
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient cleavage of aryl ether C–O linkages by Rh–Ni and Ru–Ni nanoscale catalysts operating in water†
Safak
Bulut
 a,
Sviatlana
Siankevich
a,
Antoine P.
van Muyden
a,
Duncan T. L.
Alexander
b,
Georgios
Savoglidis
a,
Jiaguang
Zhang
c,
Vassily
Hatzimanikatis
a,
Ning
Yan
c and
Paul J.
Dyson
*a
a,
Sviatlana
Siankevich
a,
Antoine P.
van Muyden
a,
Duncan T. L.
Alexander
b,
Georgios
Savoglidis
a,
Jiaguang
Zhang
c,
Vassily
Hatzimanikatis
a,
Ning
Yan
c and
Paul J.
Dyson
*a
aInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015, Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch
bInterdisciplinary Centre for Electron Microscopy (CIME), Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015, Lausanne, Switzerland
cDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117576, Singapore
First published on 6th June 2018
Abstract
Bimetallic Ru–Ni and Rh–Ni nanocatalysts coated with a phase transfer agent efficiently cleave aryl ether C–O linkages in water in the presence of hydrogen. For dimeric substrates with weaker C–O linkages, i.e. α-O-4 and β-O-4 bonds, low loadings of the precious metal (Rh or Ru) in the nanocatalysts quantitatively afford monomers, whereas for the stronger 4-O-5 linkage higher amounts of the precious metal are required to achieve complete conversion. Under the optimized, relatively mild operating conditions, the C–O bonds in a range of substituted ether compounds are efficiently cleaved, and mechanistic insights into the reaction pathways are provided. This work paves the way to sustainable approaches for the hydrogenolysis of C–O bonds.
The hydrogenolysis of aryl ether and other types of C–O bonds is highly challenging because of their relatively high bond dissociation energies and the competition with alternative hydrogenation reactions.1 However, due to increased interest in the valorization of the lignin component of biomass, an abundant renewable polymer comprising aromatic units held together by various types of C–O bonds, the need for efficient and selective hydrogenolysis catalysts is essential for biomass valorisation.2 Indeed, the enormous amount of waste lignin available offers a renewable raw material that could potentially rival coal tar and petroleum as a viable source of aromatic and cyclic hydrocarbons,3 and would therefore contribute towards reducing CO2 emissions and the associated environmental problems.4 Lignin is an integral part of cell walls of ligneous plants and has a protective function against hydrolytic and biological degradation.5 Consequently, lignin is a very stable, robust polymer that is difficult to breakdown.6 In lignin three different types of C–O linkages are dominant, i.e. α-O-4, β-O-4 and 4-O-5 linkages (Fig. 1).7
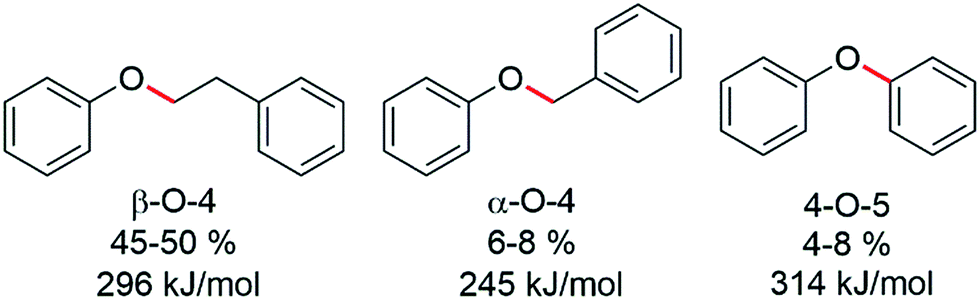 | ||
| Fig. 1 The three main C–O linkages present in lignin together with their approximate abundance8 and C–O bond dissociation energies9a illustrated using key model compounds. | ||
A wide range of heterogeneous noble metal10 and non-noble metal catalysts11 have been reported for the hydrogenolysis of the substrates illustrated in Fig. 1. These catalysts tend to operate only at elevated temperatures (typically above 120 °C for noble metal catalysts and above 200 °C for non-noble metal catalysts) and under elevated pressures of H2 (≥10 atm). Several recent reports describe catalysts that operate under somewhat less forcing conditions,11c,12 including Ni-based homogenous and heterogenous catalysts reported by Hartwig and coworkers.13 Initially, they used a homogenous system generated in situ from Ni(COD)2 and a N-heterocyclic carbene in the presence of an excess of NaOtBu.13a However, high catalyst loadings (20 mol% of Ni(COD)2 and 40 mol% of N-heterocyclic carbene) were required and, they subsequently showed that lower catalyst loadings could be achieved using Ni nanoparticles that formed in situ from Ni(COD)2 in the presence of an excess of NaOtBu.13b,13c These reactions operate under inert conditions in dry organic solvents at temperatures between 120–140 °C. Recent work from the same group demonstrates the importance of using an excess of strong base,14 even when using the pre-synthesized Ni(SIPr)2 (SIPr = 1,3-bis-(2,6-diisopropylphenyl)imidazolinium chloride) catalyst that was generated in situ in the initial report.13a It should be noted that the separation of water from raw biomass is difficult and costly and, consequently, air and moisture stable catalytic methodologies would be advantageous for the valorization of real biomass.
Here, we describe a series of Ru–Ni and Rh–Ni nanocatalysts (NCs) coated with a phase transfer agent that catalyse the hydrogenolysis of even the least reactive 4-O-5 linkage in diphenyl ether at lower temperatures, below 100 °C and under 1 atm of H2, with a relative low catalyst loading of 5 mol%, using water as the reaction medium (both for the synthesis of NCs and catalytic reactions), without using any additives, e.g. a base ruthenium–nickel Ru100−xNix and rhodium–nickel Rh100−xNix NCs, where x is the molar percentage of Ni used in the synthesis, were prepared in water from the reduction of RuCl3, RhCl3 or NiCl2, or their combinations, with NaBH4 in the presence of cetyltrimethylammonium bromide (CTAB), employed as both a stabilizing agent and a phase transfer reagent. The Ru15Ni85 and Rh15Ni85 NCs (selected as they exhibit the optimum catalytic activity, see below) were characterized using transmission electron microscopy (TEM), high angle annular dark field (HAADF) scanning transmission electron microscopy combined with energy-dispersive X-ray spectroscopy (STEM-EDS) mapping (Fig. 2), and powder X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) (Fig. S1–S3†). The material is unstable under the extended electron beam exposure of STEM-EDS mapping, for instance with a tendency of neighbouring NCs to coalesce together (Fig. S4†).
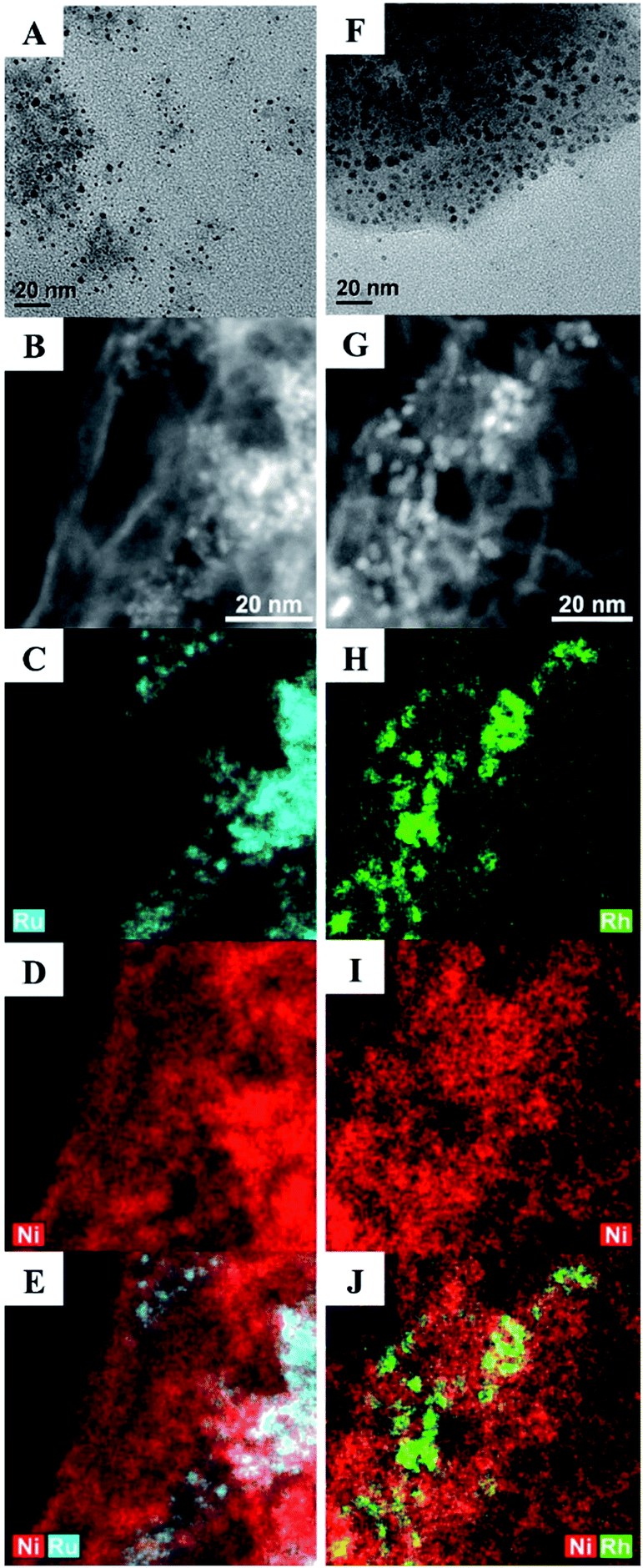 | ||
| Fig. 2 TEM (A and F), and HAADF STEM (B and G) and corresponding EDS maps (C–E and H–J) for the (left) Ru15Ni85 and (right) Rh15Ni85 materials. The NCs appear brighter on the Z-contrast HAADF images. Note that the scale of the EDS maps is the same as that in the HAADF STEM images. | ||
Consequently, the acceleration voltage and probe current were reduced to minimise this effect (80 kV and ∼0.5 nA respectively), and only data representative of the initial sample state are shown. Images and EDS maps for the Ru15Ni85 and Rh15Ni85 samples (Fig. 2) reveal a morphology comprising of NCs that are consistent with a bimetallic nature of Ru–Ni or Rh–Ni (depending on the sample), dispersed on thin, more homogeneous membrane-like structures, which contain mostly Ni (Fig. S5†). These membranes aggregate into larger, porous structures, and EDS shows that they also contain O, Cl and Br (Fig. S6†). The Ru15Ni85 NCs have an average size of 2.0 ± 0.2 nm whereas the Rh15Ni85 NCs are slightly larger, average diameter = 2.7 ± 0.3 nm (Fig. 2). Their compositions were also confirmed by inductively coupled plasma mass spectrometry (ICP-MS, see ESI†).
The catalytic activity of the NCs was evaluated in the hydrogenolysis of three lignin model substrates using water as a solvent (Fig. 1). At a catalyst loading of 5 mol% (based on metal content) quantitative cleavage of the C–O linkages was obtained at 95 °C under 1 atm of H2 in 16 hours, depending on the metal–nickel (metal = Ru or Rh) molar ratio (see Fig. 3, 4 and Tables S2–S7†). Compounds with only one C6-ring derived from the hydrogenolysis of the C–O bond are denoted as monomers whereas dimer products are generated by hydrogenation of the substrate without the cleavage of the C–O bond. As expected, conversion of the β-O-4 and α-O-4 linkage containing substrates (Fig. 3 and 4, respectively) to monomers is easier to achieve than hydrogenolysis of diphenyl ether, which contains the 4-O-5 linkage.
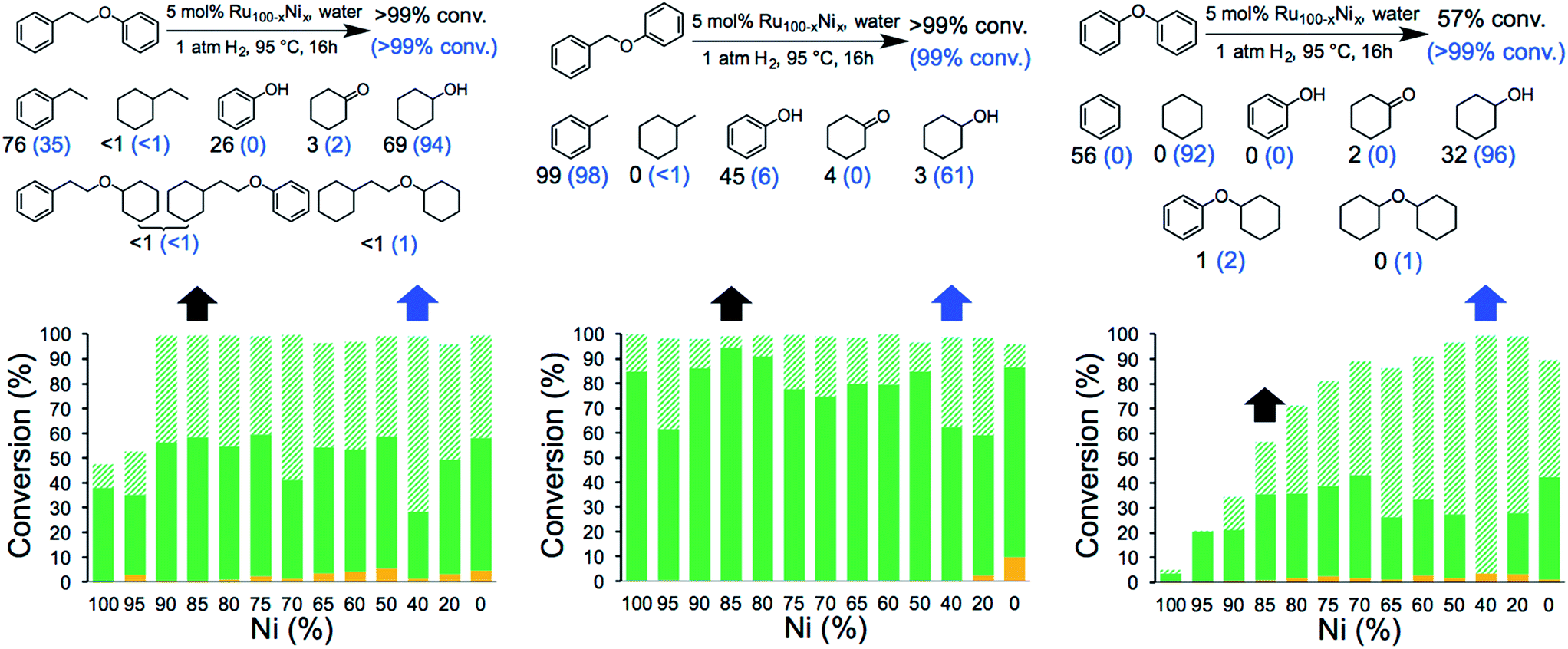 | ||
Fig. 3 Hydrogenolysis/hydrogenation of (left) 1-phenoxy-2-phenylethane (β-O-4 linkage), (middle) benzyl phenyl ether (α-O-4 linkage), (right) diphenyl ether (4-O-5 linkage) and product yield for selected metal combinations catalysed by Ru100−xNix NCs. Full details are provided in Tables S2, S4 and S6.† The black arrows refer to the M15Ni85 NCs and the corresponding yields are in black. The blue arrows refer to the M60Ni40 NCs and the corresponding yields are in blue in parenthesis. The fractions comprise partially/fully hydrogenated dimers ( ), non-hydrogenated monomers ( ), non-hydrogenated monomers ( ) and hydrogenated monomers ( ) and hydrogenated monomers ( ). ). | ||
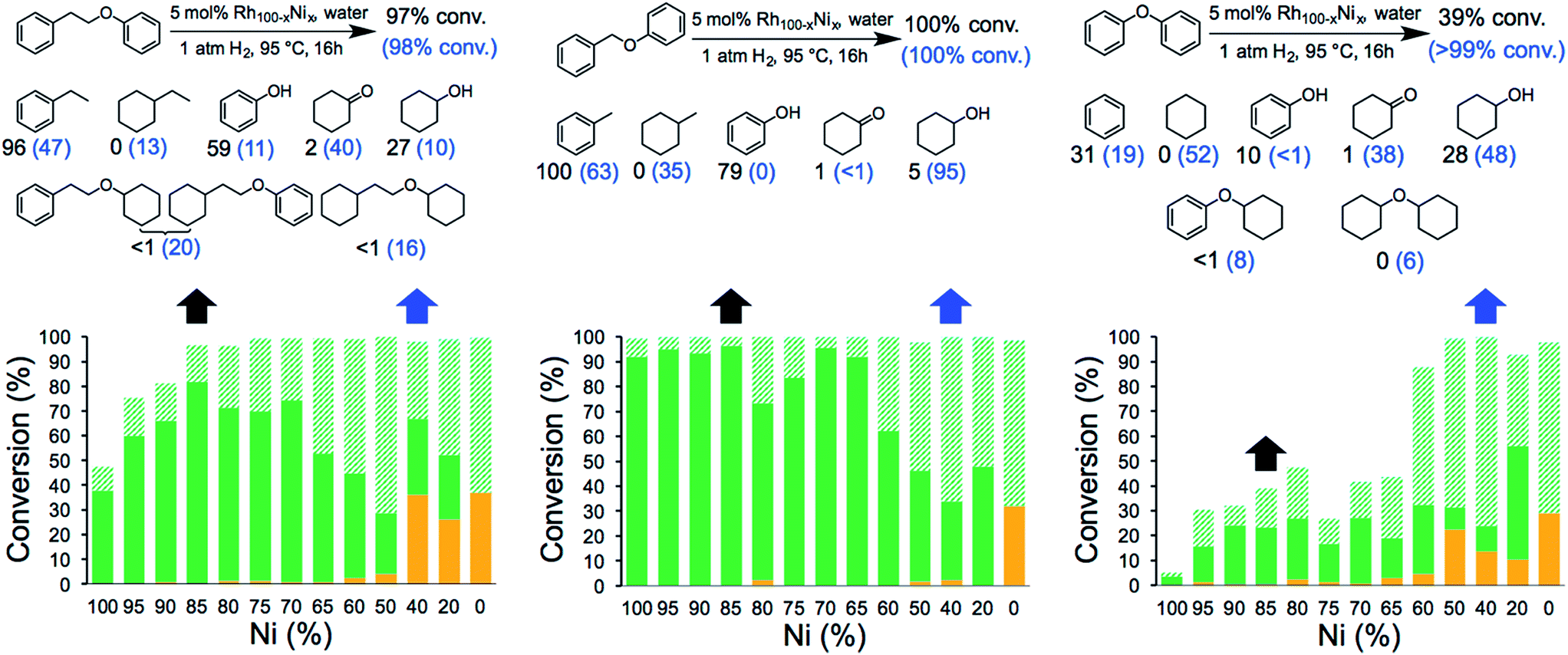 | ||
Fig. 4 Hydrogenolysis/hydrogenation of (left) 1-phenoxy-2-phenylethane (β-O-4 linkage), (middle) benzyl phenyl ether (α-O-4 linkage), (right) diphenyl ether (4-O-5 linkage) and product yield for selected metal combinations catalysed by Rh100–xNix NCs. Full details are provided in Tables S3, S5 and S7.† The black arrows refer to the M15Ni85 NCs and the corresponding yields are in black. The blue arrows refer to the M60Ni40 NCs and the corresponding yields are in blue in parenthesis. The fractions comprise partially/fully hydrogenated dimers ( ), non-hydrogenated monomers ( ), non-hydrogenated monomers ( ) and hydrogenated monomers ( ) and hydrogenated monomers ( ). ). | ||
The bimetallic NCs containing only low amounts of Ru and Rh result in complete conversion of the α-O-4 and β-O-4 linkage containing substrates. In contrast, a relative high molar ratio of the precious metal was required to achieve near-quantitative cleavage of the 4-O-5 bond. Notably, the bimetallic NCs afford higher monomer yields under milder conditions than other reported bimetallic NCs containing Ni.10c
Ethylbenzene is the major product resulting from the cleavage of the β-O-4 bond for all the bimetallic catalyst, in the best case, i.e. with the Rh15Ni85 NC catalyst, reaching close to the maximum (96%). Significant differences in the relative amounts of phenol and cyclohexanol are observed with the different catalysts. In general, the Ru15Ni85 catalyst is able to hydrogenate the phenol ring more efficiently than the Rh15Ni85 catalyst (69 vs. 27% yield, respectively). Raising the content of noble metal to 60% leads to an increase in the total amount of hydrogenated products, indicating that the noble metal is primarily responsible for hydrogenation of the aromatic rings, as observed elsewhere.15 A similar trend was noted for the α-O-4 substrate. Since the β-O-4 and α-O-4 linkages are almost completely converted after 16 h reaction time, the NCs were also evaluated at shorter reaction times, i.e. 4 hours for the β-O-4 linkage and 1 hour for α-O-4 linkage (see Fig. S13†). At these shorter reaction times, mostly monomer products were observed, implying that the hydrogenation of the rings tends to take place after the hydrogenolysis. With the substrates containing the β-O-4 and α-O-4 linkages the best activities were observed for the RuNi NCs containing 85–90 and 75–85% Ni, respectively. In contrast with the RhNi NCs higher conversions are obtained at high loadings of Rh, i.e. 60–80% Rh.
The hydrogenolysis of diphenyl ether is more challenging than the other two substrates due to the higher bond dissociation energy of the 4-O-5 linkage.16 Nevertheless, at higher ratios of noble metal, i.e. M60Ni40 (where M = Ru or Rh), both bimetallic systems favor the formation of monomers (>90%), with cyclohexane and cyclohexanol obtained in 92 and 96% yield, respectively, with the Ru60Ni40 nanocatalyst. At lower noble metal contents substrate conversion is significantly reduced (Fig. 3 and 4). Moreover, these nanocatalysts result in higher monomer yields compared to commercial Ru/C, Rh/C and RANEY® Ni catalysts (Table S1†).
The morphology and composition of the catalyst was analysed after hydrogenolysis (the samples used for this study were taken from the reaction employing diphenyl ether as the substrate). Remarkably, the morphology of Ru15Ni85 and Rh15Ni85 materials is largely preserved (Fig. S7 and S8†). ICP analysis was performed on the Ni100 NCs after washing with water revealing a Ni content of approx. 51 wt% (see ESI†), indicating that the CTAB remains attached to the Ni NCs (since CTAB is soluble in water). As a control experiment, the catalytic activity of the bimetallic NCs M100−xNix (M = Ru or Rh and x = 85 or 50) was compared to monometallic NCs mixed in the same ratio in the transformation of diphenyl ether. The mixed NC systems are significantly less effective catalysts (typically 25–75% lower conversion) compared to the bimetallic NCs (Table S8†).
The standard Gibbs free energies of formation of all substrates and products and the standard Gibbs free energies of the reactions were estimated using a group contribution method. The method is specific to aqueous solution thermodynamics and describes meso- and macroscopic phenomena in the catalytic system that operates in water. The standard Gibbs free energies of all reactants and products were used to calculate standard Gibbs free energies of all the reactions, mapping the main possible pathways with formed intermediates and the corresponding thermodynamic parameters for the catalytic transformation of the model lignin dimers as shown in Fig. 5 (also see Fig. S9 and Table S9†). From the thermodynamic calculations all hydrogenation reactions appear to be less thermodynamically favourable than the hydrogenolysis. Specifically, for the hydrogenolysis of the cyclohexyl-phenyl ether the standard Gibbs energy of reaction showed that the formation of benzene and cyclohexanol are favoured over the formation of cyclohexane and phenol, which is in agreement with the obtained catalytic results.
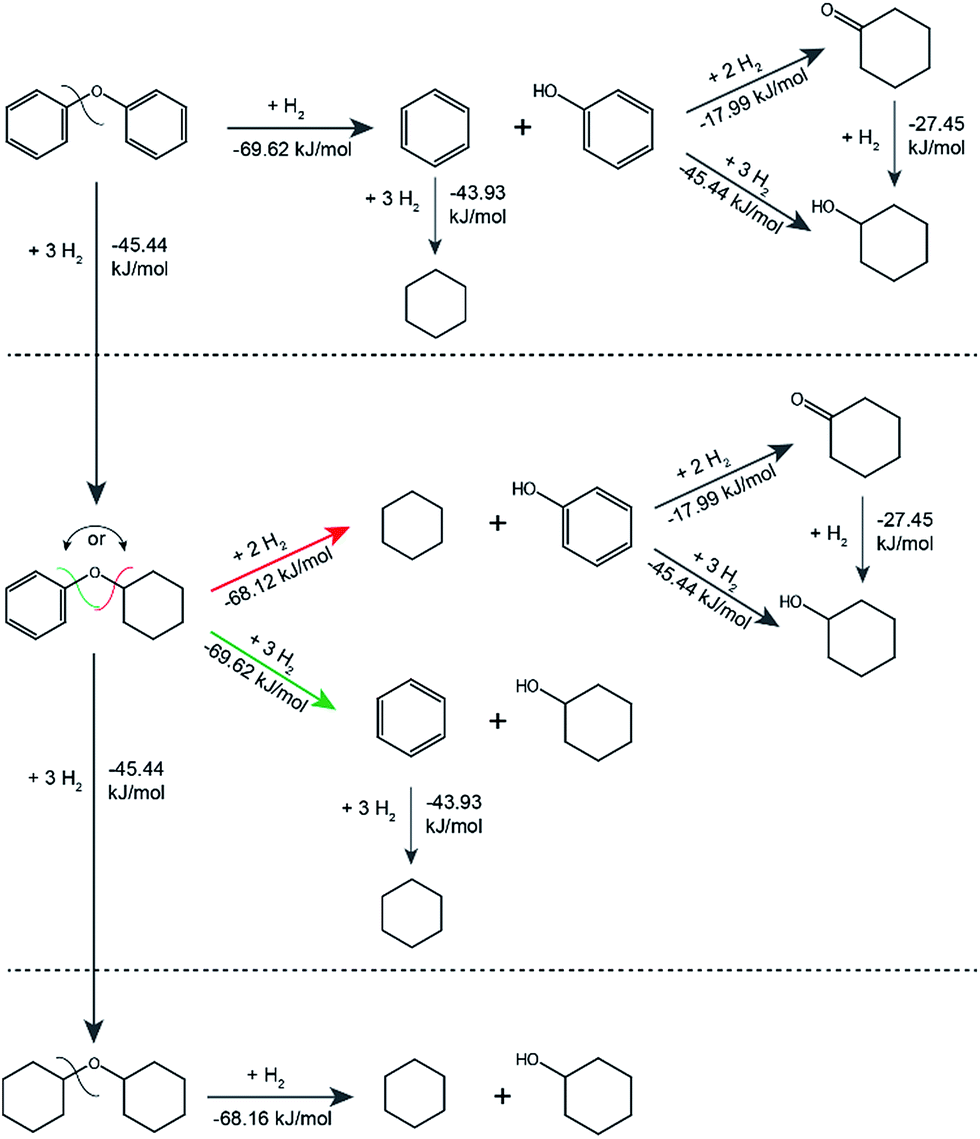 | ||
| Fig. 5 Possible pathways and standard Gibbs free energies of reaction for the hydrogenolysis/hydrogenation of the model dimers (diphenyl ether, cyclohexyl phenyl ether and dicyclohexyl ether) in aqueous solution. | ||
Several substituted ether compounds (with C2-β-O-4 linkages) were also studied and the major products isolated (Fig. 6 and Table S10†). The Ru15Ni85 and Rh15Ni85 nanocatalysts completely convert the biaryl ethers containing hydroxy, keto or methoxy groups into monomers. With the Ru15Ni85 NCs hydrogenation of the arene ring is also observed resulting in cyclohexanol as a main product in 91% and 71% yield for 2-phenoxy-1-phenylethanol and 2-phenoxy-1-phenylethanone, respectively. In contrast, Rh15Ni85 NCs afford 1-(hydroxyethyl)benzene and phenol as the major products, i.e. 78 and 67% yield for 2-phenoxy-1-phenylethanol or 88 and 86% yield for 2-phenoxy-1-phenylethanone, respectively. The presence of ortho-methoxy groups significantly reduces the extent of hydrogenation, which has been noted previously, i.e. with arene hydrogenation catalysts that compare benzene, toluene and methoxybenzene substrates.17 The ability of the M15Ni85 (M = Ru or Rh) NCs to transform beech wood was also validated, albeit under more forcing conditions, leading to a conversion of 57% with the formation of four main aromatic monomers in yields of up to 24% (Table S11†).
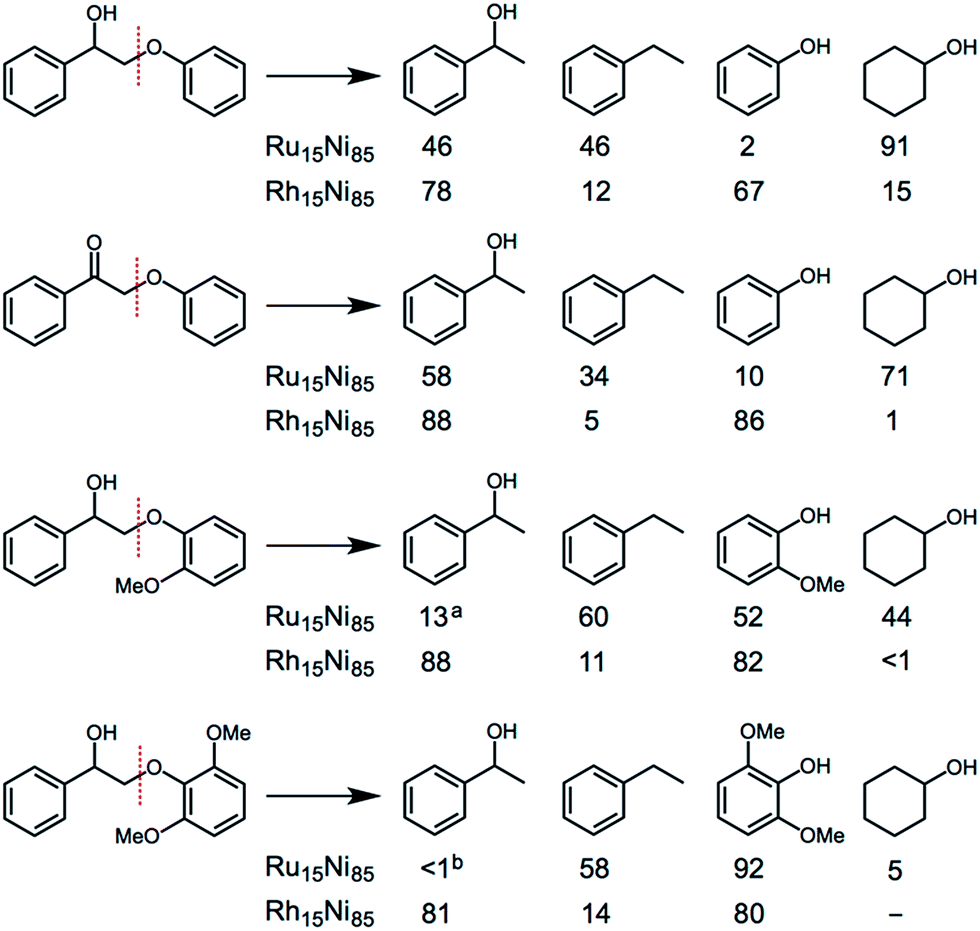 | ||
| Fig. 6 Comparison of the main products obtained from the catalytic transformation of substituted ether compounds using Ru15Ni85 and Rh15Ni85 nanocatalysts. Conditions: aromatic ether (0.189 mmol), catalyst (5 mol%, 0.00945 mmol), H2O (1 mL), H2 (1 atm), 16 h, 95 °C. a,bIn addition the hydrogenated derivative, 1-cyclohexylethanol, was also observed (12%a and 22%b). Full details are provided in Table S10.† | ||
From the catalytic data, it would appear that the Ni is largely responsible for the hydrogenolysis reaction whereas the Ru and Rh are predominantly involved in the hydrogenation of the aromatic rings, as the extent of hydrogenation is higher at higher loadings of the precious metals. Several recent reports describe the hydrogenolysis of various C–O bonds using mostly in situ generated homogenous Ni catalysts.13a,18 Some of these catalysts were used to activate aryl C(sp2)–O bonds in different substrates,18b,18c and others were able to activate both aryl C(sp2)–O and benzylic C(sp3)–O bonds.13a,18d–i It is interesting to establish the activity and selectivity of the catalysts when both bond types are present. Hartwig et al. tested their catalyst on two compounds in the same reaction, one with an aryl C(sp2)–O and the other with a benzylic C(sp3)–O bond, and observed higher performance for the activation of aryl C(sp2)–O bond.13a Similarly, Martin et al. observed the hydrogenolysis of stronger aryl C(sp2)–O bonds in preference to benzylic C(sp3)–O bonds, both present in the same substrate.18d Apparently, the selection of Ni precursors and ligands play an important role for the activation of each bond type. Accordingly, Shi et al. were able to selectively activate one of the bonds present in the same molecule, aryl C(sp2)–O or benzylic C(sp3)–O, by changing the ligands of the Ni catalyst.18i The substrates used in our work contain both type of bonds (the 4-O-5 linkage contains only aryl C(sp2)–O bonds, whereas in α-O-4 and β-O-4 linkages both C(sp2)–O and C(sp3)–O bonds are present). In the presence of both bond types in the α-O-4 and β-O-4 linkages, the Ru100−xNix and Rh100−xNix NCs preferentially catalyse the hydrogenolysis of C(sp3)–O bonds relative to C(sp2)–O bonds (see Tables S2–S5†).
Moreover, following scission of the C–O linkages in the aryl ether substrates, hydrogenation of the aromatic ring in phenol takes place more efficiently than that of ethylbenzene and toluene. This is probably not only due to the intrinsic higher reactivity of the aromatic ring in phenol, but also due to stronger substrate–catalyst interactions, i.e. coordination of the oxygen heteroatom to the metal surface,19 and enhanced mass transfer of phenol since it has a higher polarity and hence solubility in water. Combined, these effects increase the contact time of phenol with the catalyst surface and decreased activation barrier leading to increased levels of hydrogenation.
In summary, we have shown that Ru–Ni and Rh–Ni nanocatalysts coated with CTAB, a phase transfer agent, efficiently catalyze the hydrogenolysis of a variety of C–O bonds in a range of different substrates. These catalysts operate in water in the absence of additional reagents, which is advantageous in terms of sustainability,20 but also ideal for the transformation of substrates present in or derived from biomass.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Swiss National Science Foundation National Research Programme (NRP 66) for financial support.Notes and references
- (a) C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef PubMed; (b) P. J. Deuss and K. Barta, Coord. Chem. Rev., 2016, 306(Part 2), 510–532 CrossRef.
- N. Yan and P. J. Dyson, Curr. Opin. Chem. Eng., 2013, 2, 178–183 CrossRef.
- (a) P. Bi, J. Wang, Y. Zhang, P. Jiang, X. Wu, J. Liu, H. Xue, T. Wang and Q. Li, Bioresour. Technol., 2015, 183, 10–17 CrossRef PubMed; (b) H. Wang, H. Ruan, H. Pei, H. Wang, X. Chen, M. P. Tucker, J. R. Cort and B. Yang, Green Chem., 2015, 17, 5131–5135 RSC; (c) H. Wang, L. Zhang, T. Deng, H. Ruan, X. Hou, J. R. Cort and B. Yang, Green Chem., 2016, 18, 2802–2810 RSC.
- (a) J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top Value-Added Chemicals from Biomass – Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin, Report PNNL-16983, United States, 2007 CrossRef; (b) F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef PubMed; (c) D. Stewart, Ind. Crops Prod., 2008, 27, 202–207 CrossRef; (d) D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 RSC.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 Search PubMed.
- (a) T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC; (b) C. S. Lancefield and N. J. Westwood, Green Chem., 2015, 17, 4980–4990 RSC; (c) N. Yan, C. Zhao, P. J. Dyson, C. Wang, L.-t. Liu and Y. Kou, ChemSusChem, 2008, 1, 626–629 CrossRef PubMed; (d) T. vom Stein, T. den Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 5859–5863 CrossRef PubMed; (e) M. C. Haibach, N. Lease and A. S. Goldman, Angew. Chem., Int. Ed., 2014, 53, 10160–10163 CrossRef PubMed.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. d. Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef.
- (a) Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed; (b) M. W. Jarvis, J. W. Daily, H.-H. Carstensen, A. M. Dean, S. Sharma, D. C. Dayton, D. J. Robichaud and M. R. Nimlos, J. Phys. Chem. A, 2011, 115, 428–438 CrossRef PubMed; (c) A. Beste and A. C. Buchanan, J. Org. Chem., 2009, 74, 2837–2841 CrossRef PubMed; (d) S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846–2852 CrossRef.
- (a) B. Güvenatam, O. Kurşun, E. H. J. Heeres, E. A. Pidko and E. J. M. Hensen, Catal. Today, 2014, 233, 83–91 CrossRef; (b) C. R. Lee, J. S. Yoon, Y.-W. Suh, J.-W. Choi, J.-M. Ha, D. J. Suh and Y.-K. Park, Catal. Commun., 2012, 17, 54–58 CrossRef; (c) V. Molinari, G. Clavel, M. Graglia, M. Antonietti and D. Esposito, ACS Catal., 2016, 6, 1663–1670 CrossRef; (d) J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef; (e) J. Zhang, M. Ibrahim, V. Collière, H. Asakura, T. Tanaka, K. Teramura, K. Philippot and N. Yan, J. Mol. Catal. A: Chem., 2016, 422, 188–197 CrossRef; (f) J. K. Kim, J. K. Lee, K. H. Kang, J. W. Lee and I. K. Song, J. Mol. Catal. A: Chem., 2015, 410, 184–192 CrossRef; (g) Y.-C. Lin, C.-L. Li, H.-P. Wan, H.-T. Lee and C.-F. Liu, Energy Fuels, 2011, 25, 890–896 CrossRef; (h) T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttamaa, F. Ribeiro, J. T. Miller and M. M. Abu-Omar, Chem. Sci., 2013, 4, 806–813 RSC; (i) Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Nat. Commun., 2016, 7, 11162 CrossRef PubMed; (j) P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef PubMed; (k) Z. Li, R. S. Assary, A. C. Atesin, L. A. Curtiss and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 104–107 CrossRef PubMed; (l) J. Lu, M. Wang, X. Zhang, A. Heyden and F. Wang, ACS Catal., 2016, 6, 5589–5598 CrossRef.
- (a) A. K. Deepa and P. L. Dhepe, ACS Catal., 2015, 5, 365–379 CrossRef; (b) X. Huang, C. Atay, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Catal., 2015, 5, 7359–7370 CrossRef; (c) J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef PubMed; (d) S. Kasakov, H. Shi, D. M. Camaioni, C. Zhao, E. Barath, A. Jentys and J. A. Lercher, Green Chem., 2015, 17, 5079–5090 RSC; (e) S. K. Singh and J. D. Ekhe, RSC Adv., 2014, 4, 27971–27978 RSC; (f) W. Song, Y. Liu, E. Barath, C. Zhao and J. A. Lercher, Green Chem., 2015, 17, 1204–1218 RSC; (g) M. D. Karkas, B. S. Matsuura, T. M. Monos, G. Magallanes and C. R. J. Stephenson, Org. Biomol. Chem., 2016, 14, 1853–1914 RSC; (h) Y.-Y. Wang, L.-L. Ling and H. Jiang, Green Chem., 2016, 18, 4032–4041 RSC; (i) X. Wang and R. Rinaldi, Catal. Today, 2016, 269, 48–55 CrossRef; (j) C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef PubMed; (k) P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef PubMed; (l) R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 53, 7310–7315 CrossRef PubMed; (m) T. Prasomsri, M. Shetty, K. Murugappan and Y. Roman-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 RSC; (n) X. Cui, H. Yuan, K. Junge, C. Topf, M. Beller and F. Shi, Green Chem., 2017, 19, 305–310 RSC.
- L. Chen, J. Xin, L. Ni, H. Dong, D. Yan, X. Lu and S. Zhang, Green Chem., 2016, 18, 2341–2352 RSC.
- (a) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef PubMed; (b) A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef PubMed; (c) F. Gao, J. D. Webb and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 1474–1478 CrossRef PubMed.
- N. I. Saper and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 17667–17676 CrossRef PubMed.
- (a) S. Hou, C. Xie, F. Yu, B. Yuan and S. Yu, RSC Adv., 2016, 6, 54806–54811 RSC; (b) X. Cui, A.-E. Surkus, K. Junge, C. Topf, J. Radnik, C. Kreyenschulte and M. Beller, Nat. Commun., 2016, 7, 11326 CrossRef PubMed.
- H. Konnerth, J. Zhang, D. Ma, M. H. G. Prechtl and N. Yan, Chem. Eng. Sci., 2015, 123, 155–163 CrossRef.
- P. J. Dyson, Dalton Trans., 2003, 2964–2974, 10.1039/B303250G.
- (a) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC; (b) J. Cornella, E. Gómez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997–2009 CrossRef PubMed; (c) J. Cornella, P. Jackson Evan and R. Martin, Angew. Chem., Int. Ed., 2015, 54, 4075–4078 CrossRef PubMed; (d) P. Álvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352–17353 CrossRef PubMed; (e) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062–1069 CrossRef PubMed; (f) A. Correa and R. Martin, J. Am. Chem. Soc., 2014, 136, 7253–7256 CrossRef PubMed; (g) C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754–6757 CrossRef PubMed; (h) C. Zarate, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2017, 139, 1191–1197 CrossRef PubMed; (i) B.-T. Guan, S.-K. Xiang, B.-Q. Wang, Z.-P. Sun, Y. Wang, K.-Q. Zhao and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 3268–3269 CrossRef PubMed.
- D. Garcia-Pintos, J. Voss, A. D. Jensen and F. Studt, J. Phys. Chem. C, 2016, 120, 18529–18537 Search PubMed.
- (a) P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef; (b) S. Bulut, Z. Fei, S. Siankevich, J. Zhang, N. Yan and P. J. Dyson, Catal. Today, 2015, 247, 96–103 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, powder XRD and XPS analysis, details for catalytic studies and results, ICP results, TEM analysis of the catalysts (before and after reaction), catalytic results with shorter reaction times and details for thermodynamic calculations. See DOI: 10.1039/c8sc00742j |
| This journal is © The Royal Society of Chemistry 2018 |