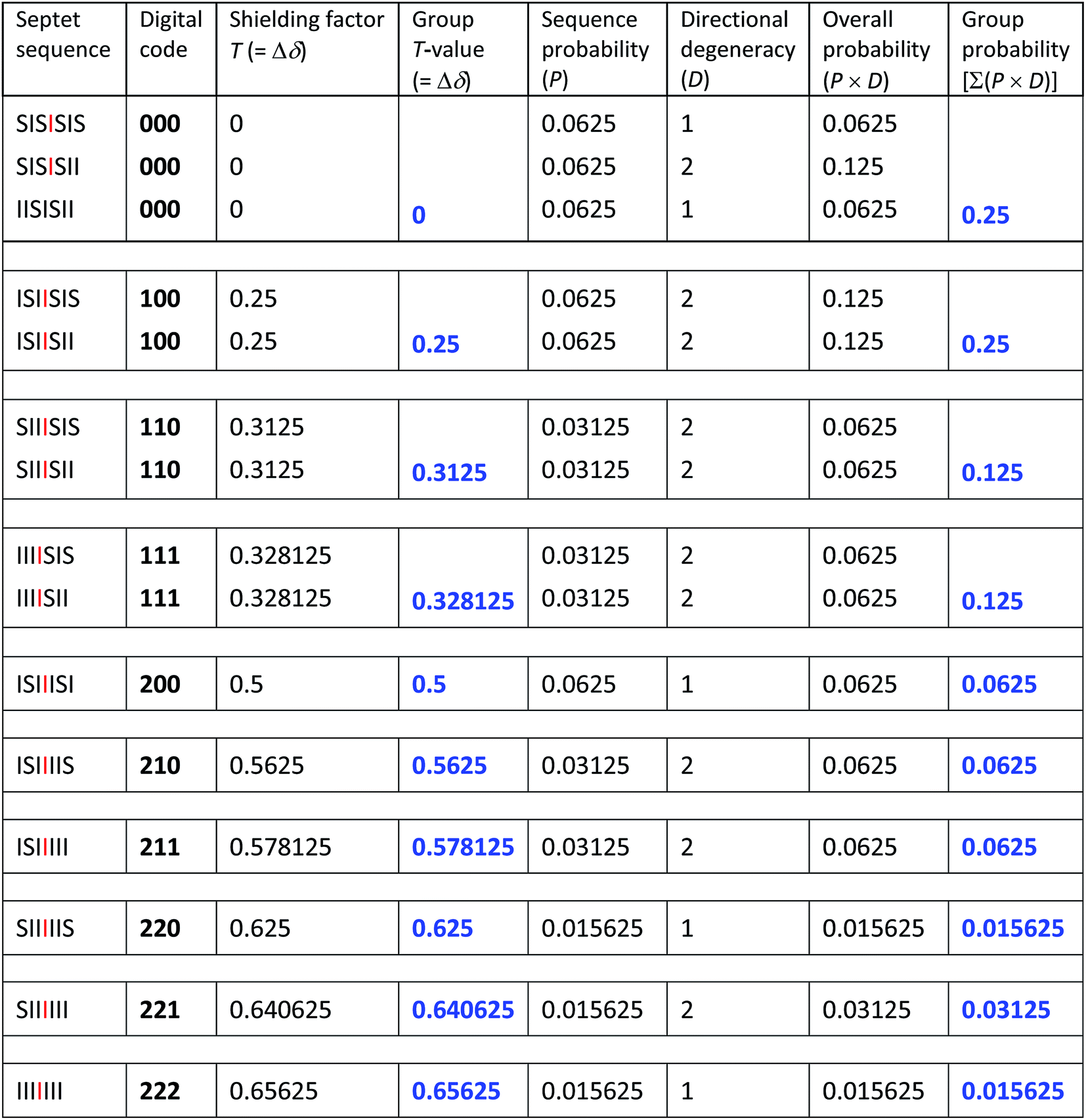
The 27 three-digit codes containing only the digits 0, 1 or 2 are shown in Table 1. Although every possible septet-sequence in copolymer 1 can be assigned a code, not every code corresponds to a realisable sequence, reducing the number of assignable codes from twenty-seven possibilities to just the ten codes shown in blue in Table 1. A number of codes show redundancy in that, from the rules of the model described above, they can represent two or even three of the 15 different sequences. There are 64 (26) “I”-centred septet combinations of “I” and “S”, but 39 of these contain “SS” pairs which are not allowed by the chemistry of the system. This reduces the total to 25, but 10 of these are unsymmetrical and so are indistinguishable in NMR terms from their reverse sequences, reducing the total to just the 15 sequences shown in Table 1.
 , and neighbouring diimide residues are shown as either “I” or Î depending on whether they are “inside” or “outside” an S residue (relative to the central
, and neighbouring diimide residues are shown as either “I” or Î depending on whether they are “inside” or “outside” an S residue (relative to the central  ). Pyrene binding to ÎÎ pairs appears to produce no additional shielding of the central
). Pyrene binding to ÎÎ pairs appears to produce no additional shielding of the central  residue, and such pairs are therefore ignored when assigning digital codes to sequences. The unused codes in this table correspond to patterns of “II” pairs for which no septet sequences can exist in copolymer 1
residue, and such pairs are therefore ignored when assigning digital codes to sequences. The unused codes in this table correspond to patterns of “II” pairs for which no septet sequences can exist in copolymer 1
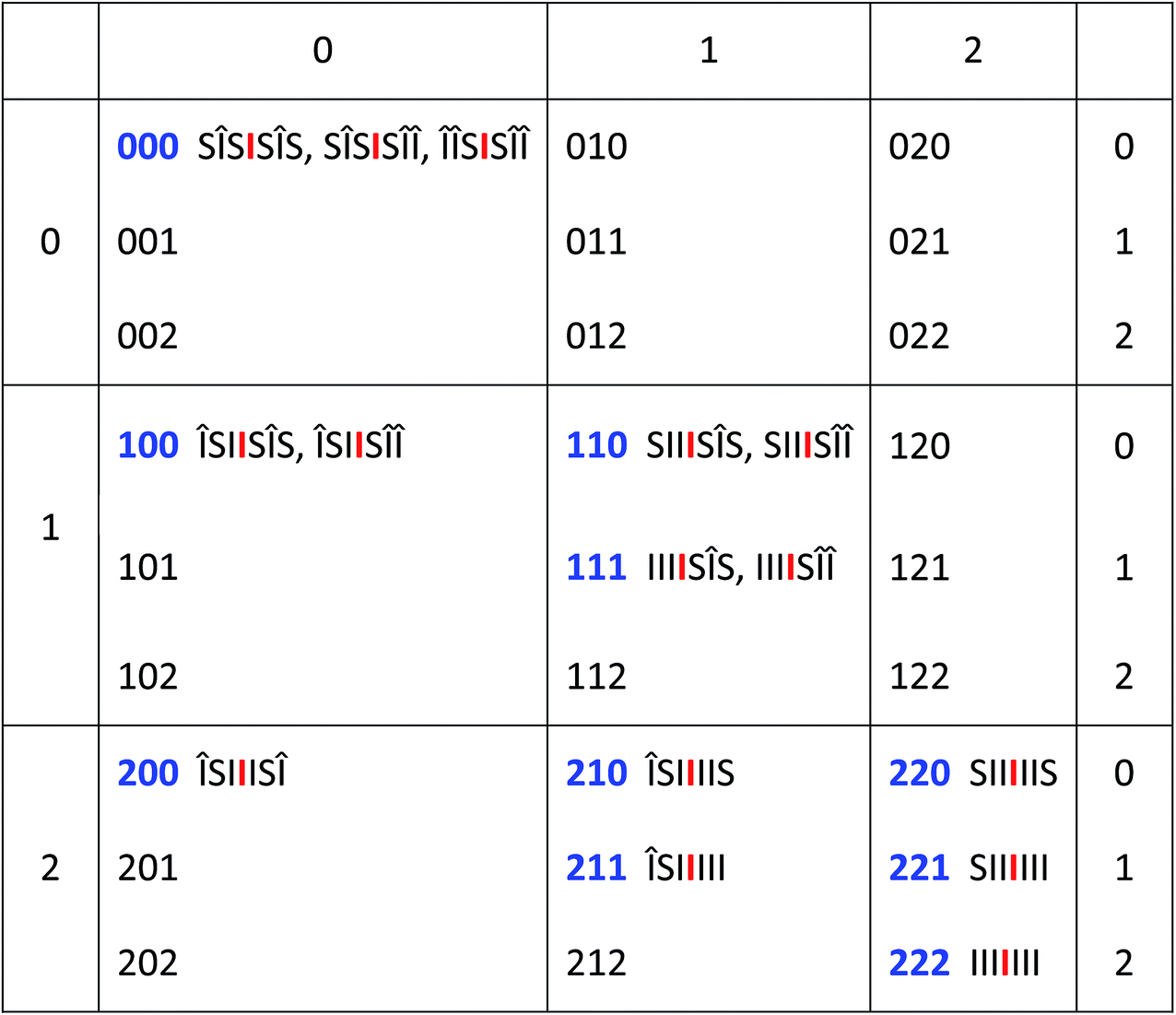
|
The magnitude of shielding of the central, “observed” diimide residue produced by each intercalating pyrene molecule diminishes progressively (we estimate, empirically, by a factor of about four per chain-fold under the conditions of the experiment) as it binds further and further out from the centre of the sequence. The sequence  , described by the code [222], thus produces the maximum possible degree of ring-current shielding of the central diimide residue when the sequence is intercalated by pyrene. The sequence
, described by the code [222], thus produces the maximum possible degree of ring-current shielding of the central diimide residue when the sequence is intercalated by pyrene. The sequence  , described by the code [210], will lead to a lesser degree of cumulative shielding, and the effect can be quantified as illustrated in Fig. 5. The integers of the digital shielding code can be shown to represent the coefficients of a quaternary expansion of a total (decimal) shielding parameter T for that sequence. In the presence of pyrene, sequences with higher values of T will produce larger complexation shifts of the central diimide resonance than sequences with lower values of T.
, described by the code [210], will lead to a lesser degree of cumulative shielding, and the effect can be quantified as illustrated in Fig. 5. The integers of the digital shielding code can be shown to represent the coefficients of a quaternary expansion of a total (decimal) shielding parameter T for that sequence. In the presence of pyrene, sequences with higher values of T will produce larger complexation shifts of the central diimide resonance than sequences with lower values of T.
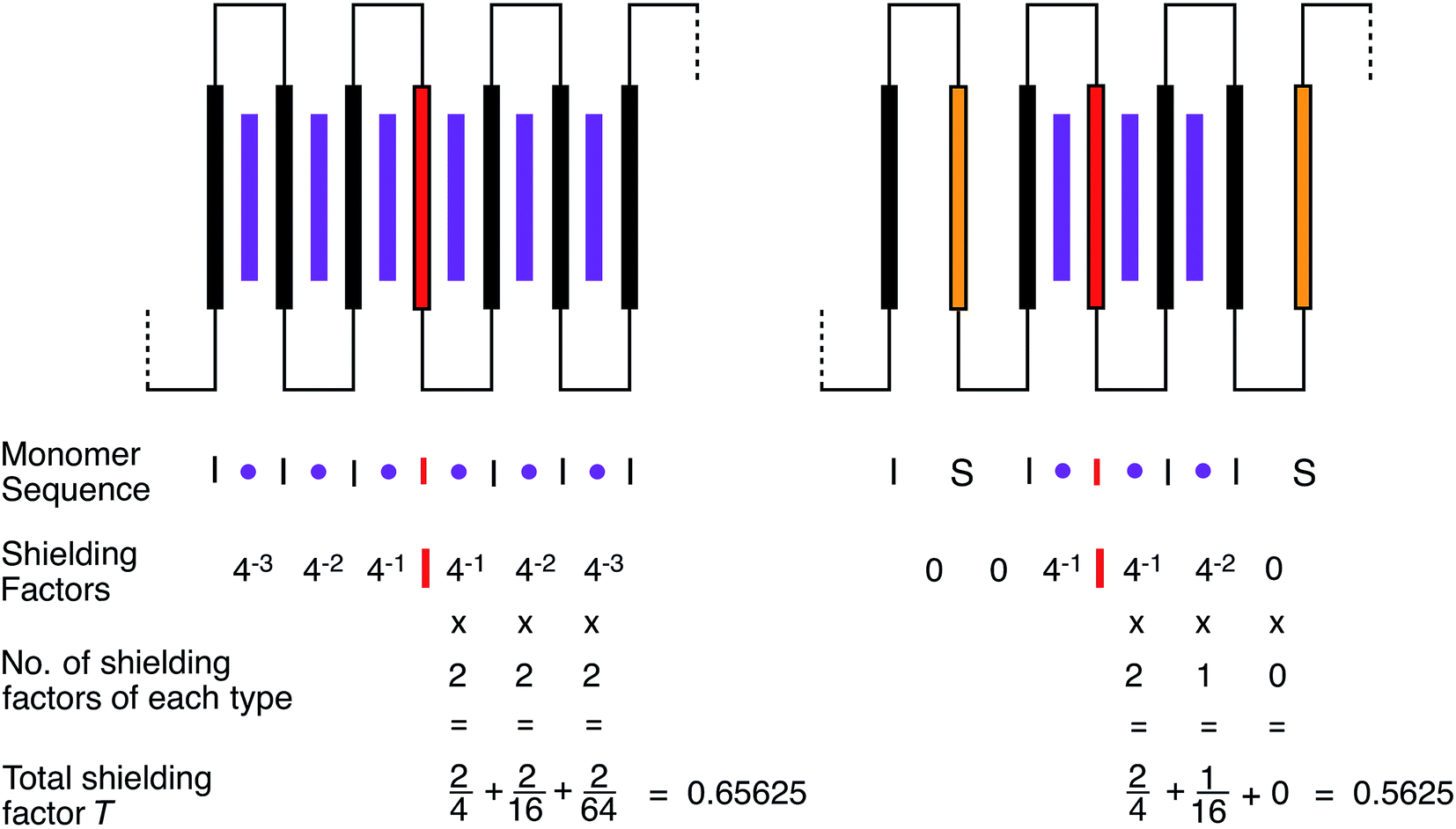 | ||
| Fig. 5 Cumulative magnetic shielding effects produced by pyrene molecules (purple) intercalating into copolymer chain-folds (black for “I” and gold for “S”) that are progressively more distant from a central, “observed” diimide residue. Two examples of septet-sequences in copolymer 1 are shown, along with their intercalated pyrene molecules. The large difference in binding constant for pyrene between the sequences “II” and “IS” is approximated by ignoring binding to “IS” (and “SI”) altogether. Ring-current shielding of the central diimide protons by intercalating pyrene molecules is proposed to fall off by a factor of approximately four at each successively more distant chain fold. Each different copolymer septet-sequence gives rise to a three-figure code representing the number of pyrene molecules (0, 1 or 2) that can intercalate at each of the three types of position in the sequence. The total magnetic shielding, T, experienced by the central diimide protons of any septet sequence is the sum of the individual shielding factors, taking account of their multiplicity (0, 1 or 2). | ||
Fig. 5 shows the summation of shielding factors for just two possible septet sequences, but the approach is valid for all allowed septets, so that in general terms:
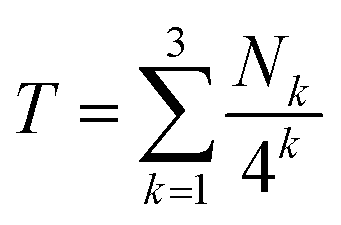 | (1) |
Then, for an infinite “I”-centred sequence in copolymer 1 we would have:
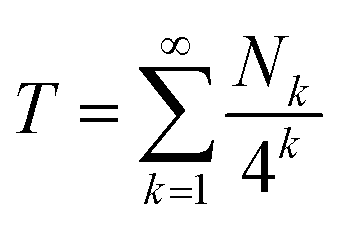 | (2) |
Eqn (2), which emerges from purely structural and magnetic considerations, defines a known, specific mathematical fractal which we term the “fourth-quarter” Cantor set. This fractal was first described (in more general terms) by Smith in 1875,32 but so far as we are aware it has not been found to underlie phenomena in the physical world until now. It is a variant of the more famous “middle-third” Cantor set,33–35 which may be defined as “those numbers in the interval [0, 1] that admit ternary expansions not containing the integer 1”.23 Geometrically, the latter set is constructed by dividing a line of unit length into three equal segments; deleting the middle segment; and repeating these operations on the remaining segments. What remains after an infinite number of such divisions and deletions is the middle-third Cantor set.25
Since the values of Nk in eqn (2) represent the coefficients of a quaternary expansion, and the possible integers of a quaternary number are 0, 1, 2 and 3, then the fourth-quarter Cantor set (eqn (2)) can be defined as “those numbers in the interval [0, 1] that admit quaternary expansions not containing the integer 3”. In the ESI† we include a formal proof of the equivalence between eqn (2) and the fourth-quarter Cantor set. The geometric construction of this set (Fig. 6) involves dividing a line of unit length into four equal segments, deleting the fourth quarter, repeating these operations on the remaining segments, and so on indefinitely. The set converges to an upper limit of 0.666…, and has a fractal (Hausdorff) dimension26 of 0.792. Unlike the middle-third set, the “remaining segments” of the fourth-quarter set are contiguous and so, in Fig. 6, are coloured to allow the construction to be visualised.
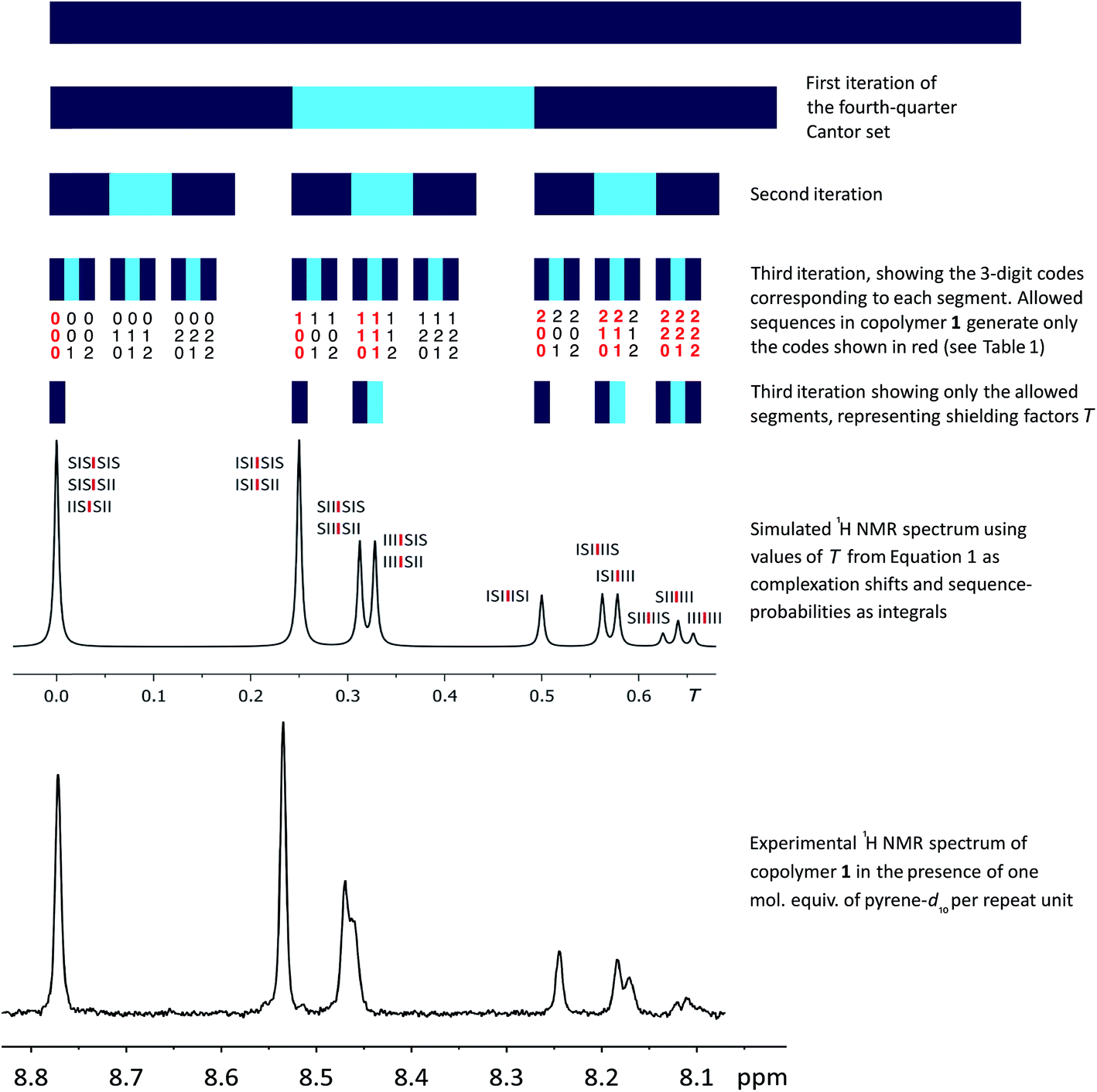 | ||
Fig. 6 First three iterations of the geometrical construction of the fourth-quarter Cantor set and comparison of the 1H NMR spectrum predicted from this set with the observed spectrum of copolymer 1 in the presence of one molar equivalent of pyrene-d10. The NMR simulation is based on all physically-permitted  -centred septet sequences present in the copolymer (Table 1), and so allows assignment of all ten resonances to the septets shown above. -centred septet sequences present in the copolymer (Table 1), and so allows assignment of all ten resonances to the septets shown above. | ||
Each iteration increases the sequence-length represented in the model, with the first iteration describing, in the present system, the induced magnetic field arising from triplet sequences, the second from quintets, the third from septets, and so on without limit. After three iterations the set contains 27 segments corresponding to the 27 septet-based codes shown in Table 1. If we now delete the segments corresponding to codes which cannot be generated by any permitted sequence in copolymer 1, a pattern of shielding factors emerges (Fig. 6; codes shown in red) that corresponds very closely to the pattern of complexation shifts observed in the NMR experiment. In the final model, complexation shifts, Δδ, displayed by the central diimide resonance in any given monomer sequence are proportional to the total shielding factor T for that sequence. As the proportionality constant between Δδ and T is very close to 1 ppm, we can set T (a dimensionless number) equal to Δδ in ppm, thus enabling simulation of a 1H NMR spectrum in which the complexation shift of each septet is given by eqn (1), using as coefficients (Nk) the digits of the codes shown in Table 1.
Of course resonance intensities (integrals) are also required for any meaningful 1H NMR simulation, and in a random copolymer these are given simply by the relative probabilities of all possible septet sequences. Values of T for each allowed septet, calculated from eqn (1), together with the corresponding sequence-probabilities, are shown in Table 2. No model is required to account for these sequence-probabilities: in a hypothetical, infinite, random copolymer chain they are rigorously defined, and a 1H NMR sample containing trillions of high molecular weight copolymer chains is a close approximation to such a chain. It is interesting that the set of sequence-probabilities for copolymer 1 shows the same type of self-similarity (a fourfold contracting transformation) as that found for the set of shielding factors T. Thus, in the diimide region, the entire NMR spectrum – in terms of both chemical shifts and resonance intensities – is self-similar. The predicted values of total shielding T (from eqn (1)) and sequence probabilities P are shown in Table 2.
|
In the present context we should emphasise the distinction between fractality and self-similarity. The self-similar character of our experimental NMR spectrum (Fig. 2) does not mean that the spectrum itself can be described as a fractal. What is a fractal is the underlying set of all shielding factors, given by eqn (1), within which the physically-permitted contributions are embedded. As seen in Fig. 2, the spectrum shows scaling about just a single point (at ca. 8.1 ppm), so that the pattern contains only one copy of itself at each scale. Mathematical or “true” fractals show scaling about several points: the classical “middle-third” Cantor set, for example, contains copies of itself in the first and last third of the set, and the fourth-quarter Cantor set described here contains copies of itself in the first, second and third quarters. The self-similar characteristics of the observed 1H NMR spectrum (Fig. 3) therefore represent only elements of the latter fractal, and the self-similarity of this spectrum is equivalent to that of the so-called logarithmic spiral, whose structure appears the same when magnified about just a single point (the origin).36 It may be noted that logarithmic spirals are found embedded within several fractals, including the well-known Mandelbrot set,15 in much the same way that the NMR spectrum under discussion here is embedded within the fourth-quarter Cantor set.
As noted above, Fig. 6 shows a simulated 1H NMR spectrum generated using total shielding factors T to predict chemical shifts and sequence-probabilities P to give predicted integrals. Correlation of the predicted spectrum with the experimental spectrum of the pyrene complex of copolymer 1 is remarkably good. Indeed, a correlation plot of T vs. Δδ is very close to being linear, as shown in Fig. 7. Moreover, eqn (2) predicts that sequences longer than septets, represented by further iterations of the fractal construction, should provide even more realistic predictions of shielding factors and complexation shifts, and this is borne out by simulation of a spectrum now based on nonet sequences (see ESI†).
 | ||
| Fig. 7 Values of T (dimensionless units) calculated from eqn (1) plotted against observed complexation shifts for the 1:1 copolymer–pyrene complex (Δδ, ppm), relative to the lowest field resonance as zero. This choice of zero removes any contribution from internal, non-complexation-related shielding within the copolymer chain. The close fit to linearity provides good evidence for the validity of the shielding model proposed in Fig. 5 and 6. | ||
Conclusions
This work describes a copolymer intercalation-complex whose 1H NMR spectrum contains almost exact copies of itself over several different length scales, demonstrating for the first time the existence of self-similarity in an NMR spectrum. The geometrical structure of the spectrum is rationalised on the basis of an atomistically-defined, supramolecular model that predicts a direct relationship between the observed spectrum and the mathematics of fractals, specifically the fourth-quarter Cantor set. A simulation of the spectrum based on this set provides a remarkably good match to the experimental data and at the same time enables all the observed resonances to be assigned to specific sequences or groups of sequences. This type of fractal can in fact provide a general, mathematical description of the environment of any monomer residue within a binary copolymer chain, and we are currently exploring its application to a wide range of copolymer systems and their supramolecular complexes.Experimental
General considerations
Synthetic procedures were carried out under an atmosphere of dry nitrogen unless otherwise specified. Solvents and reagents were obtained from Sigma-Aldrich and used without purification unless otherwise stated. N,N-Dimethylacetamide (DMAc) and N,N-dimethylformamide (DMF) were distilled over calcium hydride before use. 4,4′-Bis[4-(3-aminophenoxy)benzenesulfonyl]-1,1′-biphenyl was prepared according to a literature procedure,37 as was the model oligomer 2.31 Proton and 13C NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer, with chemical shifts referenced to residual solvent resonances and reported in ppm relative to TMS. Inherent viscosities (ηinh) were measured at 25 °C in a thermostatted water bath on 0.1% w/v polymer solutions in 1-methylpyrrolidinone (NMP) using a Schott-Geräte CT-52 semi-automated viscometer and AVS 470 measurement system. Gel permeation chromatography was carried out using a Polymer Laboratories PL-220 instrument fitted with 2× PL 10 μm mixed B columns. Computational modelling using molecular mechanics with charge-equilibration, (DREIDING force-field, Materials Studio, v. 7.0, Accelrys Inc., San Diego) was carried out in order to obtain a preliminary structural model for copolymer–pyrene interactions. Simulated NMR spectra were generated using the “Peak Table to Spectrum” script in Mestrenova (v. 9.0, Mestrelab Research, Santiago de Compostela). Single crystal X-ray data were measured for the pyrene complex 3 on an Oxford Diffraction X-Calibur Gemini diffractometer at 150 K using Cu-Kα radiation.Copolymer 1
Copolymer 1. 4,4′-Bis[4-(3-aminophenoxy)benzenesulfonyl]-1,1′-biphenyl (0.400 g, 6.2 × 10−4 mol) and 2,2′-(ethylenedioxy)bis(ethylamine) (0.090 g, 6 × 10−4 mol) were dissolved in dry DMAc (5 mL) by stirring at room temperature. When the diamines had dissolved completely, 1,4,5,8-naphthalenetetracarboxylic dianhydride (0.330 g, 1.2 × 10−3 mol) was added in one portion. The suspension was sonicated for 2 h until homogeneous. The dark brown solution was then stirred overnight before being transferred to a Petri dish and heated under vacuum to 80 °C to remove the solvent. The resulting poly(amic acid) film was imidized by heating under vacuum at 150 °C for 30 min, 180 °C for 30 min and finally at 200 °C for 1 h. The film was then dissolved in DMF (5 mL) and reprecipitated from methanol to afford copolyimide 1. Traces of a macrocyclic by-product38 were then removed by Soxhlet extraction with chloroform and subsequent re-precipitation of the copolymer from DMF solution in methanol.
Yield 0.52 g, 74%. Tg = 244 °C, inherent viscosity (ηinh) = 0.23 dL g−1; GPC (0.1% w/w LiCl in DMF as eluent with RI detection) Mn = 17800, Mw = 27500 relative to polystyrene standards. 1H NMR (CDCl3/CF3COOH 6:1 v/v) δ 8.93 (s), 8.88–8.82 (m), 8.79 (s), 8.06–7.99 (m), 7.75–7.67 (m), 7.65–7.58 (m), 7.24–7.17 (m), 7.13–7.04 (br), 4.54–4.44 (m), 4.03–3.94 (m), 3.92–3.82 (m); 13C NMR (CDCl3/CF3COOH 6:1 v/v): 163.7, 163.5, 156.0, 144.3, 140.6, 135.3, 134.1, 132.2, 131.7, 131.4, 130.1, 128.5, 128.1, 127.0, 126.8, 126.3, 126.2, 125.0, 121.5, 120.4, 69.7, 68.0, 39.6; FT-IR νmax/cm−1 1709, 1668, 1581, 1485, 1341, 1247, 1153, 1107, 767.
1H NMR titration of copolymer 1 with pyrene-d10
Equimolar stock solutions with exact concentrations of pyrene-d10 (24 mM) and copolymer 1 (24 mM) with respect to diimide residues in CDCl3/hexafluoropropan-2-ol (6:1 v/v) were prepared by weighing an approximate amount of sample and adding the required volume of solvent via a micropipette. Copolymer solution (100 μL) was added to each NMR tube using a micropipette and varying volumes of pyrene solution were then added to the tubes (e.g. 100 μL for 1:1 molar ratio of pyrene to diimide). The NMR tubes were all then filled to 600 μL total volume with the required volume of solvent (e.g. 400 μL to the 1:1 solution) and were well-mixed, thus affording a constant diimide concentration of 4 mM in each tube.
Crystal data for complex 3
C46H30N4O10·C16H10·(CF3)2CHOH, Mr = 1169.06, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.6269(3), b = 14.9210(5), c = 17.4851(7) Å, α = 108.065(3), β = 102.085(3), γ = 91.766(2)°. V = 2563.73(11) Å3, T = 150(2) K, Z = 2, Dc = 1.515 g cm−3, μ(Cu-Kα) = 1.006 mm−1, F(000) = 1204. Independent measured reflections 8183. R1 = 0.0516, wR2 = 0.0927 for 6639 independent observed reflections [2θ ≤ 126°, I > 2σ(I)]. CCDC 1503298.†
, a = 10.6269(3), b = 14.9210(5), c = 17.4851(7) Å, α = 108.065(3), β = 102.085(3), γ = 91.766(2)°. V = 2563.73(11) Å3, T = 150(2) K, Z = 2, Dc = 1.515 g cm−3, μ(Cu-Kα) = 1.006 mm−1, F(000) = 1204. Independent measured reflections 8183. R1 = 0.0516, wR2 = 0.0927 for 6639 independent observed reflections [2θ ≤ 126°, I > 2σ(I)]. CCDC 1503298.†
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Professor Kenneth Falconer of the University of St Andrews and Professor Ricardo Grau Abalo of the Universidad Central de Las Villas for valuable discussions. The work was supported by the Engineering and Physical Research Council of the UK (grant numbers EP/E00413X/1 and EP/G026203/1) and by the European Union (Marie Skłodowska-Curie Network EURO-SEQUENCES, grant number 642083). We thank Diamond Light Source Ltd, AWE plc, Cytec Aerospace Materials UK Ltd, and the University of Reading for PhD studentship funding.References
- H. F. Judson, The Eighth Day of Creation, Cape, London, 1979 Search PubMed.
- F. H. C. Crick, Symp. Soc. Exp. Biol., 1958, 12, 138–163 CAS.
- M. Nirenberg, Trends Biochem. Sci., 2004, 29, 46–54 CrossRef CAS PubMed.
- D. Thieffry and S. Sarkar, Trends Biochem. Sci., 1998, 23, 312–316 CrossRef CAS PubMed.
- C. R. Dawkins, The Blind Watchmaker, Longmans, London, 1986, p. 115 Search PubMed.
- H. M. Colquhoun and J.-F. Lutz, Nat. Chem., 2014, 6, 455–456 CrossRef CAS PubMed.
- T. T. Trinh, C. Laure and J.-F. Lutz, Macromol. Chem. Phys., 2015, 216, 1498–1506 CrossRef CAS.
- N. Zydziak, W. Konrad, F. Feist, S. Afonin, S. Weidner and C. Barner-Kowollik, Nat. Commun., 2016, 7, 13672 CrossRef PubMed.
- J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810–815 CrossRef CAS PubMed.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef PubMed.
- A. E. Stross, G. Iadevaia, D. Nunez-Villanueva and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 12655–12663 CrossRef CAS PubMed.
- G. De Bo, M. A. Y. Gall, M. O. Kitching, S. Kuschel, D. A. Leigh, D. J. Tetlow and J. W. Ward, J. Am. Chem. Soc., 2017, 139, 10875–10879 CrossRef CAS PubMed.
- G. J. Chaitin, J. ACM, 1974, 21, 403–421 CrossRef.
- J. E. R. Barrios, Principia, 2015, 19, 121–146 CrossRef.
- B. B. Mandelbrot, Fractals: Form, Chance and Dimension, W. H. Freeman and Co., San Francisco, 1977 Search PubMed.
- S. C. Zimmerman, M. Mrksich and M. Baloga, J. Am. Chem. Soc., 1989, 111, 8528–8530 CrossRef CAS.
- H. M. Colquhoun and Z. Zhu, Angew. Chem., Int. Ed., 2004, 43, 5040–5045 CrossRef CAS PubMed.
- H. M. Colquhoun, Z. Zhu, C. J. Cardin, Y. Gan and M. G. B. Drew, J. Am. Chem. Soc., 2007, 129, 16163–16174 CrossRef CAS PubMed.
- Z. Zhu, C. J. Cardin, Y. Gan and H. M. Colquhoun, Nat. Chem., 2010, 2, 653–660 CrossRef PubMed.
- Z. Zhu, C. J. Cardin, Y. Gan, C. A. Murray, A. J. P. White, D. J. Williams and H. M. Colquhoun, J. Am. Chem. Soc., 2011, 133, 19442–19447 CrossRef CAS PubMed.
- P. Lazzeretti, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36, 1–88 CrossRef CAS.
- S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS.
- J. Hutchinson, Indiana Univ. Math. J., 1981, 30, 713–747 CrossRef.
- K. J. Falconer, Fractal Geometry: Mathematical Foundations and Applications, Wiley, New York, 1990 Search PubMed.
- H.-O. Peitgen, H. Jurgens and D. Saupe, Chaos and Fractals, Springer-Verlag, Berlin, 1992 Search PubMed.
- K. J. Falconer, Fractals: A Very Short Introduction, O.U.P., Oxford, 2013, p. 116 Search PubMed.
- G. R. Newkome, P. Wang, C. N. Moorefield, T. J. Cho, P. P. Mohapatra, S. Li, S.-H. Hwang, O. Lukoyanova, L. Echegoyen, J. A. Palagallo, V. Iancu and S.-W. Hla, Science, 2006, 312, 1782–1785 CrossRef CAS PubMed.
- M. Wang, C. Wang, X.-Q. Hao, J. Liu, X. Li, C. Xu, A. Lopez, L. Sun, M. P. Song, H. B. Yang and X. Li, J. Am. Chem. Soc., 2014, 136, 6664–6671 CrossRef CAS PubMed.
- R. Sarkar, K. Guo, C. N. Moorefield, M. J. Saunders, C. Wesdemiotis and G. R. Newkome, Angew. Chem., Int. Ed., 2014, 53, 12182–12185 CrossRef CAS PubMed.
- J. Shang, Y. Wang, M. Chen, J. Dai, X. Zhou, J. Kuttner, G. Hilt, X. Shao, M. Gottfried and K. Wu, Nat. Chem., 2015, 7, 389–393 CrossRef CAS PubMed.
- M. P. Parker, C. A. Murray, L. R. Hart, B. W. Greenland, W. Hayes, C. J. Cardin and H. M. Colquhoun, Cryst. Growth Des., 2018, 18, 386–392 CAS.
- H. J. S. Smith, Proc. Lond. Math. Soc., 1875, 6, 140–153 Search PubMed.
- G. Cantor, Math. Ann., 1883, 21, 545–591 CrossRef.
- G. Cantor, Acta Math., 1884, 4, 381–392 CrossRef.
- J. F. Fleron, Math. Mag., 1994, 67, 136–140 CrossRef.
- R. C. Archibald, Am. Math. Mon., 1918, 25, 189–193 Search PubMed.
- H. M. Colquhoun, D. J. Williams and Z. Zhu, J. Am. Chem. Soc., 2002, 134, 13346–13347 CrossRef.
- H. M. Colquhoun, Z. Zhu, D. J. Williams, M. G. B. Drew, C. J. Cardin, Y. Gan, A. G. Crawford and T. B. Marder, Chem.–Eur. J., 2010, 16, 907–918 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, analytical and computational methods, tables of calculated shielding factors and sequence probabilities for allowed septet and nonet sequences in copolymer 1, simulated 1H NMR spectra over a range of length scales, atomic coordinates for the energy-minimised model shown in Fig. 3a, demonstration of the quaternary expression for the fourth-quarter Cantor set and its generalisation to non-integer bases, full details of the crystallographic analysis and structural data (CIF) for complex 3. CCDC 1503298. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00830b |
| This journal is © The Royal Society of Chemistry 2018 |