
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A redox-active diborane platform performs C(sp3)–H activation and nucleophilic substitution reactions†
Thomas
Kaese
,
Timo
Trageser
,
Hendrik
Budy
,
Michael
Bolte
 ,
Hans-Wolfram
Lerner
and
Matthias
Wagner
*
,
Hans-Wolfram
Lerner
and
Matthias
Wagner
*
Institut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Straße 7, D-60438 Frankfurt am Main, Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
First published on 19th March 2018
Abstract
Organoboranes are among the most versatile and widely used reagents in synthetic chemistry. A significant further expansion of their application spectrum would be achievable if boron-containing reactive intermediates capable of inserting into C–H bonds or performing nucleophilic substitution reactions were readily available. However, current progress in the field is still hampered by a lack of universal design concepts and mechanistic understanding. Herein we report that the doubly arylene-bridged diborane(6) 1H2 and its B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B-bonded formal deprotonation product Li2[1] can activate the particularly inert C(sp3)–H bonds of added H3CLi and H3CCl, respectively. The first case involves the attack of [H3C]− on a Lewis-acidic boron center, whereas the second case follows a polarity-inverted pathway with nucleophilic attack of the BB double bond on H3CCl. Mechanistic details were elucidated by means of deuterium-labeled reagents, a radical clock, α,ω-dihaloalkane substrates, the experimental identification of key intermediates, and quantum-chemical calculations. It turned out that both systems, H3CLi/1H2 and H3CCl/Li2[1], ultimately funnel into the same reaction pathway, which likely proceeds past a borylene-type intermediate and requires the cooperative interaction of both boron atoms.
B-bonded formal deprotonation product Li2[1] can activate the particularly inert C(sp3)–H bonds of added H3CLi and H3CCl, respectively. The first case involves the attack of [H3C]− on a Lewis-acidic boron center, whereas the second case follows a polarity-inverted pathway with nucleophilic attack of the BB double bond on H3CCl. Mechanistic details were elucidated by means of deuterium-labeled reagents, a radical clock, α,ω-dihaloalkane substrates, the experimental identification of key intermediates, and quantum-chemical calculations. It turned out that both systems, H3CLi/1H2 and H3CCl/Li2[1], ultimately funnel into the same reaction pathway, which likely proceeds past a borylene-type intermediate and requires the cooperative interaction of both boron atoms.
Introduction
For decades, organoboranes remained limited to a passive role as reagents in organic synthesis, where boryl substituents either serve as placeholders for other functional groups (e.g., halides, hydroxy, and amino groups),1 or are involved in Pd-catalyzed C–C-coupling reactions.2 Another useful asset, the potential of boron compounds to actively promote the cleavage of element–element bonds, lay dormant until the concepts of “Boron Lewis-acid catalysis“3–6 and “Frustrated Lewis pairs”7–9 were introduced about 15 years ago. Since then, it became increasingly apparent that appropriately selected main group compounds can rival transition metal complexes in mediating the transformation of organic substrates.Certain organoboranes are catalytically active not only in their Lewis-acidic neutral forms, but also in their exhaustively reduced states. As prominent examples, 9,10-dihydro-9,10-diboraanthracenes (DBAs) catalyze inverse electron-demand Diels–Alder reactions of 1,2-diazines3 as well as the dehydrogenation of ammonia-borane.5 Upon reduction, the corresponding [DBA]2− anions readily add C(sp)–H or H–H bonds across the two boron atoms; the latter reaction can be exploited for the economic conversion of chlorosilanes into hydrosilanes.10,11
With the triad 1H2/Li[1H]/Li2[1] (Scheme 1), we recently developed a system of ditopic boranes, which is comparable to the DBA/[DBA]2− pair, because it encompasses a Lewis-acidic (1H2) together with a dianionic species ([1]2−). As a decisive difference, however, the boron atoms in [DBA]2− are linked by two o-phenylene rings, whereas in [1]2− they are directly connected by a double bond. Both systems thus possess different frontier orbitals and should exhibit different reactivities.
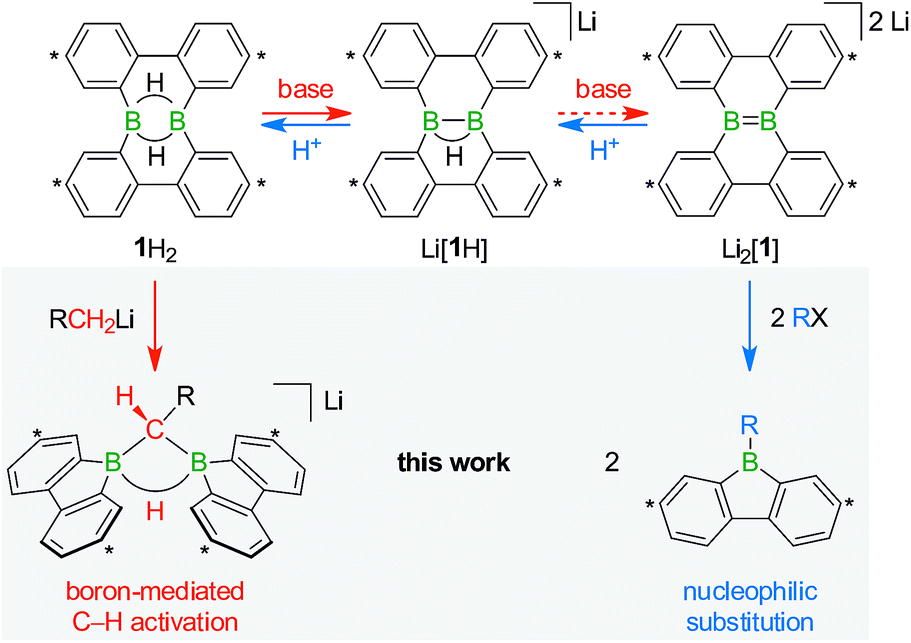 | ||
| Scheme 1 The members of the triad 1H2/Li[1H]/Li2[1] are linked through redox processes as well as protonation/deprotonation reactions. Treatment of 1H2 with RCH2Li leads to C(sp3)–H activations and skeletal rearrangements to furnish 1,1-bis(9-borafluorenyl)methanes (together with Li[1H]; R = H, C3H7). The addition of haloalkanes RX to Li2[1] results in nucleophilic substitution reactions and again skeletal rearrangements to afford 9-R-9-borafluorenes (in some cases accompanied by C(sp3)–H activations; X = Cl, Br, I). Carbon atoms marked with asterisks bear tBu substituents. | ||
The anions [1H]− and [1]2− are accessible in good yields via alkali-metal reduction of 1H2.12–14 Stepwise protonation with ethereal HCl cleanly takes [1]2− back to [1H]− and finally 1H2.14 The reverse deprotonation reaction of 1H2 to afford [1H]− is also quantitative, provided that the sterically demanding bases (Me3Si)2NLi and (Me3Si)3CLi are used. In case of the smaller nBuLi, the deprotonation reaction (20%) is accompanied by the formation of an anionic diborylmethane featuring a boron-bridging hydrogen atom (30%; Scheme 1, R = C3H7).14 These remarkable results immediately raise the following questions: (i) can 1H2 activate C(sp3)–H bonds of added alkyllithium reagents RCH2Li? (ii) Will [1]2− show nucleophilic behavior also toward electrophiles other than the proton (i.e., RX)?
Derivatization reactions of the inert C(sp3)–H bond are as topical as they are challenging – even if transition-metal catalysts are present.15–18 The few known boron-promoted examples fall into the three categories compiled in Scheme 2: (1) Braunschweig performed the reductive dechlorination of a dichloroborane precursor to generate an intermediate borylene, which inserted into the H3C group of a nearby mesityl substituent.19 (2) Wang et al. observed hydrogen-atom abstraction from a H3C group with concomitant formation of B–H and B–C bonds when they reduced 2,6-bis(BMes2)mesitylene to its diradical state.20 (3) Fontaine exploited an intramolecular deprotonation step on an FLP platform to establish an NCH2–B bond; subsequent H2 liberation provided the necessary thermodynamic driving force.21
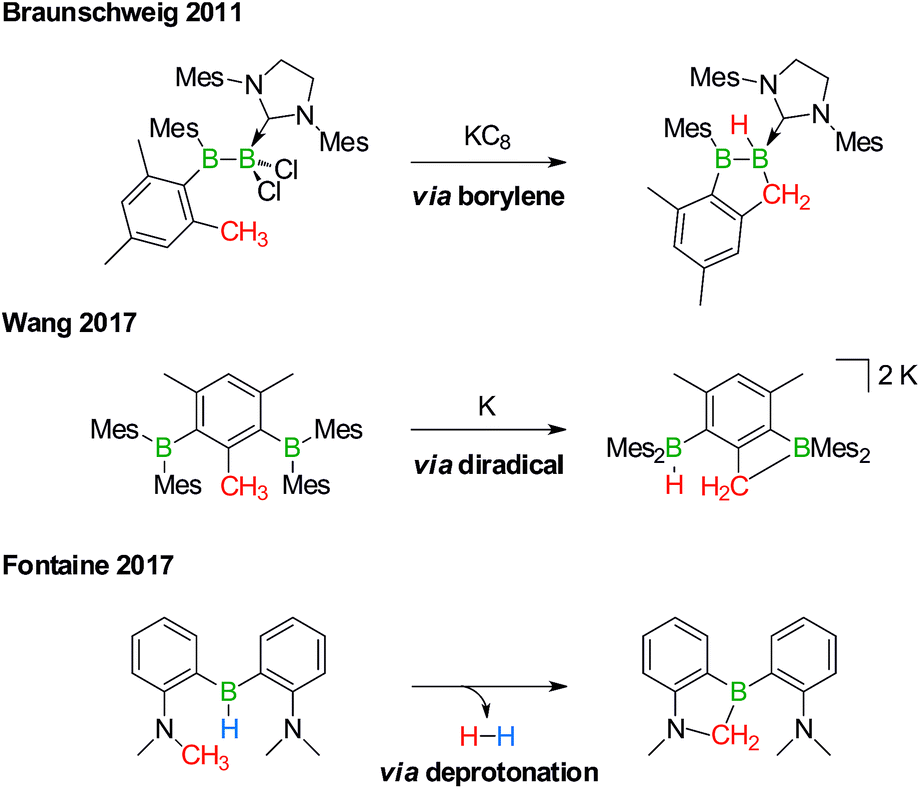 | ||
| Scheme 2 Selected examples of transition metal-free intramolecular C(sp3)–H activations through borylene (top), diradical (middle), and deprotonation reactions (bottom). Mes = 2,4,6-(H3C)3C6H2. | ||
The umpolung of carbon electrophiles through their conversion in, e.g., nucleophilic organolithium or Grignard reagents was one of the most important breakthroughs for the laboratory synthesis of organic compounds. A comparably high impact on the future progress of boron chemistry can be expected from the development of efficient tools to accomplish a polarity inversion of the intrinsically electrophilic boron center.22
In 2006, Yamashita and Nozaki pioneered the field of nucleophilic boron compounds by disclosing a lithium boryl isostere of stable N-heterocyclic carbenes (NHCs; Fig. 1).23 More than 10 years later, Hill expanded the class of compounds to include an isolable magnesium pinacolatoboryl complex.24 In the intervening period, a wealth of chemistry had already been developed based on the in situ generation of pinacolatoboryl nucleophiles via the alkoxide-induced heterolytic cleavage of bis(pinacolato)diboron (Lin, Kleeberg, Marder and others).25 Boryl nucleophiles can also be stabilized through π delocalization of the boron lone pair, as exemplified by Braunschweig's NHC-adduct of a borolyl salt (which may in fact react via radical pathways),26 the cyclic (alkyl)(amino)carbene-coordinated BH fragment of Kinjo/Bertrand,27 as well as Willner's/Finze's alkali metal tricyanoborate (Fig. 1).28
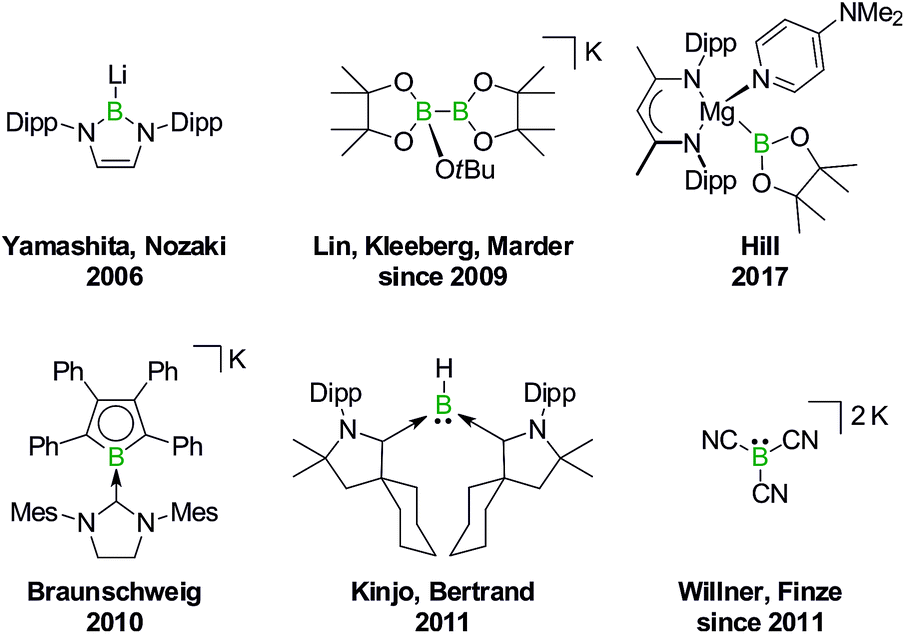 | ||
| Fig. 1 Selected isolable boron compounds showing formal nucleophilic behavior. Dipp = 2,6-(iPr)2C6H3. | ||
Before the background provided by the literature and our own previous results, we regarded the triad 1H2/Li[1H]/Li2[1] as a perfect platform for further studies into boron-promoted C–H-activation processes and boron-centered nucleophiles. Herein we present evidence that the reactions of 1H2 with RCH2Li indeed proceed through C(sp3)–H-cleavage steps and that the boron-bridging H atoms in the diborylmethane products stem from the organolithium reagents and are not remains of 1H2 (cf.Scheme 1; R = H, C3H7). We also show that the BB double bond of the dianion [1]2− behaves as a closed-shell nucleophile toward organohalides and that specifically H3CCl/Li2[1] and H3CLi/1H2 funnel into the same reaction channel. When H3CCl is replaced by an excess of H3C–I, C–H-activation is completely suppressed by a second nucleophilic substitution reaction to afford 2 equiv. of 9-methyl-9-borafluorene (Scheme 1; R = H3C). Employing α,ω-dihaloalkanes X(CH2)nX and Li2[1], we gained further insight into the competition between the nucleophilic substitution and C–H-activation scenarios as well as the cooperativity of the two adjacent boron centers (X = Cl, Br).
Results and discussion
We started our study by addressing the question: why and how does the reaction of 1H2 with nBuLi furnish not only the deprotonation product Li[1H], but also the diborylmethane-hydride adduct shown in Scheme 1 (R = C3H7)?First, we confirmed that a simplified system using H3CLi in place of nBuLi maintains the same general reactivity (Scheme 3). From equimolar mixtures of 1H2 and H3CLi, the products Li[1H] and Li[2] are formed in slightly varying relative amounts but constant combined yields of close to 50% (the analogous finding holds for the nBuLi case). The 1H NMR spectroscopic monitoring of the reaction in a sealed NMR tube (THF-d8, room temperature) showed no free H2 (δ 4.55 ppm),29 which is an important observation considering that the starting materials 1H2 and H3CLi contain a sum of five BHB/H3CLi protons, of which only three remain in the product Li[2].
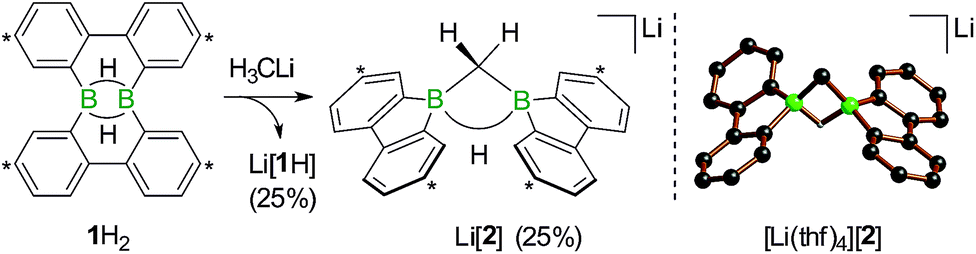 | ||
| Scheme 3 The addition of H3CLi to 1H2 furnishes the C–H activation product Li[2] together with the deprotonated compound Li[1H] (left; carbon atoms marked with asterisks bear tBu substituents). Molecular structure of [Li(thf)4][2] in the solid state (right). The solvent-separated [Li(thf)4]+ cation, all tBu groups, and all CH atoms are omitted for clarity. Selected atom···atom distance [Å] and bond angle [°]: B⋯B = 1.974(6); B–CH2–B = 76.8(3). | ||
Deuterium-labeling experiments with D3CLi/1H2 or H3CLi/1D2 combinations furnished isotopically pure Li[2-d3] or Li[2], respectively (Scheme 4). Thus, not only the methylene linker (δ(1H) 0.49 ppm, d), but also the boron-bridging hydrogen atom (δ(1H) 1.94 ppm, br) in Li[2] originate from the organolithium reagent. None of the two BHB atoms of 1H2 is still present in the product Li[2-d3] (see the ESI† for more information). We also note the appearance of two sets of aryl-proton signals that neither belong to Li[1H] nor Li[2] (or their partly deuterated counterparts) and are consequently accountable for the missing 50% product yield (see below).
 | ||
| Scheme 4 The reactions D3CLi/1H2 (top) or H3CLi/1D2 (bottom) give the C–D- or C–H-activation products Li[2-d3] or Li[2], respectively. Carbon atoms marked with asterisks bear tBu substituents. | ||
In the following, a plausible mechanistic model for the conversion of 1H2 with H3CLi will be described (black arrows in Scheme 5), which accounts for all available experimental evidence. It explains (i) the C–H activation of [H3C]−, (ii) the fate of the boron-bonded hydrogen atoms of 1H2, and (iii) the combined yield of only 50% for Li[1H] and Li[2]: similar to the case (Me3Si)3CLi/1H2, the reaction H3CLi/1H2 starts with the deprotonation of 1H2 to afford Li[1H]. The byproduct CH4 was detected by 1H and 13C{1H} NMR spectroscopy; when D3CLi was employed as the Brønsted base, we instead observed the formation of D3CH (sealed NMR tubes; see the ESI† for more details).
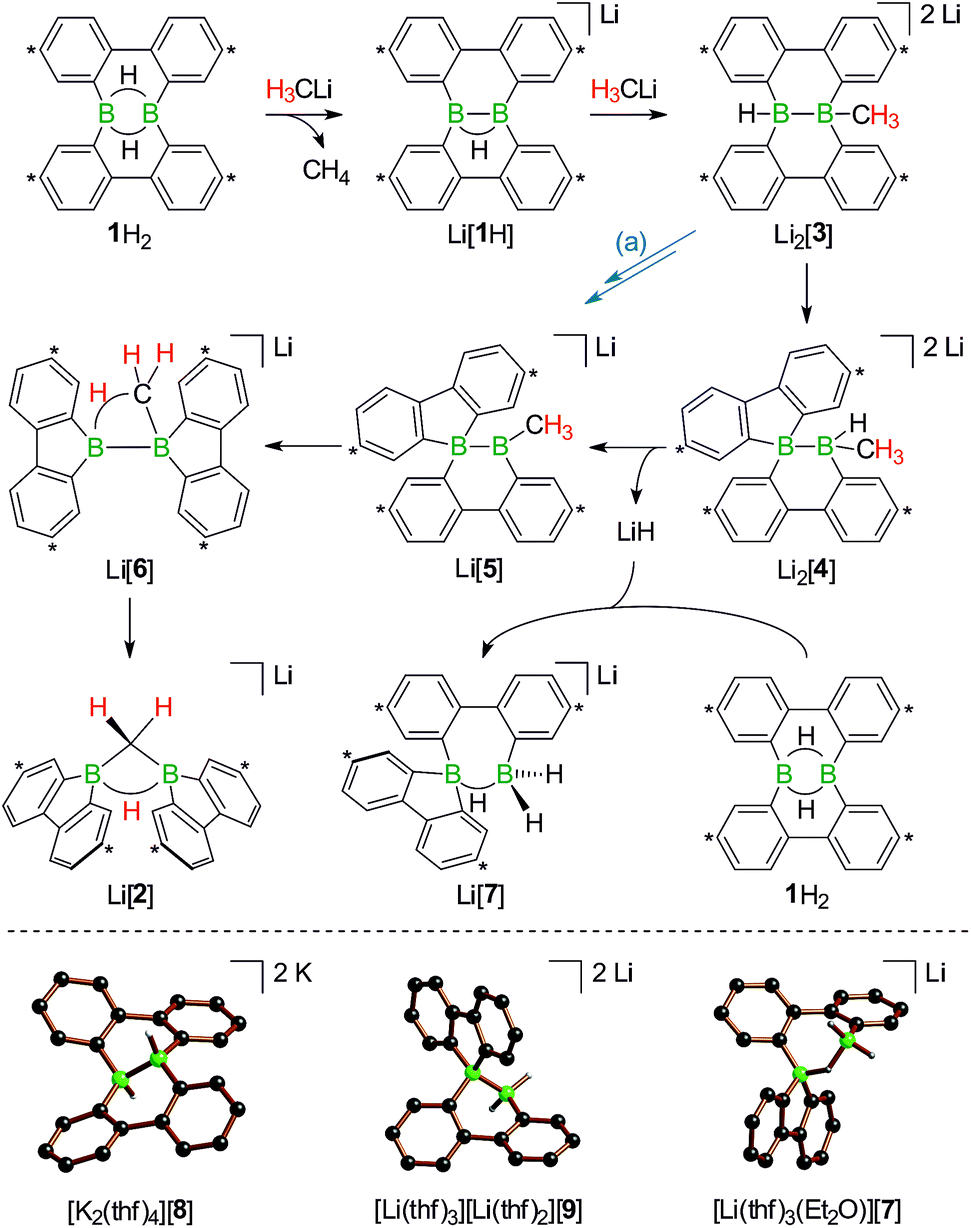 | ||
| Scheme 5 Proposed reaction mechanism explaining the formation of Li[1H], Li[2], and Li[7] from an equimolar mixture of H3CLi and 1H2 (top; carbon atoms marked with asterisks bear tBu substituents). The alternative pathway (a) leads from Li2[3] to Li[5], first via hydride elimination and second via a 1,2-phenyl shift. Molecular structures of [K2(thf)4][8], [Li(thf)3][Li(thf)2][9], and [Li(thf)3(Et2O)][7] in the solid state (bottom). The solvent-separated cations, all tBu groups, and all CH atoms are omitted for clarity. | ||
Contrary to the case (Me3Si)3CLi/1H2, the reaction involving H3CLi does not necessarily stop at the stage of Li[1H], because the small [H3C]− ion also has the potential to act as a Lewis base. Nucleophilic attack of H3CLi on a boron atom of Li[1H] establishes a B–CH3 bond and shifts the boron-bridging hydrogen atom to a terminal position. The structural motif of the resulting intermediate [3]2− has precedence in the crystallographically characterized dianion [8]2−,13 which carries a further hydrogen atom rather than a boron-bonded methyl group (Scheme 5, top and bottom). Li2[3] rearranges to Li2[4] through a 1,2-phenyl shift, accompanied by a 1,2-hydride shift. Again, a comparable hydrogen-containing species Li2[9] exists (Scheme 5, bottom), and its molecular structure has been confirmed by X-ray analysis.13 Li2[9] can isomerize to Li2[FluB(H)–(H)BFlu] (BFlu = 9-borafluorenyl),13 thereby providing an example of a 1,2-phenyl/1,2-hydride-shift cascade closely related to the isomerization of Li2[3] to Li2[4]. The latter reaction continues with an LiH-elimination step to generate Li[5], which possesses a three-coordinate boron atom with a vacant pz orbital and therefore easily undergoes a 1,2-phenyl shift to produce Li[6]. The anion [6]− can be viewed as the [H3C]− adduct of a diborane(4) containing two 9-borafluorene units that are linked by a B–B single bond. Only the sp3-hybridized boron atom has acquired an electron octet, however, also the B(sp2) center might gain some electron density from an agostic interaction with the methyl group and thereby reduce its strong Lewis acidity.30 Finally, this interaction turns into C–H-bond activation accompanied by B–B-bond cleavage and ultimately results in the formation of Li[2]. It is well known that B(sp2)–B(sp3) diboranes readily undergo B–B-bond heterolysis and thereby act as mild sources of nucleophilic boron.31 Moreover, the core parts of [2]− and [6]− are isoelectronic with protonated cyclopropane [C3H7]+. This cation has been thoroughly investigated by experimental32–34 and theoretical35,36 methods and found to be a highly fluctional system,37 which supports the idea of [6]− rearranging to [2]−. At this stage, the dynamic behavior comes to an end, because, contrary to the case of [C3H7]+, the three corners of [2]− are not equivalent and the BHB bridge should be thermodynamically favored over alternative BHC bridges.
In addition to the qualitative comparison with the all-carbon model system [C3H7]+, we studied the key C–H-activation step of the organoboron anion [6]− by quantum-chemical calculations (Fig. 2). Apart from the Li+ counterion, which likely is solvent-separated in THF solution (cf. the solid-state structure of [Li(thf)4][2]; Scheme 3, right), we also omitted the tBu substituents. The computed parent systems will be denoted with a superscript ‘c’ (e.g., [5c]− represents Li[5]). The 1,2-phenyl shift in [5c]− proceeds viaTS1 with an activation barrier of ΔG‡ = 9.9 kcal mol−1 and is endoergic by ΔGR = 5.9 kcal mol−1. The resulting open-chain rearrangement product [6c-open]− features a large B–B–CH3 bond angle of 121° and the vacant pz orbital of the B(sp2) atom is oriented almost orthogonal to the B–CH3-bond vector, which precludes an agostic interaction in this isomer. To establish the B–H–C bridge proposed above, the tricoordinate borafluorene fragment must be rotated by approximately 70° and the B–B–CH3 bond angle contracted – ultimately to a value of 68° in the local-minimum structure [6c]−. The conversion of [6c-open]− to the cyclic isomer [6c]−viaTS2 (ΔG‡ = 7.0 kcal mol−1) is associated with a moderate energy penalty of ΔGR = 4.6 kcal mol−1. The actual C–H-activation process involves the transition state TS3 in which the B–B bond and one C–H bond are concertedly cleaved and a new B–C bond is formed (ΔG‡ = 4.4 kcal mol−1).
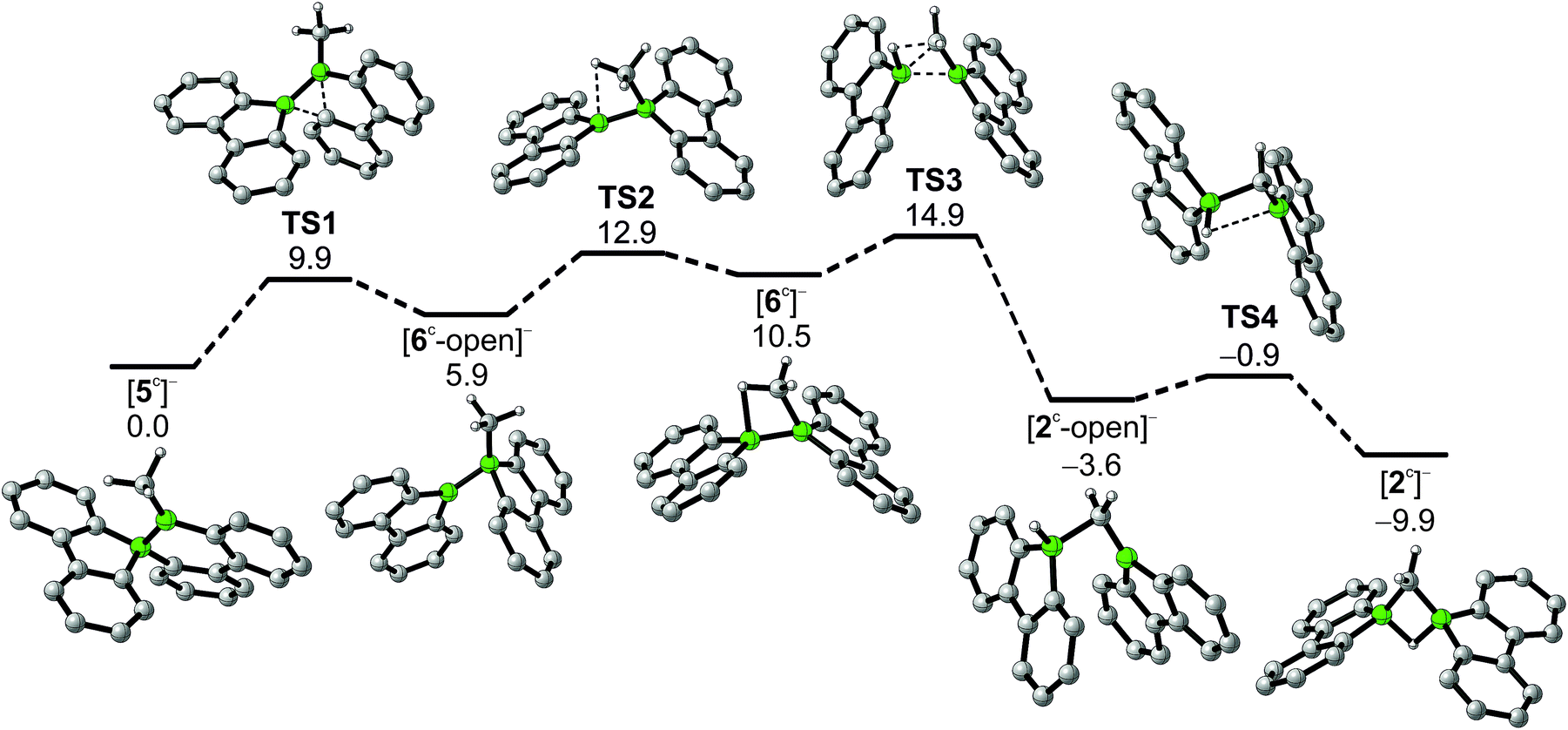 | ||
| Fig. 2 Reaction pathway for the conversion of [5c]− to [2c]−, calculated at the PBE0D/TZVP level of theory with the SMD polarized continuum model for solvation in THF. Gibbs free energies at 298 K (ΔG) are given in kcal mol−1 relative to [5c]−. | ||
The primary, open-chain activation product [2c-open]− is thermodynamically favored by −14.1 kcal mol−1 and −3.6 kcal mol−1 compared to [6c]− and [5c]−, respectively. A further stabilization is achievable through rotation about a B–C bond and placement of the hydrogen atom into a boron-bridging position to obtain the final product [2c]− (TS4: ΔG‡ = 2.7 kcal mol−1; ΔGR = −6.3 kcal mol−1). In summary, the reaction cascade from [5c]− to [2c]− possesses an overall activation barrier of ΔG‡ = 14.9 kcal mol−1, which is easily surmountable at room temperature. An appreciable thermodynamic driving force is provided by the exergonicity of the [2c]− formation (ΔGR = −9.9 kcal mol−1).
To experimentally substantiate the role of Li[1H] as the first intermediate along the pathway from 1H2 to Li[2], we treated an isolated sample of Li[1H] with 1 equiv. of H3CLi in THF. Even though the reaction started as expected, it stopped at the stage of Li2[4] (which enabled us to record a 1H NMR spectrum of this compound). The elimination of LiH from Li2[4] is thus not a spontaneous process, but apparently requires a hydride-trapping reagent. Compound 1H2 constitutes an ideal candidate for this purpose and, indeed, after the addition of 1 equiv. of 1H2, Li2[4] quantitatively vanished and Li[2] formed instead. Moreover, we found two sets of proton resonances that are assignable to two isomeric hydride-trapping products of 1H2 (cf. Li[7], Li[10]; Schemes 5 and 6).
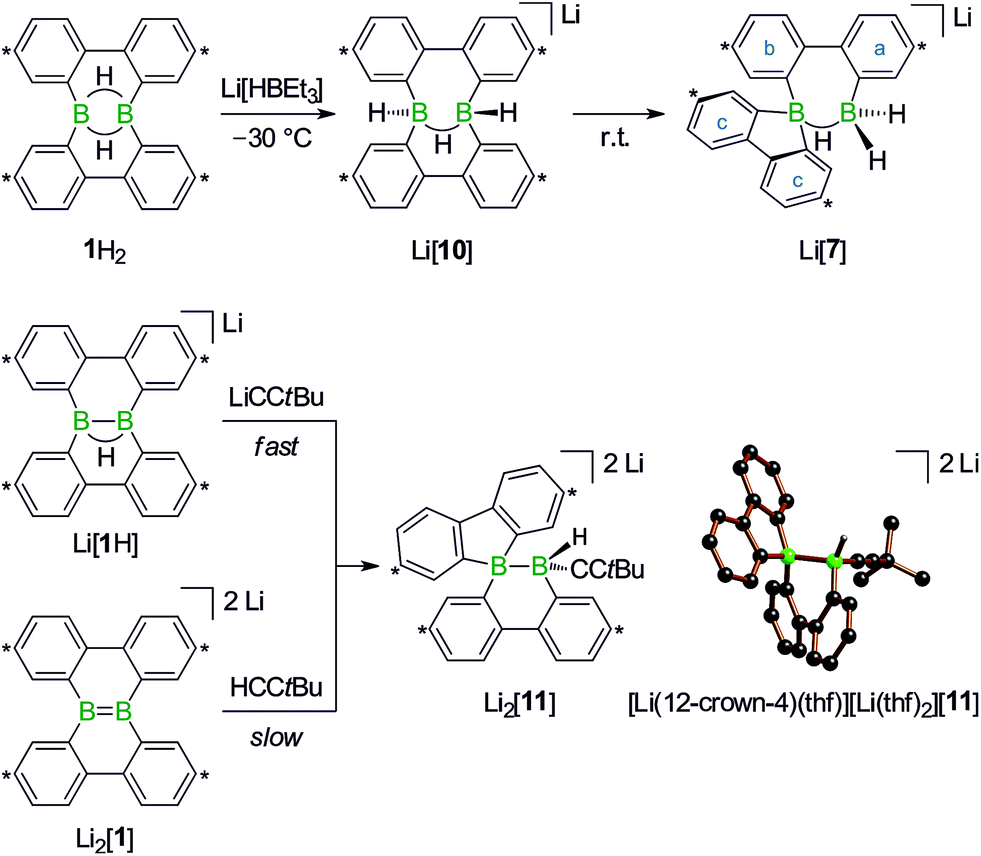 | ||
| Scheme 6 Reaction of 1H2 with Li[HBEt3] at −30 °C to give Li[10], which isomerizes to Li[7] at room temperature (top). Compound Li2[11] forms in both reactions, tBuCCLi/Li[1H] and tBuCCH/Li2[1] (bottom; carbon atoms marked with asterisks bear tBu substituents). Molecular structure of [Li(12-crown-4)(thf)][Li(thf)2][11] in the solid state. The solvent-separated cations, phenyl-bonded tBu groups, and all CH atoms are omitted for clarity. | ||
As a caveat we emphasize that the reaction from 1H2 to Li[2] may bypass the intermediate Li2[4] if hydride transfer from Li2[3] to 1H2 is faster than the rearrangement from Li2[3] to Li2[4] (blue path (a) in Scheme 5). Arguments in favor of this alternative route include: (i) the 1,2-phenyl shift required to generate intermediate Li[5] should be more facile on a B(sp2)–B(sp3) rather than a B(sp3)–B(sp3) scaffold (cf. Li2[3] → Li2[4]; Scheme 5). (ii) Li2[4] was observed only when the reaction was started from Li[1H], i.e., when the hydride trap 1H2 was absent, thus rendering the blue path impassable.
After the above discussion of a plausible mechanistic picture underlying the overall reaction scenario, we now present analytical data of key intermediates and products. The reaction H3CLi/1H2 furnishes Li[1H] and Li[2] besides the isomeric hydride-trapping products Li[7] and Li[10]. The first species, Li[1H], is a known compound and therefore does not require further discussion.14 The second species, Li[2], is reminiscent of the published C–H-activation product obtained from the reaction nBuLi/1H2 (cf.Scheme 1, R = C3H7).14 The main difference between both compounds relates to the fact that Li[2] possesses an average C2v symmetry in solution, whereas a pending C3H7 substituent reduces the symmetry to Cs. Consequently, the 1H NMR spectrum of Li[2] contains only one set of signals for all four tBu-C6H3 rings. The corresponding spectrum of its Cs-symmetric congener features two sets of resonances,14 one of them with chemical shift values almost identical to those of Li[2] and thus likely assignable to those halves of the 9-borafluorene subunits, which point into the same direction as the proton residing on the methylene bridge. A similar interpretation is valid for the 13C{1H} NMR spectrum of Li[2]. Single crystals of [Li(thf)4][2] suitable for X-ray analysis were grown from THF-hexane (Scheme 3). Like its C3H7 derivative,14 [Li(thf)4][2] forms solvent-separated ion pairs in the crystal lattice, and all key geometric parameters of the two anions are identical within the experimental error margins. We also note a pleasingly good agreement between the experimentally determined structure of [2]− and the computed structure of [2c]− (cf. the ESI† for full details).
1H NMR spectra measured on H3CLi/1H2 mixtures reproducibly showed resonances pointing toward a primary hydride-trapping product Li[10], which features a BHB bridge and two terminal hydrogen substituents in mutual trans arrangement (Scheme 6). For comparison, we prepared an authentic sample of Li[10] from 1H2 and 1 equiv. of the ‘superhydride’ Li[HBEt3]. At low temperatures, Li[10] forms quantitatively; since the compound is thermolabile, its NMR spectra had to be recorded at −30 °C. Li[10] gives rise to a double set of proton resonances in THF solution. On average, the two 2,2′-biphenylylene fragments of the anion [10]− should be related by a mirror plane containing the B2H3 core. The two phenylene rings of each individual 2,2′-biphenylylene moiety, however, are chemically inequivalent (as confirmed by 2D NMR experiments).
At room temperature, Li[10] readily isomerizes to the secondary hydride-trapping product Li[7], which we have isolated and characterized by NMR spectroscopy as well as X-ray crystallography. The anion of [Li(thf)3(Et2O)][7] consists of one 9-borafluorenyl and one BH2 fragment that are linked by a μ-H atom and a 2,2′-biphenylylene bridge (Scheme 5, bottom). As a result, both boron atoms are tetracoordinate and placed at a distance of B⋯B = 2.382(8) Å. In the solid state, the central seven-membered HB2C4 ring is non-planar and the anion possesses C1 symmetry (the torsion angle of the bridging 2,2′-biphenylylene amounts to 36°).
The molecular scaffolds of [7]− and the known anion [9]2− are essentially superimposable, apart from the fact that the latter features a covalent B–B bond (1.810(5) Å) instead of the μ-H atom (Scheme 5, bottom).13 In line with their marked structural resemblance, both anions exhibit similar 1H NMR spectra: in each case, three sets of aryl resonances are detectable. Two of those are well resolved at room temperature (H-a, H-b), whereas the third set consists of very broad signals, each of them integrating 2H (H-c; Scheme 6). This points toward a dynamic behavior of the compounds in solution, which likely arises from conformational changes of the twisted boron heterocycles. The 11B NMR spectrum of [7]− is characterized by two resonances with chemical shift values of δ −3.6 and −10.1 ppm, testifying to the presence of two magnetically inequivalent, tetracoordinate boron nuclei.38
Turning our attention from the products of the reaction CH3Li/1H2 to its intermediates, we note that the 1H NMR spectrum of Li2[4] shows the same peculiarities as those of its structural congeners Li[7] and Li2[9]: well resolved resonances coexist with severely broadened signals. Together with a BCH3 resonance at δ −0.1 ppm, this can be taken as a support for our structural proposal of Li2[4], but the motional broadening precludes the measurement of meaningful 13C{1H} NMR and 2D correlation spectra. Despite numerous efforts, we have not succeeded in growing crystals of Li2[4] and therefore considered replacing the H3C group with an alternative sterically undemanding organic substituent: The reaction tBuCCLi/Li[1H] provided the alkynyl analogue Li2[11] of Li2[4] in single-crystalline form ([Li(12-crown-4)(thf)][Li(thf)2][11]; Scheme 6). X-ray crystallography confirmed the proposed ring-contracted, H-shifted structure of [11]2−.
NMR spectroscopy reproduced the characteristic distribution of well-resolved and motionally broadened line shapes; the chemical shift values of the aryl protons of Li2[11] are reasonably close to those of Li2[4] (cf. the ESI† for an overlay of the respective 1H NMR spectra). Remarkably, Li2[11] is also accessible via a different approach, starting from the doubly boron-doped dibenzo[g,p]chrysene Li2[1] and tBuCCH, the conjugate weak acid of [tBuCC]− (Scheme 6).
The facile protonation14 of Li2[1] prompted us to investigate whether an umpolung approach to synthesize compounds of the type Li[2] might also be successful, which would provide fundamentally interesting insights into the reactivities of BB double-bonded species. As mentioned above, the intermediate Li[6] of the reaction H3CLi/1H2 can be regarded as the [H3C]− adduct of a diborane(4). Conceptually, it should be possible to arrive at the same molecule by formally transferring two electrons from the carbon nucleophile to the redox-active organoborane and thus starting from methylium-ion sources and the anion [1]2− (Fig. 3).39
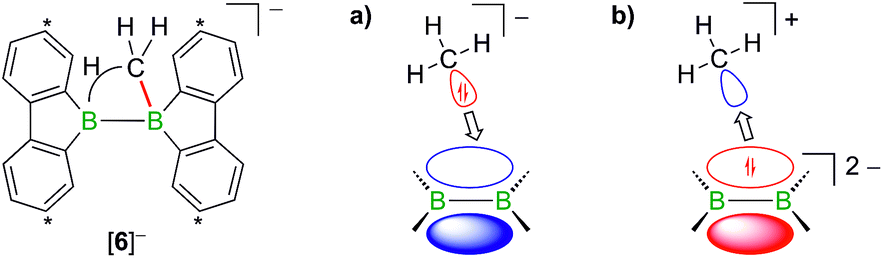 | ||
| Fig. 3 Two borderline cases to describe the bonding situation in [6]− as (a) the [H3C]− adduct of a diborane(4) and (b) the [H3C]+ adduct of a [1]2− anion. Carbon atoms marked with asterisks bear tBu substituents. | ||
Indeed, when a THF solution of Li2[1] is stirred at room temperature under a blanket of H3CCl gas (1 atm), a quantitative conversion to Li[2] occurs (Scheme 7).40 This approach is far more atom- and time-economic than the previous access route via the polarity-inverted couple H3CLi/1H2, because we avoid wasting 50% of 1H2 as a hydride-trapping reagent and do no longer have to separate the resulting hydride-trapping products. Mechanistically, the electron-rich BB fragment of Li2[1] likely acts as a nucleophile toward H3CCl to form [12]−, which carries a boron-bonded methyl substituent and contains a central B–B single bond. The B(sp2)–B(sp3) species Li[12] then undergoes a 1,2-phenyl shift to afford Li[5] and thereby funnels into the reaction cascade outlined above for the formation of Li[2] from H3CLi/1H2 (Scheme 7).
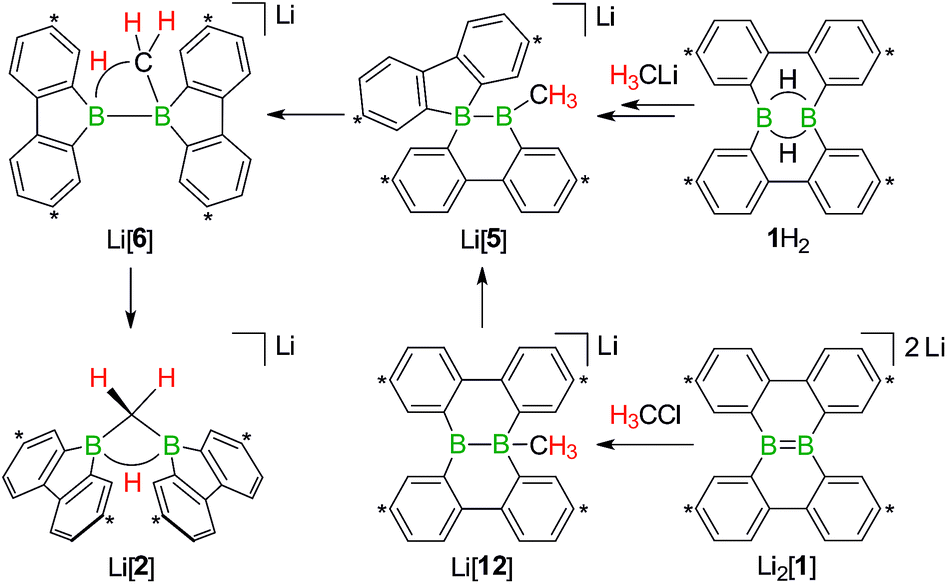 | ||
| Scheme 7 The addition of H3CCl to Li2[1] quantitatively furnishes Li[2]. The reaction pathways to Li[2], starting from either 1H2 or Li2[1], merge at the stage of Li[5] (cf. also Scheme 5). Carbon atoms marked with asterisks bear tBu substituents. | ||
When H3CCl is replaced by 1 equiv. of iodomethane (H3C–I), the outcome is a mixture of Li[2], 9-methyl-9-borafluorene (13), and residual Li2[1] (Scheme 8). After increasing the relative amount of H3C–I to 3 equiv., we almost exclusively obtained 13. The different behaviors of the two halomethanes can be rationalized by viewing the intermediate Li[6] as an adduct between the 9-borafluorenyl anion ([BFlu]−) and (H3C)BFlu (13; Scheme 9). [BFlu]− is isoelectronic to the carbene 9-fluorenylidene. A formal carbene-like reactivity is reflected by the intramolecular insertion of [BFlu]− into the C–H bond of the 9-methyl-9-borafluorene moiety to afford Li[2]. When the strong electrophile H3C–I with its excellent iodide leaving group is present, also the nucleophilic character of [BFlu]− comes into play and opens a competing intermolecular pathway, which ultimately leads to 13. As the relative amount of H3C–I is increased, the substitution reaction becomes dominant (we note in passing that the reaction with H3C–I can alternatively be viewed as a carbene-like insertion of [BFlu]− into the C–I bond with subsequent elimination of LiI).
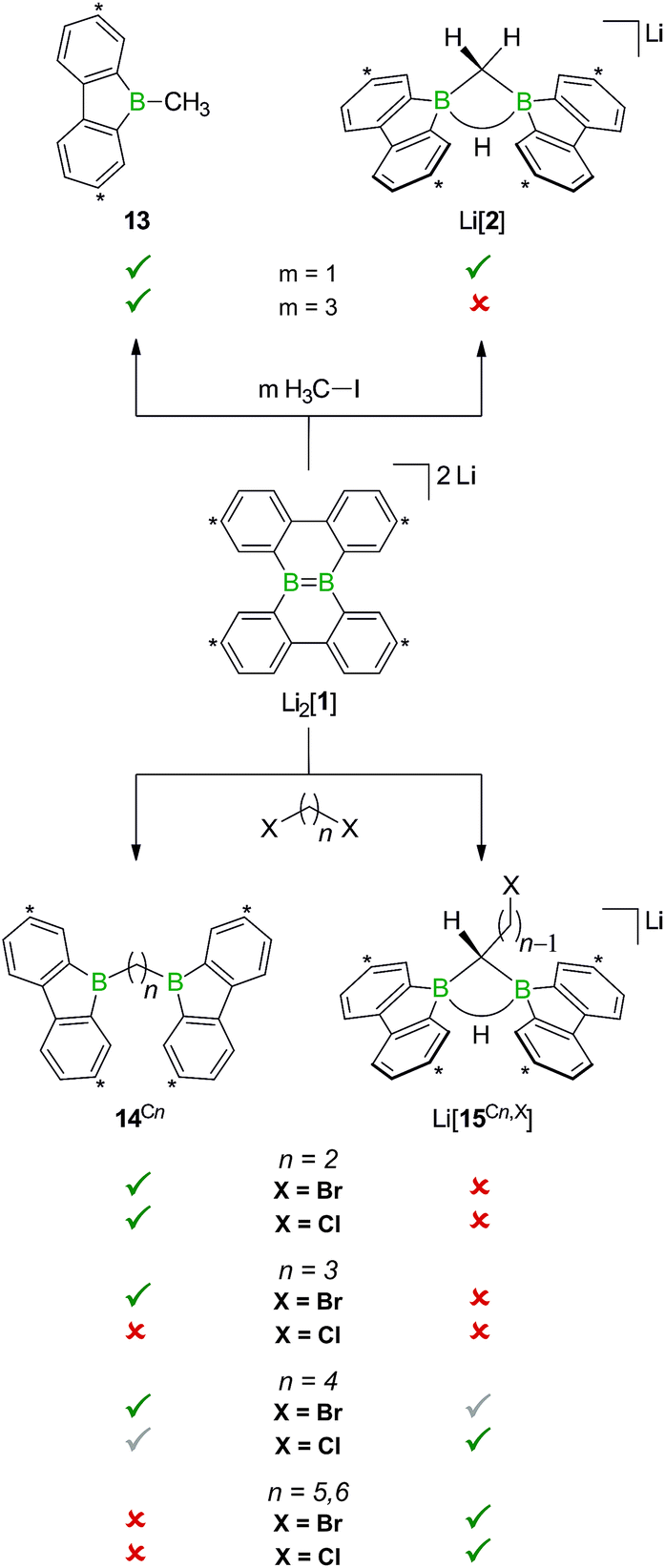 | ||
Scheme 8 The outcome of the reaction H3C–I/Li2[1] depends on the stoichiometries employed. While 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures give 13 together with Li[2], 3:1 mixtures exclusively furnish 13. Use of α,ω-dihaloalkanes X(CH2)nX instead of H3C–I affords ditopic boranes 14Cn (n = 2–4) and/or Li[15Cn,X] (n = 4–6; X = Cl, Br). Carbon atoms marked with asterisks bear tBu substituents. :1 mixtures give 13 together with Li[2], 3:1 mixtures exclusively furnish 13. Use of α,ω-dihaloalkanes X(CH2)nX instead of H3C–I affords ditopic boranes 14Cn (n = 2–4) and/or Li[15Cn,X] (n = 4–6; X = Cl, Br). Carbon atoms marked with asterisks bear tBu substituents. | ||
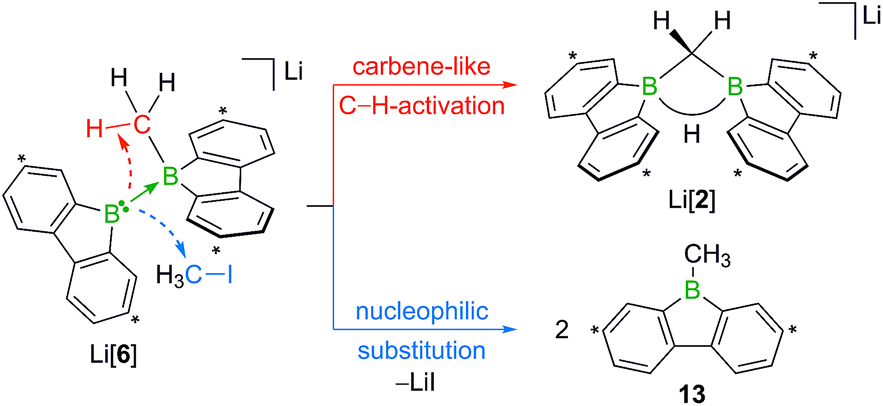 | ||
| Scheme 9 The intermediate Li[6] can be interpreted as an adduct between the 9-borafluorenyl anion ([BFlu]−) and 9-methyl-9-borafluorene (13). Intramolecular C–H insertion of the carbene-like [BFlu]− furnishes Li[2]; intermolecular nucleophilic attack on H3C–I affords 2 equiv. of 13. Carbon atoms marked with asterisks bear tBu substituents. | ||
In case of the system H3C–I/Li2[1], the methyl group initially gets attached to only one of the symmetry-related boron centers, but the other is equally important for the subsequent C–H-activation and nucleophilic substitution steps. The degree of B–B cooperativity in Li2[1] as well as the insertion vs. nucleophilic behavior of [BFlu]− thus deserve a detailed assessment. To this end, we conducted a systematic study using 1:1 mixtures of Li2[1] and α,ω-dihaloalkanes X(CH2)nX with chain lengths in the range of n = 2–6 and leaving groups of different qualities (e.g., X = Cl, Br). In these experiments, smaller alkylidene linkers are supposed to mimic higher local concentrations of the electrophile. As summarized in Scheme 8, clean twofold substitution reactions are observed with the short-chain substrates (n = 2 and 3, cf.14C2 and 14C3; 1,3-dichloropropane leads to a complex mixture of products). Clean C–H-activation reactions occur with the long-chain substrates (n = 5 and 6) to afford the haloalkyl species Li[15C5,Cl]/Li[15C5,Br] and Li[15C6,Cl]/Li[15C6,Br]. The medium-chain substrates (n = 4) mark the switching point between both scenarios: with the worse chloride leaving group, C–H-activation is preferred over the twofold substitution. The reverse is true in the case of the better bromide leaving group. The solid-state structures of 14C2·thf, 14C3 (Fig. 4), 14C4, and [Li(12-crown-4)2][15C5,Cl] (Fig. 4) were characterized by X-ray crystallography (cf. the ESI† for full information). Also the connectivities of [Li(thf)4][15C4,Cl], [Li(thf)4][15C6,Cl], and [Li(thf)4][15C6,Br] are supported by X-ray diffraction studies, however, due to disordered haloalkyl chains, tBu groups, and THF molecules, the quality of these three structures prevents their inclusion into this publication.41
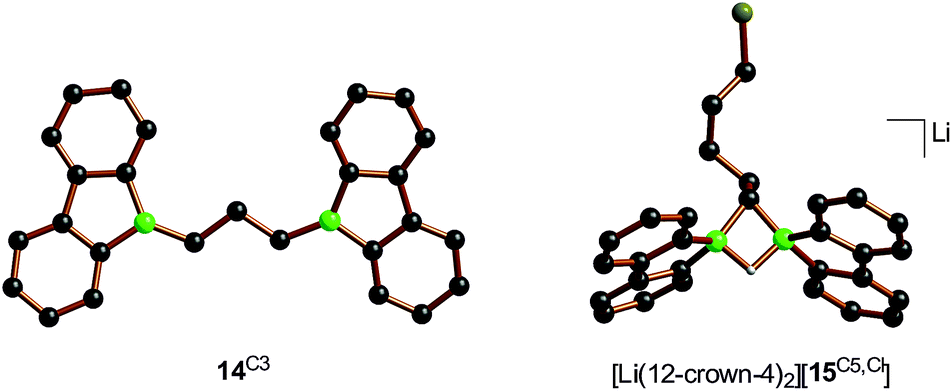 | ||
| Fig. 4 Molecular structures of 14C3 and of the terminally chlorine-substituted [Li(12-crown-4)2][15C5,Cl] in the solid state. The solvent-separated [Li(12-crown-4)2]+ cation, all tBu groups, and all CH atoms are omitted for clarity. | ||
The observed chain-length dependence of the product distribution suggests that the carbene-type insertion and the second nucleophilic substitution both follow an intramolecular pathway involving two cooperating boron atoms.
If the remaining CH2X center and the BCH2 group are similarly close to the B–B bond, the nucleophilic process occurs at a higher rate than the carbene-type C–H-activation. As the alkylidene spacer grows, the second electrophilic functionality moves further apart whereas the reactive α-CH2 unit stays in place such that the C–H-activation becomes more and more relevant until it finally takes over.
Although the reaction between Li2[1] and, e.g., H3C–I can convincingly be rationalized by assuming a nucleophilic pathway, the possible operation of a radical mechanism remains to be ruled out. We first note in this context that 1,2-dihaloethane in the presence of Li2[1] did not undergo reductive dehalogenation with ethene formation. Yamashita, Nozaki et al. have treated their boryllithium compound with methyl trifluoromethanesulfonate (H3COTf)42 on the one hand and benzyl bromide (BnBr) on the other (Scheme 10, top). In the first case, they observed the corresponding methyl borane in yields of 85%, whereas in the second case exclusively the bromoborane was obtained.43 To explain the different outcomes, they proposed halogenophilic attack of the boryllithium or single electron transfer to the benzyl halide. We repeated Nozaki's experiments with Li2[1] (Scheme 10, middle): H3COTf showed the same reactivity as described above for H3C–I (cf. Li[2] and 13); BnBr (as well as BnCl) gave the C–H-activation product Li[16] rather than any haloboranes, as confirmed by NMR spectroscopy and X-ray crystallography on [Li(thf)4][16].
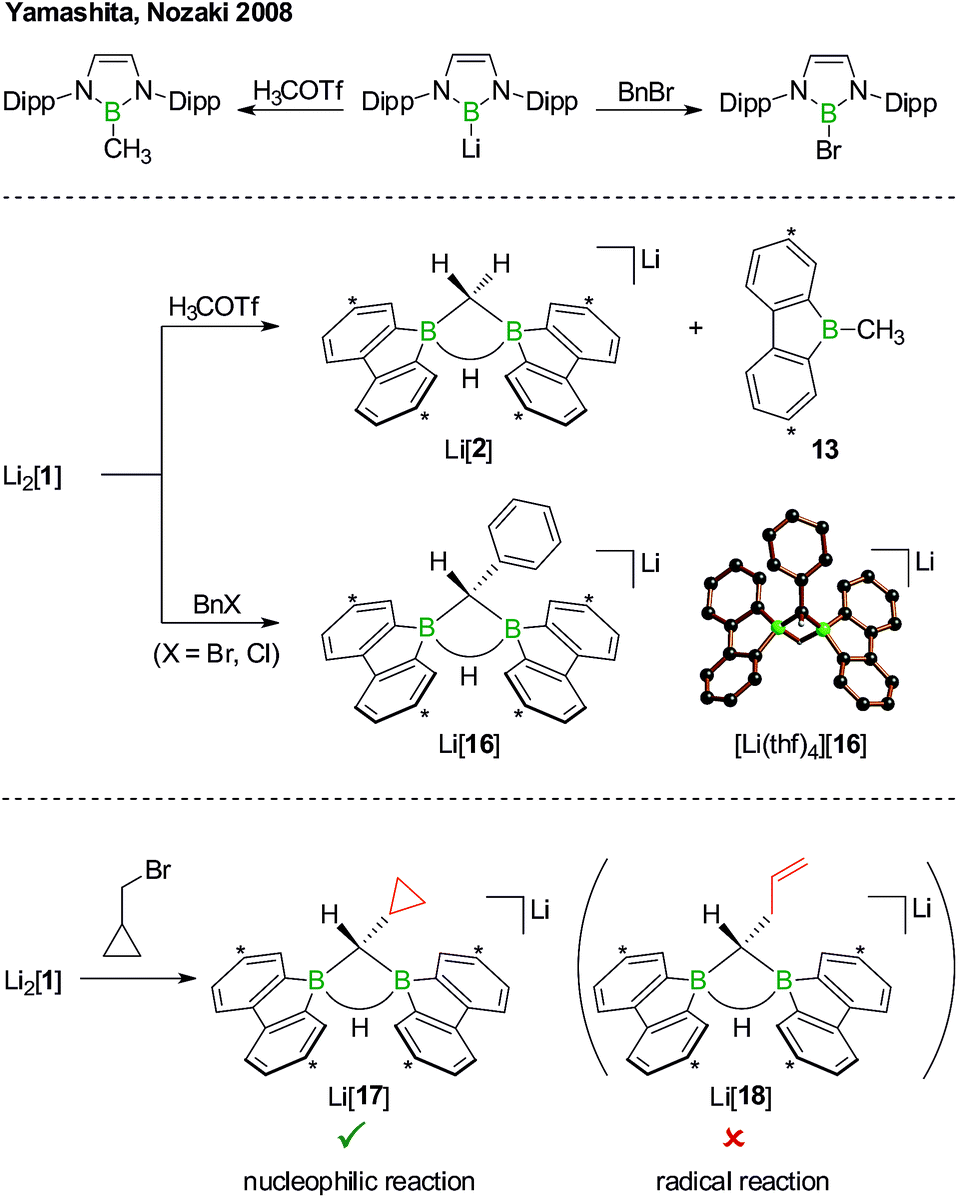 | ||
| Scheme 10 The reactions of Yamashita's and Nozaki's boryllithium compound with H3COTf or BnBr furnish the corresponding methyl borane or bromoborane, respectively (top). In the analogous reactions with Li2[1], only the organyl moieties are transferred to boron (middle; cf. Li[2]/13 and Li[16]). The reaction of Li2[1] with the radical clock (bromomethyl)cyclopropane quantitatively furnishes Li[17], which is a strong indication for a closed-shell, nucleophilic pathway (bottom). Dipp = 2,6-(iPr)2C6H3, Bn = CH2C6H5, H3COTf = H3COSO2CF3; carbon atoms marked with asterisks bear tBu substituents. In the crystal structure plot of [Li(thf)4][16], the solvent-separated cation, the tBu groups, and all C(sp2)–H atoms are omitted for clarity. | ||
As the ultimate test, we added Li2[1] to 1 equiv. of (bromomethyl)cyclopropane, a well-established radical clock (Scheme 10, bottom).44–46 A quantitative conversion to the C–H-activation product Li[17], still carrying an intact cyclopropyl substituent, occurred (NMR-spectroscopic control). The absence of the ring-opened olefin derivative Li[18] in the reaction mixture strongly supports the proposal of a closed-shell scenario in contrast to an open-shell process.
The results collected thus far are not only fundamentally interesting with respect to the reactivities of electron-rich BB double bonds, but open new access routes to ditopic boranes of high Lewis acidity. Molecules containing two or more potentially cooperating boron sites are of great current interest, inter alia, as organocatalysts5,11,47 or electron-storage media.48,49 Compounds of the class 14Cn already constitute free Lewis acids, but do not contain functional groups amenable to further derivatization.
The opposite is true for the salts Li[15Cn,X]. Here, the terminal halogen atoms provide ample opportunities, e.g., for grafting the organoboron units onto polymers, dendrimers, or surfaces, but the Lewis acids need to be activated through LiH elimination prior to use.
While the bulky hydride scavenger (H3C)3SiCl failed in this respect, the smaller electrophile H3C–I efficiently transformed the model compound Li[2] to its conjugate acid 14C1 (Scheme 11). As important diagnostic criteria, the BHB proton resonance vanishes in the course of the reaction, and the 11B NMR signal shifts from the tetracoordinate (Li[2]: δ −14 ppm) to the tricoordinate spectral region (14C1: δ 45 ppm).49,50
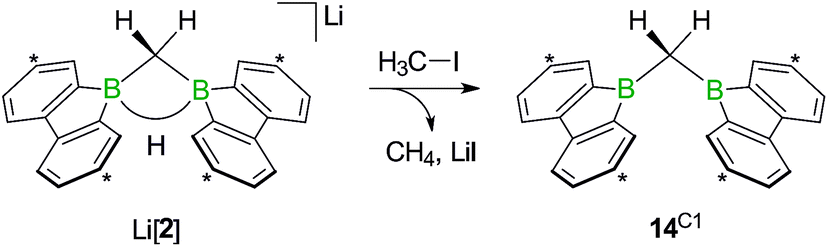 | ||
| Scheme 11 The addition of H3C–I to Li[2] furnishes the bis(9-borafluorenyl)methane 14C1. Carbon atoms marked with asterisks bear tBu substituents. | ||
In line with the reaction H3C–I/Li[2], the haloalkyl derivatives Li[15Cn,X] are not long-term stable in THF at room temperature: 1H NMR monitoring of the solutions revealed in each case a gradual decrease of the CH2X resonance and a concomitant increase of a signal assignable to a terminal CH3 group, which leads to the conclusion that the pending haloalkyl substituent can take a similar role as added H3C–I. It is important to note in this context that the follow-up X/H exchange reactions are completely suppressed at −78 °C and even at room temperature slow enough not to interfere with targeted derivatizations of the CH2X termini.
Conclusion
In summary, C(sp3)–H activation and nucleophilic substitution reactions have been performed on the same redox-active diborane platform. We propose that the doubly 2,2′-biphenylylene-bridged diborane(6) 1H2 reacts with H3CLi to furnish the rearranged B(sp2)–B(sp3) intermediate Li[FluB–BFlu(CH3)] (Li[6]; BFlu = 9-borafluorenyl). Li[6] also forms via an umpolung approach starting from H3CX and the BB bonded, nucleophilic Li2[1], a compound which can be regarded as the product of a double deprotonation of 1H2 (X = Cl, I). Li[6] readily undergoes B–B-bond heterolysis to formally give the [BFlu]− anion and (H3C)BFlu (13). The final product distribution depends on the relative amount of H3CX and the leaving-group qualities of X, because [BFlu]− can either insert into a C(sp3)–H bond of 13 or replace the halogen atom of a second equivalent of H3CX. The product of the carbene-type C–H insertion is Li[FluB(μ-CH2)(μ-H)BFlu] (Li[2]) while the nucleophilic substitution on C–X generates 2 equiv. of 13. Further insight into the competition between the two scenarios was gained with the help of α,ω-dihaloalkanes X(CH2)nX (X = Cl, Br). In the resulting intermediates Li[FluB–BFlu((CH2)nX)], both possible follow-up reactions should be intramolecular processes. A longer alkylidene chain corresponds to a lower local concentration of the electrophile, while the BCH2 groups are always similarly close to the reactive B–B bond. Consequently, short chains (n = 2,3) result in double substitution products FluB(CH2)nBFlu and long chains (n = 5,6) in C–H-activation products Li[FluB(μ-C(H)(CH2)n−1X)(μ-H)BFlu]. In the case of the intermediate chain length n = 4, a mixture of both compounds is obtained: the worse leaving group X = Cl leads to a higher proportion of the C–H-activated species, the better leaving group X = Br furnishes more FluB(CH2)4BFlu. We finally note that the B–B-bond heterolysis of Li[6] with concomitant transfer of a reactive [BFlu]− moiety is reminiscent of the reactivity patterns of the widely used alkoxy-diborane(4) adducts [pinB–Bpin(OR)]−.25 As a decisive difference, however, [BFlu]− appears to be considerably more reactive than in situ-generated [Bpin]−, because C–H-insertion reactions of the latter are so far unknown.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. K. thanks the Fonds der Chemischen Industrie for a Ph.D. grant. The authors are grateful to Dr M. Diefenbach for helpful discussions regarding the quantum-chemical calculations. We acknowledge Prof. E. D. Jemmis and Sagar Ghorai for discussions. We thank Albemarle Lithium GmbH (Frankfurt) for the generous gift of chemicals. Credit for the TOC-background picture: European Southern Observatory, ESO/L. Calçada.Notes and references
- Organoboranes for Syntheses, ed. P. V. Ramachandran and H. C. Brown, American Chemical Society, ACS Symposium Series, Washington D.C., 2001 Search PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- S. N. Kessler and H. A. Wegner, Org. Lett., 2010, 12, 4062–4065 CrossRef CAS PubMed.
- J. B. Grande, T. Urlich, T. Dickie and M. A. Brook, Polym. Chem., 2014, 5, 6728–6739 RSC.
- L. Schweighauser and H. A. Wegner, Chem.–Eur. J., 2016, 22, 14094–14103 CrossRef CAS PubMed.
- E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS.
- Frustrated Lewis Pairs I & II, ed. G. Erker and D. W. Stephan, Springer, Heidelberg, 2013 Search PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2010, 29, 5762–5765 CrossRef CAS.
- E. von Grotthuss, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067–14071 CrossRef CAS PubMed.
- (a) A. Hübner, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2014, 53, 10408–10411 CrossRef PubMed; (b) A. Hübner, A. M. Diehl, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2013, 32, 6827–6833 CrossRef.
- T. Kaese, A. Hübner, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2016, 138, 6224–6233 CrossRef CAS PubMed.
- T. Kaese, H. Budy, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2017, 56, 7546–7550 CrossRef CAS PubMed.
- K. M. Waltz and J. F. Hartwig, Science, 1997, 277, 211–213 CrossRef CAS.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- K. M. Waltz and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 11358–11369 CrossRef CAS.
- I. A. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- (a) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044–19047 CrossRef CAS PubMed. Related examples include: (b) T. Mennekes, P. Paetzold and R. Boese, Angew. Chem., Int. Ed., 1990, 29, 899–900 CrossRef; (c) W. J. Grigsby and P. P. Power, J. Am. Chem. Soc., 1996, 118, 7981–7988 CrossRef CAS; (d) W. J. Grigsby and P. P. Power, Chem.–Eur. J., 1997, 3, 368–375 CrossRef CAS; (e) Y. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326–12337 CrossRef CAS PubMed; (f) D. P. Curran, A. Boussonnière, S. J. Geib and E. Lacôte, Angew. Chem., Int. Ed., 2012, 51, 1602–1605 CrossRef CAS PubMed; (g) Y.-L. Rao, L. D. Chen, N. J. Mosey and S. Wang, J. Am. Chem. Soc., 2012, 134, 11026–11034 CrossRef CAS PubMed.
- L. Wang, Y. Fang, H. Mao, Y. Qu, J. Zuo, Z. Zhang, G. Tan and X. Wang, Chem.–Eur. J., 2017, 23, 6930–6936 CrossRef CAS PubMed.
- (a) É. Rochette, M.-A. Courtemanche and F.-G. Fontaine, Chem.–Eur. J., 2017, 23, 3567–3571 CrossRef PubMed. Related examples include: (b) G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4409–4412 CrossRef PubMed; (c) G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 6446–6449 CrossRef PubMed.
- The following literature survey focuses entirely on free boron-centered nucleophiles and does not cover corresponding transition-metal complexes. For an overview of these and related compound classes, see: (a) H. Braunschweig and T. Wagner, Angew. Chem., Int. Ed., 1995, 34, 825–826 CrossRef CAS; (b) H. Braunschweig, M. Burzler, R. D. Dewhurst and K. Radacki, Angew. Chem., Int. Ed., 2008, 47, 5650–5653 CrossRef CAS PubMed; (c) T. Kajiwara, T. Terabayashi, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2008, 47, 6606–6610 CrossRef CAS PubMed; (d) H. Braunschweig, R. D. Dewhurst, T. Herbst and K. Radacki, Angew. Chem., Int. Ed., 2008, 47, 5978–5980 CrossRef CAS PubMed; (e) T. Terabayashi, T. Kajiwara, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14162–14163 CrossRef CAS PubMed; (f) H. Braunschweig, P. Brenner, R. D. Dewhurst, M. Kaupp, R. Müller and S. Östreicher, Angew. Chem., Int. Ed., 2009, 48, 9735–9738 CrossRef CAS PubMed; (g) Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef CAS PubMed; (h) A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Coord. Chem. Rev., 2010, 254, 1950–1976 CrossRef CAS; (i) R. Frank, J. Howell, J. Campos, R. Tirfoin, N. Phillips, S. Zahn, D. M. P. Mingos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 9586–9590 CrossRef CAS PubMed; (j) H. Braunschweig, J. O. C. Jimenez-Halla, K. Radacki and R. Shang, Chem. Commun., 2015, 51, 16569–16572 RSC.
- (a) Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed; (b) Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS PubMed; (c) M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282–10292 CrossRef CAS PubMed; (d) L. Weber, Eur. J. Inorg. Chem., 2017, 3461–3488 CrossRef CAS. Related examples include: (e) M. Unverzagt, G. Subramanian, M. Hofmann, P. von Ragué Schleyer, S. Berger, K. Harms, W. Massa and A. Berndt, Angew. Chem., Int. Ed., 1997, 36, 1469–1472 CrossRef; (f) S. Robinson, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Chem. Commun., 2012, 48, 5769–5771 RSC; (g) W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef CAS PubMed; (h) B. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7, 11871 CrossRef CAS PubMed; (i) M. M. Morgan, J. M. Rautiainen, W. E. Piers, H. M. Tuononen and C. Gendy, Dalton Trans., 2018, 47, 734–741 RSC.
- (a) A.-F. Pécharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef PubMed; (b) A.-F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Angew. Chem., Int. Ed., 2017, 56, 16363–16366 CrossRef PubMed.
- (a) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef CAS PubMed; (b) R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC; (c) S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem.–Eur. J., 2015, 21, 7082–7098 CrossRef CAS PubMed; (d) E. C. Neeve, S. J. Geier, I. A. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (e) J. Cid, H. Gulyás, J. J. Carbó and E. Fernández, Chem. Soc. Rev., 2012, 41, 3558–3570 RSC.
- (a) H. Braunschweig, C.-W. Chiu, K. Radacki and T. Kupfer, Angew. Chem., Int. Ed., 2010, 49, 2041–2044 CrossRef CAS PubMed; (b) R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453–5457 CrossRef CAS PubMed. For an overview of N-heterocyclic carbene boranes, see: (c) D. P. Curran, A. Solovyev, M. Makhlouf Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed.
- (a) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed. Related examples include: (b) T. Imamoto and T. Hikosaka, J. Org. Chem., 1994, 59, 6753–6759 CrossRef CAS; (c) J. Monot, A. Solovyev, H. Bonin-Dubarle, É. Derat, D. P. Curran, M. Robert, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2010, 49, 9166–9169 CrossRef CAS PubMed; (d) D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590–7592 CrossRef CAS PubMed; (e) D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837–7839 RSC; (f) L. Kong, Y. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280–9283 CrossRef CAS PubMed; (g) L. Kong, R. Ganguly, Y. Li and R. Kinjo, Chem. Sci., 2015, 6, 2893–2902 RSC.
- (a) E. Bernhardt, V. Bernhardt-Pitchougina, H. Willner and N. Ignatiev, Angew. Chem., Int. Ed., 2011, 50, 12085–12088 CrossRef CAS PubMed; (b) J. Landmann, J. A. P. Sprenger, R. Bertermann, N. Ignat'ev, V. Bernhardt-Pitchougina, E. Bernhardt, H. Willner and M. Finze, Chem. Commun., 2015, 51, 4989–4992 RSC; (c) J. Landmann, J. A. P. Sprenger, M. Hailmann, V. Bernhardt-Pitchougina, H. Willner, N. Ignat'ev, E. Bernhardt and M. Finze, Angew. Chem., Int. Ed., 2015, 54, 11259–11264 CrossRef CAS PubMed; (d) H. Braunschweig, R. D. Dewhurst, L. Pentecost, K. Radacki, A. Vargas and Q. Ye, Angew. Chem., Int. Ed., 2016, 55, 436–440 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- (a) L. Kaufmann, H. Vitze, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2008, 27, 6215–6221 CrossRef CAS; (b) A. Iida, A. Sekioka and S. Yamaguchi, Chem. Sci., 2012, 3, 1461–1466 RSC.
- See ref. 25b.
- M. Saunders and E. L. Hagen, J. Am. Chem. Soc., 1968, 90, 6881–6882 CrossRef CAS.
- G. A. Olah and A. M. White, J. Am. Chem. Soc., 1969, 91, 5801–5810 CrossRef CAS.
- G. A. Olah and A. M. White, J. Am. Chem. Soc., 1969, 91, 6883–6885 CrossRef CAS.
- L. Radom, J. A. Pople, V. Buss and P. v. R. Schleyer, J. Am. Chem. Soc., 1972, 94, 311–321 CrossRef CAS.
- P. C. Hariharan, L. Radom, J. A. Pople and P. v. R. Schleyer, J. Am. Chem. Soc., 1974, 96, 599–601 CrossRef CAS.
- M. Saunders, P. Vogel, E. L. Hagen and J. Rosenfeld, Acc. Chem. Res., 1973, 6, 53–59 CrossRef CAS.
- H. Nöth and B. Wrackmeyer, Nuclear Magnetic Resonance Spectroscopy of Boron Compounds, in NMR Basic Principles and Progress, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer Verlag, Berlin, Heidelberg, New York, 1978 Search PubMed.
- Scattered precedence for comparable reactivity of electron-precise B–B single and multiple bonds exists: (a) A. Hübner, T. Kaese, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2015, 137, 3705–3714 CrossRef PubMed; (b) H. Braunschweig, P. Constantinidis, T. Dellermann, W. C. Ewing, I. Fischer, M. Hess, F. R. Knight, A. Rempel, C. Schneider, S. Ullrich, A. Vargas and J. D. Woollins, Angew. Chem., Int. Ed., 2016, 55, 5606–5609 CrossRef CAS PubMed; (c) M. Frick, E. Kaifer and H.-J. Himmel, Angew. Chem., Int. Ed., 2017, 56, 11645–11648 CrossRef CAS PubMed; (d) M. Arrowsmith, H. Braunschweig and T. E. Stennett, Angew. Chem., Int. Ed., 2017, 56, 96–115 CrossRef CAS PubMed; (e) A. Widera, E. Kaifer, H. Wadepohl and H.-J. Himmel, Chem.–Eur. J., 2018, 24, 1209–1216 CrossRef CAS PubMed; (f) J. Horn, A. Widera, S. Litters, E. Kaifer and H.-J. Himmel, Dalton Trans., 2018, 47, 2009–2017 RSC.
- A comparable selectivity toward C–H activation was also observed for EtBr (cf. Li[19] in the ESI†).
- T. Kaese, T. Trageser, H. Budy, M. Bolte, H.-W. Lerner and M. Wagner, 2018, Private communication to the Cambridge Structural Database; deposition numbers: CCDC 1819625 ([Li(thf)4][15C4,Cl]·THF), CCDC 1819626 ([Li(thf)4][15C6,Cl]·THF), and CCDC 1819627 ([Li(thf)4][15C6,Br]).†.
- S. J. Tremont and H. U. Rahman, J. Am. Chem. Soc., 1984, 106, 5759–5760 CrossRef CAS.
- See ref. 23b.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS.
- D. C. Nonhebel, Chem. Soc. Rev., 1993, 22, 347–359 RSC.
- C. Chen, L. Ouyang, Q. Lin, Y. Liu, C. Hou, Y. Yuan and Z. Weng, Chem.–Eur. J., 2014, 20, 657–661 CrossRef CAS PubMed.
- A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2010, 46, 3592–3594 RSC.
- A. Hübner, A. M. Diehl, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2014, 53, 4832–4835 CrossRef PubMed.
- See ref. 39a.
- A. Hübner, Z.-W. Qu, U. Englert, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2011, 133, 4596–4609 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1819687–1819695. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00743h |
| This journal is © The Royal Society of Chemistry 2018 |