
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of microfluidic platforms for the synthesis of metal complexes and evaluation of their DNA affinity using online FRET melting assays†
Viktoria
Rakers
ab,
Paolo
Cadinu
 ab,
Joshua B.
Edel
*ab and
Ramon
Vilar
*ab
ab,
Joshua B.
Edel
*ab and
Ramon
Vilar
*ab
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: joshua.edel@imperial.ac.uk; r.vilar@imperial.ac.uk
bInstitute of Chemical Biology, Imperial College London, London SW7 2AZ, UK
First published on 1st March 2018
Abstract
Guanine-rich DNA sequences can fold into quadruple-stranded structures known as G-quadruplexes. These structures have been proposed to play important biological roles and have been identified as potential drug targets. As a result, there is increasing interest in developing small molecules that can bind to G-quadruplexes. So far, these efforts have been mostly limited to conventional batch synthesis. Furthermore, no quick on-line method to assess new G-quadruplex binders has been developed. Herein, we report on two new microfluidic platforms to: (a) readily prepare G-quadruplex binders (based on metal complexes) in flow, quantitatively and without the need for purification before testing; (b) a microfluidic platform (based on FRET melting assays of DNA) that enables the real-time and on-line assessment of G-quadruplex binders in continuous flow.
Introduction
Microfluidic synthesis offers significant advantages over more conventional bulk strategies in part due to the fluid properties becoming increasingly controlled by viscous forces rather than inertial forces.1,2 Advantages include an increase in the surface-to-volume ratios, possibility to implement machine learning algorithms for real-time monitoring and feedback, high thermal transfer efficiencies, and simple control over reaction conditions using parameters such as reagent flow rates, concentration, temperature and pressure.3 Many examples have been previously reported for both gas and liquid phase microfluidic reactions including fluorination,4,5 Suzuki coupling,6 photochemical reactions7,8 and Swern oxidation9 amongst others. Furthermore, parallel screening of several reactions can also be performed which could have important applications in high-throughput synthesis/screening of new drug candidates.10–12 Herein, we build on these advantages by reporting on a novel in-flow platform for the synthesis and testing of G-quadruplex DNA binders based on nickel(II)–salphen complexes.DNA is a well-established target for a range of different drugs such as antibiotics and anticancer agents.13 Most ligands are exclusively designed to target the canonical form of DNA, namely the double helix (B-DNA).14,15 However, duplex DNA is not particularly accessible since in its ‘resting’ state it is tightly coiled around proteins and packed in chromosomes. The most likely time when small molecules can interact with DNA and disrupt its functions, is when it is being processed, for example during replication and transcription. In these processes, the double helix partially disassembles and when this happens other secondary structures can form. One of these non-canonical DNA structures that has attracted significant interest is the guanine-quadruplex (G4)16,17 which has been associated with essential biological processes including regulation of gene expression, telomere maintenance and genomic instability.18–22In vitro studies identified over 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 distinct sequences in the human genome with the ability to form G4 structures.23 More recently, ChIP sequencing studies have indicated that, from this vast number, approximately 10000 G4 DNA structures can form in vivo, a good proportion of which are present in gene promoters.24 Therefore, there has been significant effort to develop small molecules that can selectively bind and stabilize G4 DNA structures resulting in modulation of biological functions and thus enabling a pharmacological effect and giving rise to a pharmacological effect.25–29 Amongst these molecules, metal complexes have attracted interest due to their unique structural and electronic properties which often make them good G4 DNA binders.30–33 One family of compounds that have been studied as G4 binders are metal salphen and salen complexes; they have been shown to bind G-quadruplex DNA with high affinity and selectivity, and to act as telomerase inhibitors as well as target G4 structures in the promoter of the c-Myc oncogene.34–43
000 distinct sequences in the human genome with the ability to form G4 structures.23 More recently, ChIP sequencing studies have indicated that, from this vast number, approximately 10000 G4 DNA structures can form in vivo, a good proportion of which are present in gene promoters.24 Therefore, there has been significant effort to develop small molecules that can selectively bind and stabilize G4 DNA structures resulting in modulation of biological functions and thus enabling a pharmacological effect and giving rise to a pharmacological effect.25–29 Amongst these molecules, metal complexes have attracted interest due to their unique structural and electronic properties which often make them good G4 DNA binders.30–33 One family of compounds that have been studied as G4 binders are metal salphen and salen complexes; they have been shown to bind G-quadruplex DNA with high affinity and selectivity, and to act as telomerase inhibitors as well as target G4 structures in the promoter of the c-Myc oncogene.34–43
To take full advantage of the benefits of microfluidic synthesis towards such compounds it is important to obtain real-time feedback in order to minimize formation of side products. To date, microfluidic synthesis has only been shown for one synthetic step of an organic G4 binder, namely a triarylpyridine derivative.44 To overcome a series of competing reactions, the authors utilized a spinning disc strategy; however, analysis of the G4 binder was performed offline outside of the microfluidic platform. There have also been some reports where microfluidic platforms have exploited the interaction between G4 DNA and an optical probe for sensing, as well as examples where microfluidic platforms have been used to assess G4 DNA–ligand interactions.45,46
Herein, we present protocols and microfluidic platforms for the efficient one-pot synthesis of metal-salphen DNA binders, followed by on site assessment of their G4-DNA binding capabilities (Fig. 1). To assess the DNA affinity of these compounds, we developed a microfluidic Förster Resonance Energy Transfer (FRET) melting assay with excellent temperature control (i.e. it can be heated and cooled rapidly with ±0.1 °C precision) and real-time fluorescence read-out using laser-induced fluorescence. We show that this strategy enables on-line assessment of G4 binding strength for both commercially available G4 binders and new potent binders which can be prepared in flow in quantitative yields, at lower costs and without the need for purification.
 | ||
| Fig. 1 (a) Schematic representation of the microfluidics-based DNA FRET melting platform. A pre-mixed solution of 0.2 μM FAM-TAMRA labelled c-Myc and 1 μM G4 ligand was allowed to flow through polyethylene tubing at 1–5 μL min−1 (FAM = 6-carboxyfluorescein; TAMRA = 5-carboxy-tetramethylrhodamine). After sufficient time for full mixing and binding, the solution was heated by a Peltier element which was placed on top of a 4 × 6 cm large aluminum block into which the tubing was imbedded. During heating (regulated by a proportional–integral–derivative (PID) controller), the solution was imaged through a quartz capillary (internal diameter = 150 μm). The flowing solution was excited at 488 nm by a 10 mW continuous-wave solid state laser, which is part of a custom-built confocal microscope set-up. A Pt1000 sensor (depicted in red) imbedded in a hole close to the imaging area probes the temperature of the aluminium block. (b) Synthetic scheme for the in-flow preparation of nickel(II) salen and salphen complexes 2–4. A mixture of 2 equivalents of 1, one equivalent of the diamine of choice and 1.1 equivalents of nickel(II) acetate tetrahydrate was injected into PTFE tubing coiled around a heating block set to 90 °C. | ||
Results and discussion
Synthesis of new nickel(II)–salphen complexes 2–4
Since metal salphen complexes have been previously shown to be very potent G4 binders,34,35 a series of three new, pH-independent salphen ligands containing trimethyl ammonium substituents (to increase their water solubility and electrostatic affinity to DNA) were synthesized and used to prepare complexes 2–4 (see Fig. 1). These three metal complexes were initially made following a conventional synthetic protocol as reported earlier for analogous metal–salphen complexes.35 They were characterized by 1H and 13C NMR spectroscopy which showed the expected number of signals with chemical shifts and integration values matching the predictions. Especially indicative of the metal complex formation is the ligand's imine resonance around 8.70 ppm and the disappearance of the aldehyde signal at 10.05 ppm. The formulation and purity of the compounds were further confirmed by mass spectrometry and elemental analyses (see Experimental details).One-pot and in-flow synthesis of complexes 2–4
As the formation of the salphen/salen ligands and the corresponding nickel(II) complexes does not generate any side products (other than water produced in the condensation reaction to form the ligand), they are ideal candidates for synthesis under microfluidic conditions. Prior to carrying out the reaction, we first investigated their one-pot batch synthesis by heating aldehyde 1 with the corresponding diamine for 5 min between 90 and 100 °C, before adding Ni(OAc)2·4H2O in DMSO. These one-pot reactions were carried out in NMR tubes and monitored by 1H NMR spectroscopy, which showed the formation of complexes 2–4 in practically 100% yields and without the need for further purification (see Fig. S5–S7†).Having established that complexes 2–4 can be obtained quantitatively in a one-pot reaction, we investigated their synthesis under microfluidic conditions. Details for the synthesis of 3 as an example are discussed: a mixture of aldehyde 1, 1,2-diaminobenzene and Ni(OAc)2·4H2O was injected into thermostable polytetrafluoroethylene (PTFE) tubing from a syringe and heated between 90 and 100 °C using a solid-state heater around which the tubing was coiled (Fig. 1b). A constant flow rate between 24.5 μL h−1 and 1320 μL h−1 allowed us to probe different reaction times ranging from 10 h to 10 min. The progress of the reactions was monitored by 1H NMR spectroscopy. As can be seen in Fig. 2c, the singlet corresponding to the aldehyde in the starting material (at 10.05 ppm) decreases over time while the resonance associated with the imine present in complex 3 (at 8.75 ppm) increases. After 540 minutes, the only resonances present in the 1H NMR spectrum are those associated with 3 (i.e. all starting materials have been fully consumed). Furthermore, as the reaction progresses, all five aromatic peaks sharpen and the final spectrum does not differ from that of the same product obtained and purified by the conventional synthetic method (see above). The ratio between the characteristic imine and aldehyde resonance in the 1H NMR spectra was used to calculate the product formation and the conversion over time was plotted (see Fig. 2b). This shows a significant conversion in the first few minutes and with the reaction nearing completion after 2.5 h. Fluctuations for the conversion were mostly seen at low reaction times (10 min and 30 min), whereas no deviations between individual repeats were observed for longer reaction times due to the fact that almost full conversion was achieved.
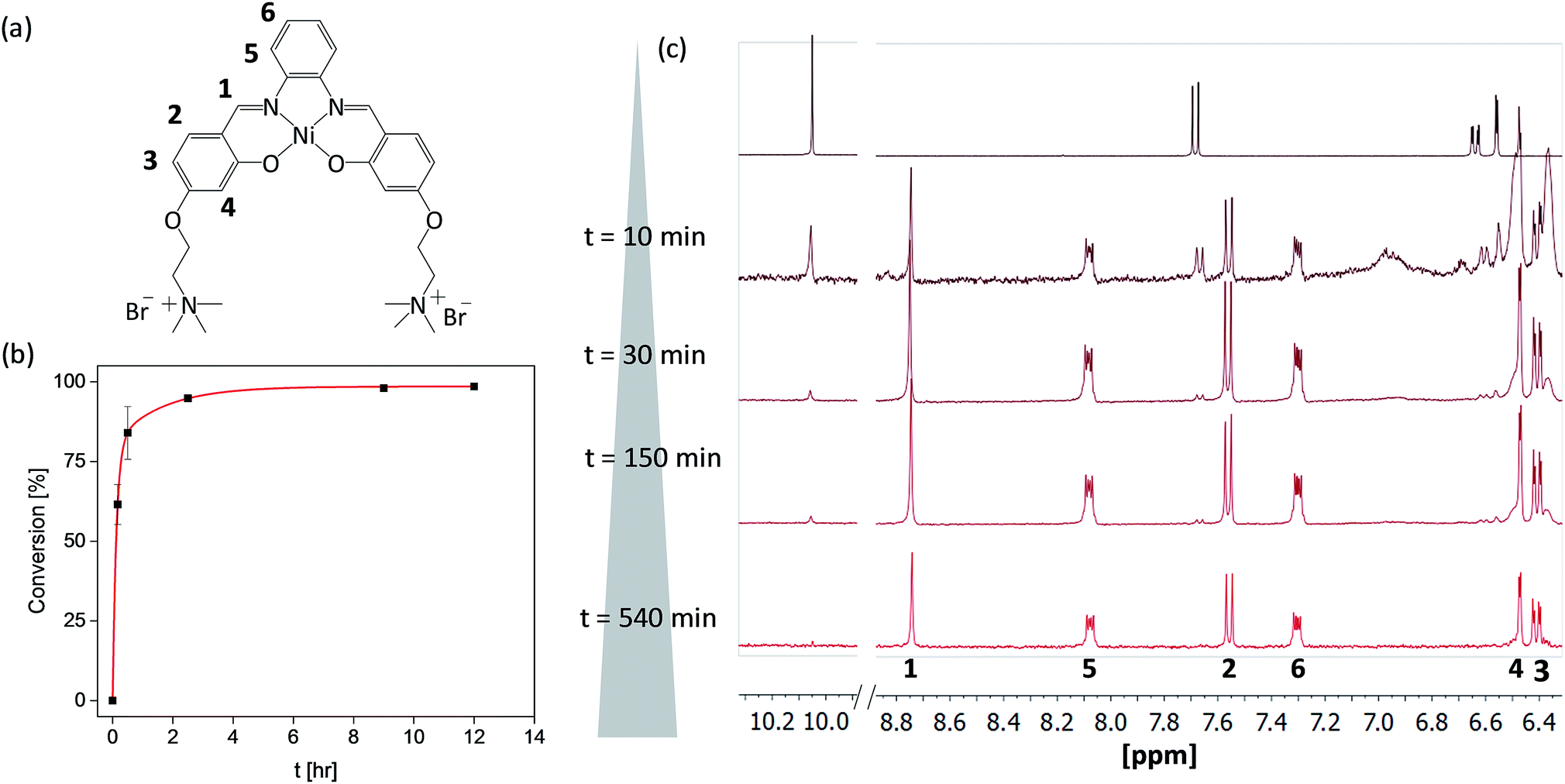 | ||
| Fig. 2 Reaction progress of the in-flow synthesis monitored by 1H NMR spectroscopy (in d6-DMSO), at 296 K. (a) Annotated chemical structure of complex 3; (b) the reaction progress of the in-flow reaction is plotted over time for five independent repeats, the curve is fitted with a biexponential function. (c) 1H NMR spectra – showing starting material 1 (top) and the in-flow reaction mixture after 10, 30, 150 and 540 minutes. | ||
DNA binding properties of complexes 2–4
The affinities of the new nickel(II) complexes 2–4 towards duplex DNA (ds26) and seven different common G-quadruplex sequences, namely c-Myc, CEB26, c-kit1 and c-kit2 (parallel conformation), HTelo (in K+) and Bcl-2 (mixed/hybrid conformation), and 22CTA (antiparallel conformation), were evaluated by performing FRET melting assays. This was carried out using samples of 2–4 prepared by the conventional method. As can be seen in Fig. 3, complexes 3 and 4 have high affinity for G4 DNA structures – in particular complex 3. The thermal stabilization induced by this complex varies between 9.4 °C (for CEB26) and 23.4 °C (for ckit2). Compound 4 appears to stabilize parallel or hybrid structures more strongly than the antiparallel sequence 22CTA (see also Fig. S39–S44†). More importantly, none of the complexes appear to bind to duplex DNA (ds26) under these conditions. The selectivity of complex 3 was investigated further via a FRET melting competition assay with CT DNA (Fig. S38†) and showed that 3 binds selectively to HTelo DNA even in the presence of a 60-fold excess of CT DNA. The nickel(II)–salen complex 2 showed generally low affinity towards G4 DNA (with ΔTm ranging between 0.4 and 9.0 °C), which is likely to be due to its smaller planar delocalised system as compared to 3 and 4. It only stabilized the oncogene promoter derived sequences c-Myc, c-kit2 and Bcl-2 with low to moderate affinity but did not exhibit any significant binding to the G4 sequences CEB26, 22CTA and HTelo (all taken from the telomeric region). | ||
| Fig. 3 ΔTm (°C) values for eight different DNA sequences (including G4 and duplex DNA) in the presence of the three new metal complexes 2–4 synthesised using the conventional method. The ΔTm values were determined (in triplicate) by conventional FRET melting assays using 0.2 μM of oligonucleotide and 1 μM of the compound being tested. | ||
Having established the affinities of 2–4 (prepared by conventional methods) for a range of G4-DNA structures, we carried out similar studies with samples of the three complexes prepared via the one-pot synthesis (Fig. 4). For these studies we used a selection of DNA structures (i.e. c-Myc, HTelo and ds26 DNA). Samples of 2–4 were prepared using the crude reaction mixture (in DMSO) which was diluted with aqueous buffer (pH 7.4) to the appropriate concentration (2 μM, i.e. five-fold as concentrated as the doubly labelled DNA solution). As a control, it was investigated if G4 DNA (c-Myc in this case) is stabilized by residual traces of Ni(OAc)2 or starting material 1. No stabilization was detected by FRET melting experiments (see Fig. S33†) demonstrating that the crude mixture can be used to assess the compounds' DNA affinity.
 | ||
| Fig. 4 ΔTm (°C) values for c-Myc, HTelo and ds26 G4 DNA in the presence of the new metal complexes 2–4 synthesised using the one-pot reaction approach (a) or conventional methods (b). The ΔTm values were determined by conventional FRET melting assays using 0.2 μM of oligonucleotide and 1 μM of the compound being tested. The error bars are shown for at least three repeat measurements. | ||
The FRET melting assays showed that complexes 2–4 have very similar affinities to DNA whether they had been prepared and isolated by the conventional method (Fig. 4b) or prepared in a one-pot batch reaction and used directly from the reaction mixture (Fig. 4a). While there is a slight difference in the absolute values for complexes 4 (but practically none for 2 and 3), the overall trend in behavior is the same for the complexes regardless of the method of synthesis. This validates our approach in which metal salphen complexes can be prepared and tested for DNA binding directly from the “crude” one-pot mixture without the need for purification.
Development of a new microfluidic FRET-melting platform
To enable on-line assessment of the ligand's DNA binding affinity, we developed a microfluidics based FRET melting platform (for orthographic drawing see Fig. S27†). Further details of the design are given in Fig. 1 and are described in the Experimental details.A solution of FAM-TAMRA labelled c-Myc in the presence or absence of 5 equivalents of compound being tested was allowed to flow into the microfluidic channel at a flow rate of 60–300 μL h−1 and a concentration of 200 nM. The flow rate was optimized to ensure thermal stability at the point of detection and to minimize any temperature gradients between the sensor and detection point. As the flow rate correlates with the residence time, low flow rates ensure that the solution can fully equilibrate to the temperature of the Al block. Even at higher temperatures (above ca. 85 °C), low flow rates of 60–300 μL h−1 were still sufficient, and a constant fluorescence signal could be obtained. Control experiments were performed between these flow rates to ensure that there was minimal DNA adsorption to the wall of the channel. This flow regime was also used in a microdroplet based study47 where the FRET efficiency was measured to obtain binding curves between streptavidin and biotin. A concentration of 200 nM of the labelled G4 was used to ensure direct comparisons of the sample with original qPCR based FRET melting studies.48 The data presented in Fig. 5 show minimal fluctuations of the fluorescence signal during the whole recording time at three different temperatures: 29.4 °C and 66.1 °C (i.e. below the G4-DNA melting temperature) and at 88.8 °C (i.e. after the unfolding of G4-DNA). Such high signal stability can only be provided in this type of platform when a homogeneously mixed solution is allowed to flow through an evenly tempered system with strong heat insulation. The temperature sensor indicated a temperature stability of ±0.1 °C. Overall, the standard deviation of the absolute fluorescence intensity for the shown melting curve amounts to 0.58 ± 0.20% of the measured signal.
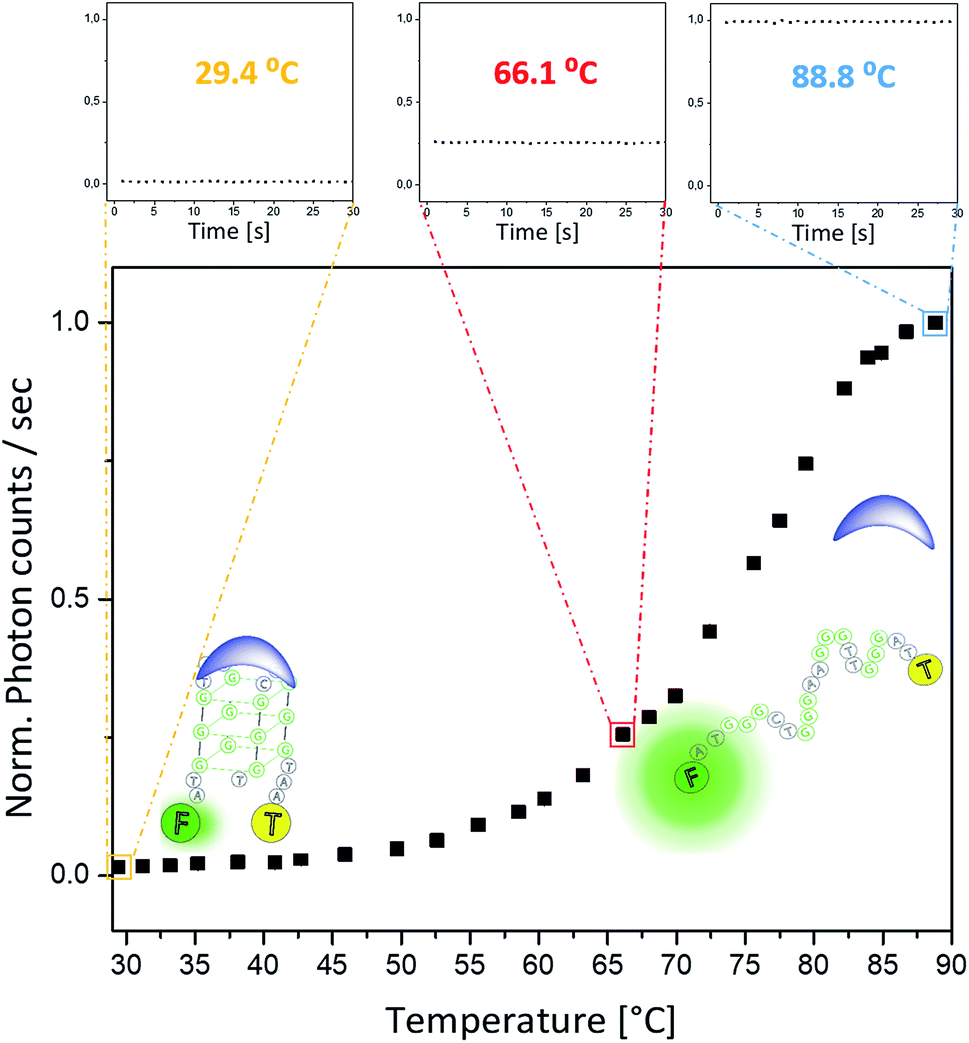 | ||
| Fig. 5 FRET melting curve of 0.2 μM FAM-TAMRA labelled c-Myc G4 DNA (with 1 μM of compound 3) obtained with the new microfluidic platform. At each temperature the fluorescence signal was recorded for 30 s, this is illustrated for three different temperatures reflecting the strong signal stability over time. For all experiments, a buffer containing 1 mM KCl, 99 mM LiCl and 10 mM Li cacodylate (pH 7.4) was used. The normalized fluorescence data are shown here. | ||
Eight different small molecules were tested to validate the platform: four previously reported G4-DNA binders (i.e. BRACO19, TMPyP4, PDS and PhenDC3 – see Fig. 6 for chemical structures), our three new nickel–salphen complexes (2–4) and, as a negative control, a compound known not to bind to G4 DNA (i.e. TMPyP2 – see Fig. 6 for the chemical structure). In all experiments, a 1:5 ratio of DNA to ligand was used and the Tm values were determined by normalizing the fluorescence signal, fitting them to a dose–response curve and reading out the melting temperature where the normalized photon counts are 0.5. All FRET melting curves are sigmoidal and, for technical repeats, Tm values showed an average standard deviation of 1.5 °C (3 to 9 repeats). While this is higher than the average standard deviation (ca. 0.5 °C) for conventional FRET melting experiments, it is sufficient to rank ligands for their affinity to G4-DNA. The ΔTm values obtained with our platform for the five previously reported compounds are consistent with the recent FRET melting data reported by Mergny, where the best G4 DNA binders are TMPyP4 and PhenDC3, followed by BRACO19 and PDS.49 As expected, TMPyP2 – negative control – did not display stabilization of G4 DNA. Regarding the new nickel(II) complexes (prepared using the ‘one pot’ synthesis and, in the case of 3, also by conventional methods), 3 and 4 show high G4-DNA affinity (as expected for square-planar metal salphen complexes) while complex 2 is a poor G4 DNA binder (Fig. 6d).
 | ||
| Fig. 6 Platform for FRET melting data. (a) FRET melting curves with 0.2 μM FAM-TAMRA labelled c-Myc and 1 μM of six different ligands (i.e. TMPyP4, BRACO19, PDS, compound 3, PhenDC3 and TMPyP2 as a negative control); (b) bar chart showing ΔTm values; (c) FRET melting curves with 0.2 μM c-Myc and 1 μM of compounds 2–4 prepared via the one-pot method; (d) bar chart showing the ΔTm values which were obtained either by conventional (PCR) methods or our microfluidic platform for compounds 2–4 (prepared via the one-pot synthesis). Error bars are shown for three repeat measurements. | ||
We used our three new nickel(II) complexes – prepared via the “one-pot” method – to compare the FRET melting data in continuous flow with that obtained via conventional qPCR methods. As can be seen in Fig. 6c and d, the ΔTm values are very similar using either of the two FRET melting assay methods. Deviations were only observed for compound 2 which did not show any stabilization of c-Myc DNA when analysed with our microfluidic platform, whereas conventional methods suggest a low stabilization of G4 DNA of ca. 4 °C. However, the overall trend is clearly in accordance with conventional methods and proves that our novel platform facilitates a real-time assessment of the binding strength of nickel(II) salphen-based G4 binders which were synthesized in a one-pot manner without the need for any purification.
Conclusions
We have synthesized three new square-planar nickel(II)–salphen complexes two of which are very good G-quadruplex DNA binders and display high selectivity over duplex DNA. Furthermore, we demonstrate that these G4 binders can be prepared via a one-pot synthesis without the need for purification, which has allowed us to prepare the compounds quantitatively in continuous flow. This in-flow platform utilizes very small volumes (ca. 350 μL per synthesis) and operates over a large temperature range. These advantages open the possibility of synthesizing large libraries of compounds over a short period of time and with very small amounts of reagents. Furthermore, the reaction time can be easily fine-tuned by adjusting the flow rate. Independent of the method of synthesis, similar G4-DNA ΔTm values (assessed by FRET melting assays) were obtained for all three nickel(II)–salphen complexes. While there are some differences in the ΔTm values determined for the compounds synthesized by the conventional and microfluidic approaches, our new in-flow platform can be reliably used as a first semiquantitative test to rank the affinity of small molecules towards G4-DNA. We have also developed a novel DNA FRET-melting microfluidics platform which combines laser induced fluorescence spectroscopy with microfluidics at various temperatures; excellent stability of both temperature and fluorescence signal was achieved. This is also the first example where a microfluidic platform has been used to carry out G4-DNA FRET melting assays in continuous flow, which we have successfully applied to assess the DNA affinity of eight compounds (five previously reported and the three new nickel(II) complexes 2–4). However, further developments will be required to reduce the average standard deviation of the ΔTm values and, consequently, realize the full potential of our new microfluidics approach to assessing G4-DNA binding.In summary, we have successfully developed two microfluidic platforms to synthesize new small molecules and assess their DNA binding affinity. Further development will focus on combining these platforms and introducing feedback control to optimize the synthesis of stronger DNA binders.
Experimental details
Materials and general protocols
All reagents and solvents were purchased from commercial suppliers and used without further purification. All DNA sequences (labelled or unlabelled) were purchased RP-cartridge purified from Eurogentec (Belgium).1H and 13C NMR spectra were recorded using a 400 MHz Bruker Avance Ultrashield NMR spectrometer (Imperial College London, Department of Chemistry, NMR Service Facility) at 296 K. Chemical shifts are given in parts per million (ppm, δ) and referenced to the residual deuterated solvent. Coupling constants were measured in Hz. Splitting patterns are designated as s for singlet, d for doublet, t for triplet, q for quartet, m for multiplet and br for broad. Electrospray (ES) and Liquid Chromatography (LC-ES) mass spectra were obtained on a Waters LCT Premier (ES-TOF)/Acquity i-Class spectrometer by Dr Lisa Haigh (Imperial College, Chemistry Department). Elemental (C, H, N) analyses were performed by Mr A. Dickerson; the values reported are the average of two separate measurements (Cambridge University). Platform fabrication was carried out by the technical staff in the Department of Bioengineering, Imperial College London.
Conventional FRET measurements48,49
All DNA strands tested in the fluorescence resonance energy transfer (FRET) assay were labelled with a fluorescent donor (FAM: 6-carboxyfluorescein) at their 5′ end and a fluorescent acceptor (TAMRA: 5-carboxy-tetramethylrhodamine) at their 3′ end. The solid stock was dissolved in milli-Q water at a concentration of 100 μM, as determined by UV-vis spectroscopy at 260 nm and diluted to 20 μM in the appropriate buffer. The sequences shown in Table 1 were used.| Sequence name | Sequence 5′–3′ | ε/L mol−1 cm−1 |
|---|---|---|
| c-Myc | FAM-GAG-GGT-GGG-GAG-GGT-GGG-GAA-G-TAMRA | 232000 |
| HTelo | FAM-AGG-GTT-AGG-GTT-AGG-GTT-AGG-G-TAMRA | 228500 |
| Ds26 | FAM-CAA-TCG-GAT-CGA-ATT-CGA-TCC-GAT-TG-TAMRA | 253200 |
| c-kit1 | FAM-AGG-GAG-GGC-GCT-GGG-AGG-AGG-G-TAMRA | 226700 |
| c-kit2 | FAM-CGG-GCG-GGC-GCG-AGG-GAG-GGG-TAMRA | 205600 |
| CEB26 | FAM-AAG-GGT-GGG-TGT-AAG-TGT-GGG-TGG-GT-TAMRA | 265100 |
| 22CTA | FAM-AGG-GCT-AGG-GCT-AGG-GCT-AGG-G-TAMRA | 220400 |
| bcl-2 | FAM-GGG-CGC-GGG-AGG-AAG-GGG-GCG-GG-TAMRA | 231300 |
The FAM-TAMRA labelled G4-DNA (0.4 μM) was obtained by heating the oligonucleotide at 95 °C for 5–10 min in annealing buffer (1 mM KCl, 99 mM LiCl, 10 mM LiCacodylate, pH 7.4 for c-Myc and ds26, 100 mM KCl, 10 mM LiCacodylate, pH 7.3 for bcl-2 and 10 mM KCl, 90 mM LiCl, 10 mM LiCacodylate, pH 7.4 for c-kit1, c-kit2, CEB26, 22CTA and h-Telo), followed by slow cooling over 4 h. All compounds were stored as DMSO stock solutions at 0.5 mM. For all binding studies samples were prepared freshly by diluting the corresponding DMSO stock solution at least 100 times in cacodylate buffer (see above) on the day of use. The labelled DNA was used at a concentration of 200 nM. To each well in a 96 well plate (Agilent Technologies, UK), 20 μL of 0.4 μM FAM-TAMRA labelled DNA and 20 μL of the 1 μM ligand solution were added. As a control, the FRET melting assays were also carried out in the absence of the complex solutions. Measurements were carried out in triplicates on a Stratagene Mx3005P qPCR machine (Agilent Technologies) equipped with a 96 well plate, a quartz tungsten halogen lamp as the excitation source, a single photomultiplier tube for detection and a Peltier-based thermal cycler. For the FAM-TAMRA system an excitation range of 492–516 nm and an emission range of 556–580 nm were used. With a temperature increase of 1 °C min−1 fluorescence readings were taken at 1 °C intervals from 25 °C to 95 °C. Data analysis was carried out with the program OriginPro 9.1 (OriginLab Corp., Northampton, MA 01060 USA). The melting temperature was calculated by fitting the normalized fluorescence data with a sigmoidal function using a dose-response model and checking the temperature value for y = 0.5. ΔTm was reported as the difference between the DNA melting temperature in the absence of any compound and the DNA melting temperature at a fivefold excess of the compound.
FRET competition assay
A FRET competition assay, adapted from the literature,50 was carried out to shed light on the selectivity for G4 over duplex DNA. A stock solution of the annealed FAM-TAMRA labelled G4 (c-Myc or HTelo) (0.4 μM) was prepared. The solid ctDNA stock (Sigma) was dissolved in the appropriate buffer (1 mM KCl, 10 mM LiCac, 99 mM LiCl, pH 7.4 for competition with c-Myc; 10 mM KCl, 10 mM LiCac, 90 mM LiCl, pH 7.4 for competition with HTelo), the ctDNA concentration was determined per base pair (ε(260 nm) = 13200 L mol−1 cm−1) by UV-vis spectroscopy. Ligand 3 was diluted to 4 μM in the appropriate buffer. Five solutions of ctDNA mixed with the ligand were prepared (2 μM ligand + 1.2 μM ctDNA; 2 μM ligand + 12 μM ctDNA; 2 μM ligand + 120 μM ctDNA; 2 μM ligand + 240 μM ctDNA in the appropriate buffer). This solution was then mixed with FAM-TAMRA labelled G4 to give a G4 concentration of 0.2 μM, a final ligand concentration of 1 μM and varying ctDNA concentrations (0 to 120 μM). Measurements were performed in triplicates under the same conditions as the FRET melting assay.
One-pot synthesis
All one-pot reactions were performed in NMR tubes which were heated up in an oil bath on a hot plate, whereas all in-flow reactions were performed in Microbore PTFE tubing (0.022′′ ID × 0.042′′ OD, Cole Parmer, UK) coiled around a solid state heater. For the two-step one-pot approach, 2 eq. of 4-(4-formyl-3-hydroxyphenoxy)-N,N,N-trimethylethan-1-ammonium bromide (1) (400 μL of a 30 mM stock solution) and 1 eq. of the corresponding diamine (6 μL of a 1 M stock solution) were heated at 90 °C for 5–30 min in DMSO; subsequently, 1.1 eq. Ni(OAc)2·4H2O (6.6 μL of a 1 M stock solution) were added. The final mixture was incubated for at least another 10 h at 90 °C and monitored at different time points by 1H NMR.In the case of the one-step in-flow approach, all three reagents, i.e. 200 μL of a 80 mM stock solution of 4-(4-formyl-3-hydroxyphenoxy)-N,N,N-trimethylethan-1-ammonium bromide (1) with 8 μL of a 1 M stock solution of the diamine and 8.8 μL of a 1 M stock solution of Ni(OAc)2·4H2O, were premixed in DMSO at the described ratio and transferred into a 1 mL syringe (VWR, UK). The mixture was injected into PTFE tubing which was coiled around a solid state heater and fixed with Kapton tape. The temperature was monitored with a K-type sensor inside the heater and controlled with a PID controller (ESM-4420, EMKO). 1 m of the PTFE tubing corresponds to a volume of 245.3 μL, so the heating time of the reaction mixture could be set by adjusting the flow rate (usually between 5 and 15 μL h−1). The reaction mixture was left overnight. After heating, all “one pot” or in-flow reactions could be monitored by 1H NMR spectroscopy. The disappearance of the singlet at ca. 10 ppm corresponding to the aldehyde proton in the starting material indicated full conversion.
Synthesis of 4-(2-bromoethoxy)-2-hydroxybenzaldehyde
This compound was prepared as previously reported in the literature.51 The product was obtained as a white powder (2.10 g, 8.6 mmol, 11.8%). 1H NMR (400 MHz, CDCl3) δH (ppm) = 3.67 (t, 3JHH = 6 Hz, 2H, CH2–Br), 4.37 (t, 3JHH = 6 Hz, 2H, OCH2), 6.45 (d, 3JHH = 2 Hz, 1H, ArH), 6.60 (dd, 3JHH = 8.4 Hz, 4JHH = 2.4 Hz, 1H, ArH), 7.49 (d, 3JHH = 8.4 Hz, 1H, ArH), 9.76 (s, 1H, CHO), 11.49 (s, 1H, OH). MS (EI+) m/z 243.97 [M+], where M = C9H9BrO3, calc. mass for C9H9BrO3: 243.97, found: 243.97.Synthesis of 4-(4-formyl-3-hydroxyphenoxy)-N,N,N-trimethylethan-1-ammonium bromide (1)
This compound was prepared by small modifications of a previously reported procedure.51 4-(2-Bromoethoxy)-2-hydroxybenzaldehyde (2.10 g, 8.6 mmol, 1 eq.) was dissolved in a 4.2 M ethanolic solution of trimethylamine (110 mL). The yellow solution was heated at 50 °C with stirring overnight. The solvent was removed and the obtained orange-yellow solid was washed with chloroform (3 × 20 mL) to obtain the product as a pale yellow solid (2.74 g, 8.5 mmol, 98.8%). Elem. anal. C12H18BrNO3(H2O) calc. % C 44.73, % H 6.26, % N 4.35 found. % C 44.35, % H 6.17, % N 4.36. 1H NMR (400 MHz, DMSO-d6) δH (ppm) = 3.18 (s, 9H, (CH3)3), 3.81 (t, 3JHH = 4.4 Hz, 2H, CH2–N), 4.54 (t, 3JHH = 4.4 Hz, 2H, OCH2), 6.56 (d, 3JHH = 2.4 Hz, 1H, ArH), 6.64 (dd, 3JHH = 8.8 Hz, 4JHH = 2.4 Hz, 1H, ArH), 7.68 (d, 3JHH = 8.8 Hz, 1H, ArH), 10.05 (s, 1H, CHO), 11.08 (s, 1H, OH). 13C NMR (400 MHz, DMSO-d6): δC 53.6, 62.5, 64.4, 102.3, 108.2, 117.2, 132.7, 163.4, 164.4, 191.7. MS (ES+) m/z 224.1 [M − Br−]+, where M = C12H18Br2NO3.Synthesis of complex 2
A mixture of compound 1 (0.17 g, 0.60 mmol, 2 eq.) and ethylenediamine (0.02 g, 0.29 mmol, 1 eq.) in methanol (20 mL) was stirred at reflux for 1 h under a nitrogen atmosphere. Ni(OAc)2·4H2O (0.07 g, 0.29 mmol, 1 eq.) was added to the orange mixture. The reaction mixture was heated to reflux overnight under nitrogen, during which time it turned red. The reaction was then cooled down to room temperature and diethyl ether (ca. 80 mL) was added to precipitate out an orange-red solid. The precipitate was filtered and washed with copious amounts of diethylether (ca. 100 mL) to afford 2 as an orange-red solid (yield: 0.13 g, 0.2 mmol, 62%). C26H38Br2N4NiO4 (3.5H2O) calc. % C 41.52, % H 6.03, % N 7.45 found. % C 41.54, % H 5.64, % N 7.34.1H NMR (400 MHz, MeOD): δH (ppm) = 3.17 (s, 18H; N(CH3)3+), 3.37 (t, 3JHH = 0.8 Hz, 4H; CH2-bridge), 3.77 (t, 3JHH = 4.8 Hz, 4H; CH2–N(CH3)3+), 4.43 (t, 3JHH = 4.8 Hz, 4H; OCH2), 6.23 (dd, 3JHH = 8.8 Hz, 4JHH = 2.8 Hz, 2H; ArH), 6.30 (d, 3JHH = 2.4 Hz, 2H; ArH), 7.23 (d, 3JHH = 8.8 Hz, 2H; ArH), 7.79 (s, 2H; –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–). 13C NMR (400 MHz, DMSO-d6): δC 53.6, 58.3, 61.8, 64.4, 103.0, 105.4, 115.7, 134.4, 162.0, 162.3, 166.2. MS (ES+) m/z 264.3 [M − 2Br−]2+, where M = C26H38Br2N4NiO4. ε432 nm (in DMSO) = 3374.67 ± 244.84 L (mol−1 cm−1).
N–). 13C NMR (400 MHz, DMSO-d6): δC 53.6, 58.3, 61.8, 64.4, 103.0, 105.4, 115.7, 134.4, 162.0, 162.3, 166.2. MS (ES+) m/z 264.3 [M − 2Br−]2+, where M = C26H38Br2N4NiO4. ε432 nm (in DMSO) = 3374.67 ± 244.84 L (mol−1 cm−1).
Synthesis of complex 3
A mixture of compound 1 (0.11 g, 0.37 mmol, 2 eq.) and 1,2-diaminobenzene (0.02 g, 0.19 mmol, 1 eq.) in methanol (30 mL) was stirred at reflux for 1 h in nitrogen. Ni(OAc)2·4H2O (0.05 g, 0.19 mmol, 1 eq.) was added to the orange mixture. The reaction mixture was heated to reflux overnight under nitrogen, during which time it turned red. The reaction mixture was then cooled down to room temperature and the solvent was removed under reduced pressure. The remaining solid was dissolved in a small amount of methanol (10 mL) and the product was precipitated out with diethyl ether (ca. 60 mL). The precipitate was filtered and washed with copious amounts of diethyl ether (ca. 100 mL) and pentane (ca. 50 mL) to afford 3 as a red solid (yield: 0.09 g, 0.12 mmol, 63%). Elem. anal. C30H38Br2N4NiO4·(2H2O) calc. % C 46.60, % H 5.48, % N 7.25 found. % C 46.50, % H 5.47, % N 7.03. 1H NMR (400 MHz, DMSO-d6): δH (ppm) = 3.20 (s, 18H; N(CH3)3+), 3.83 (t, 3JHH = 4 Hz, 4H; CH2– N(CH3)3+), 4.51 (t, 3JHH = 4 Hz, 4H; OCH2), 6.4 (dd, 3JHH = 8.8 Hz, 4JHH = 2.4 Hz, 2H; ArH), 6.47 (d, 3JHH = 2 Hz, 2H; ArH), 7.26–7.31 (m, 2H; ArH), 7.57 (d, 3JHH = 8 Hz, 2H; ArH), 8.07–8.11 (m, 2H; ArH), 8.76 (s, 2H; –CHN–). 13C NMR (400 MHz, DMSO-d6): δC 53.6, 62.1, 64.3, 102.7, 107.6, 115.8, 116.3, 127.4, 136.0, 142.8, 155.4, 163.7, 167.6. MS (ES+) m/z 288.3 [M − 2Br−]2+, where M = C30H38Br2N4NiO4. ε452 nm (in DMSO) = 16183.33 ± 366.89 L (mol−1 cm−1).
Synthesis of complex 4
A mixture of compound 1 (0.14 g, 0.42 mmol, 2 eq.) and 2,3-diaminonaphthalene (0.03 g, 0.21 mmol, 1 eq.) in methanol (20 mL) was stirred at reflux for 1 h in a nitrogen atmosphere. Ni(OAc)2·4H2O (0.06 g, 0.23 mmol, 1 eq.) was added to the orange mixture. The reaction mixture was heated up to reflux overnight under nitrogen, during which time it turned dark red. The reaction mixture was then cooled down to room temperature and the solvent was removed under reduced pressure. The solid was re-dissolved in a small amount of methanol (10 mL) and the product was precipitated out with diethyl ether (ca. 60 mL). The precipitate was filtered and washed with copious amounts of diethyl ether (ca. 100 mL) to obtain the product 4 as a brownish/black solid (yield: 0.13 g, 0.2 mmol, 93%). Elem. anal. C34H40Br2N4NiO4·(4H2O) calc. % C 47.52, % H 5.63, % N 6.52 found. % C 47.89, % H 5.44, % N 6.34. 1H NMR (400 MHz, DMSO-d6): δH (ppm) = 3.19 (s, 18H; N(CH3)3+), 3.82 (t, 3JHH = 4.4 Hz, 4H; CH2–N(CH3)3+), 4.52 (t, 3JHH = 4 Hz, 4H; OCH2), 6.43 (dd, 3JHH = 8.8 Hz, 4JHH = 2.4 Hz, 2H; ArH), 6.47 (d, 3JHH = 2.4 Hz, 2H; ArH), 7.53–7.56 (m, 2H; ArH), 7.58 (d, 3JHH = 9.2 Hz, 2H; ArH), 7.88–7.90 (m, 2H; ArH), 8.56 (s, 2H; ArH), 8.92 (s, 2H; –CHN–). 13C NMR (400 MHz, DMSO-d6): δC 53.6, 62.1, 64.3, 102.7, 107.8, 113.4, 116.1, 127.1, 128.2, 132.0, 135.9, 142.1, 155.9, 163.9, 167.8. MS (ES+) m/z 313.3 [M − 2Br−]2+, where M = C34H40Br2N4NiO4. ε449 nm (in DMSO) = 18944.46 ± 165.67 L (mol−1 cm−1).
FRET melting platform experiments
An aluminum mould was designed with the AutoCad software (Autodesk Inventor, 2015) and fabricated at the Department of Bioengineering at Imperial College London. A serpentine shaped deepening allowed inserting the polyethylene tubing (TUB3656, ID = 0.38 mm, OD = 1.09 mm, Smiths Medical International, UK) which was connected to a quartz tubing (TSP150375, ID = 150 μm, Polymicro Technologies, CM Scientific, BD200DL Silsden, UK) at the imaging position. The polyimide coating of the quartz tubing was removed with a flame beforehand, followed by thorough washing with hexane and acetone, and plasma cleaning (PDC-001, Harrick Plasma, USA). Due to its strong auto-fluorescence it is crucial that the coating is removed completely. The quartz tubing was imaged through a hole with a 10× objective at the end of the serpentine. A PT1000 sensor (Farnell, SENSOR, PT1000, 600 °C, CLASS A) was embedded in a small cavity in the Al mould and covered with PDMS to make sure it stayed at the same position during all experiments. A thermoelectric module (Peltier, sealed, 20 V, 6.7 A, 40 × 40 × 3.3, 71C) together with a heat sink was placed on the top of the Al mould. Both the PT1000 sensor and the thermoelectric module were connected to a PID controller (Liard Technologies, TC-XX-PR-592, 926-1066-ND), which controlled the temperature. It was connected to a computer via RS232 standard and supplied with DC current via a power supply. For temperature regulation PD control was used, with U = 14.1 V and max. power output between 6% and 60%. The temperature of the flowing fluid was increased at intervals of ca. 2–3 °C within a range of 25 °C to 90 °C. For all continuous flow measurements, the analyte flow rate was set to 1–5 μL min−1 by a syringe pump (P-2000, Harvard Apparatus, USA). All FAM-TAMRA labelled DNA was diluted to a concentration of 50–200 nM in annealing buffer (1 mM KCl, 99 mM LiCl, 10 mM LiCac, pH 7.4). For all continuous flow measurements with various compounds, a DNA concentration of 0.2 μM and a compound concentration of 1 μM were used, unless stated otherwise. The DNA–compound mixture was incubated for 10 min at RT before being allowed to flow through the platform. Measuring one FRET melting curve took about 60 min which corresponds to a sample usage of less than 100 μL. The most time- and sample-consuming facet of our platform experiments is the initial equilibration which can take up to 2 h. This is a common disadvantage in microfluidics, whereas equilibration times of no more than 5 min are normally used on a quantitative PCR apparatus. In order to avoid hysteresis phenomena, a slow temperature ramp up of about 2 °C min−1 was chosen. This is higher than in conventional methods which normally increase the temperature at a rate of 0.5 °C/30 s. However, hysteresis is not an issue in the regime we employed. Between different experiments, the tubing was washed with copious amounts of DI water and mild H2SO4 acid. All measurements were conducted using a previously reported custom-built confocal microscope.52 Briefly the setup consists of a 488 nm, 10 mW continuous-wave solid state laser (Sapphire 488LP, Coherent) coupled to an Olympus IX71 inverted microscope. The laser is reflected onto a 498 nm dichroic mirror into the back aperture of the objective (10×). The fluorescence is collected through the same objective and transmitted through the dichroic mirror and a 505 nm long pass emission filter before being focused onto a 75 μm pinhole (P75S, ThorLabs) which ensured the blockage of out-of-focus light. A further dichroic mirror (630DCXR, Chroma) was used to split fluorescence emission into two bands, 500–580 nm and 640–800 nm before detection by two avalanche photodiodes (SPCM-AQR-14, PerkinElmer). For all measurements a laser power between 2 and 10 μW was used. LabView 8.5 was used for optical data acquisition and a custom-written Matlab program (developed in-house) was used to extract the intensity data and their standard deviation. Further data analysis was carried out with OriginPro 9.1 (OriginLab Corp., Northampton, MA 01060 USA). The melting temperature was calculated by fitting the normalized fluorescence data with a sigmoidal function using a dose–response model and checking the temperature value for y = 0.5.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Imperial College for a President's PhD Scholarship (V. R.) and Dr James Bannock for advice on the heating element of the microfluidic platform. J. B. E. has been funded in part by an ERC Consolidator grant (NanoPD).References
- C. Wiles and P. Watts, Eur. J. Org. Chem., 2008, 10, 1655–1671 CrossRef.
- A. J. Andrew, Nature, 2006, 442, 394–402 CrossRef PubMed.
- Y. Liu and X. Jiang, Lab Chip, 2017, 17, 3960–3978 RSC.
- R. D. Chambers and R. C. H. Spink, Chem. Commun., 1999, 2, 883–884 RSC.
- R. D. Chambers, M. a. Fox and G. Sandford, Lab Chip, 2005, 5, 1132–1139 RSC.
- G. M. Greenway, S. J. Haswell, D. O. Morgan, V. Skelton and P. Styring, Sens. Actuators, B, 2000, 63, 153–158 CrossRef CAS.
- H. Lu, M. A. Schmidt and K. F. Jensen, Lab Chip, 2001, 1, 22–28 RSC.
- R. C. R. Wootton, R. Fortt and A. J. De Mello, Org. Process Res. Dev., 2002, 6, 187–189 CrossRef CAS.
- T. Kawaguchi, H. Miyata, K. Ataka, K. Mae and J. I. Yoshida, Angew. Chem., Int. Ed., 2005, 44, 2413–2416 CrossRef CAS PubMed.
- H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704–5708 CrossRef CAS PubMed.
- J. Wang, G. Sui, V. P. Mocharla, R. J. Lin, M. E. Phelps, H. C. Kolb and H. R. Tseng, Angew. Chem., Int. Ed., 2006, 45, 5276–5281 CrossRef CAS PubMed.
- E. Garcia-Egido, V. Spikmans and B. H. Warrington, Lab Chip, 2003, 3, 73–76 RSC.
- A. Ali and S. Bhattacharya, Bioorg. Med. Chem., 2014, 22, 4506–4521 CrossRef CAS PubMed.
- P. A. Holt, R. Buscaglia, J. O. Trent and J. B. Chaires, Drug Dev. Res., 2011, 72, 178–186 CrossRef CAS PubMed.
- P. G. Baraldi, A. Bovero, F. Fruttarolo, D. Preti, M. A. Tabrizi, M. G. Pavani and R. Romagnoli, Med. Res. Rev., 2004, 24, 475–528 CrossRef CAS PubMed.
- E. Y. N. Lam, D. Beraldi, D. Tannahill and S. Balasubramanian, Nat. Commun., 2013, 4, 1796–1798 CrossRef PubMed.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS PubMed.
- N. Maizels, Nat. Struct. Mol. Biol., 2006, 13, 1055–1059 CAS.
- N. Maizels and L. T. Gray, PLoS Genet., 2013, 9, e1003468 CAS.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- A. Bugaut and S. Balasubramanian, Nucleic Acids Res., 2012, 40, 4727–4741 CrossRef CAS PubMed.
- H. J. Lipps and D. Rhodes, Trends Cell Biol., 2009, 19, 414–422 CrossRef CAS PubMed.
- V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, Nat. Biotechnol., 2015, 33, 877–881 CrossRef PubMed.
- R. Hänsel-Hertsch, D. Beraldi, S. V. Lensing, G. Marsico, K. Zyner, A. Parry, M. Di Antonio, J. Pike, H. Kimura, M. Narita, D. Tannahill and S. Balasubramanian, Nat. Genet., 2016, 48, 1267–1272 CrossRef PubMed.
- D. Monchaud and M.-P. Teulade-Fichou, Org. Biomol. Chem., 2008, 6, 627–636 CAS.
- S. Neidle, FEBS J., 2010, 277, 1118–1125 CrossRef CAS PubMed.
- S. A. Ohnmacht and S. Neidle, Bioorg. Med. Chem. Lett., 2014, 24, 2602–2612 CrossRef CAS PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- S. Müller and R. Rodriguez, Expert Rev. Clin. Pharmacol., 2014, 7, 663–679 CrossRef PubMed.
- Q. Cao, Y. Li, E. Freisinger, P. Z. Qin, R. K. O. Sigel and Z.-W. Mao, Inorg. Chem. Front., 2017, 4, 10–32 RSC.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS PubMed.
- R. Vilar, Met. Ions Life Sci., 2018, 18, 325–349 Search PubMed.
- S. F. Ralph, Curr. Top. Med. Chem., 2011, 11, 572–590 CrossRef CAS PubMed.
- J. E. Reed, A. A. Arnal, S. Neidle and R. Vilar, J. Am. Chem. Soc., 2006, 128, 5992–5993 CrossRef CAS PubMed.
- A. Arola-Arnal, J. Benet-Buchholz, S. Neidle and R. Vilar, Inorg. Chem., 2008, 47, 11910–11919 CrossRef CAS PubMed.
- C. Q. Zhou, T. C. Liao, Z. Q. Li, J. Gonzalez-Garcia, M. Reynolds, M. Zou and R. Vilar, Chem.–Eur. J., 2017, 23, 4713–4722 CrossRef CAS PubMed.
- P. Wang, C. H. Leung, D. L. Ma, S. C. Yan and C. M. Che, Chem.–Eur. J., 2010, 16, 6900–6911 CrossRef CAS PubMed.
- A. Ali, M. Kamra, S. Roy, K. Muniyappa and S. Bhattacharya, Bioconjugate Chem., 2017, 28, 341–352 CrossRef CAS PubMed.
- L. Lecarme, E. Prado, A. De Rache, M. L. Nicolau-Travers, G. Gellon, J. Dejeu, T. Lavergne, H. Jamet, D. Gomez, J. L. Mergny, E. Defrancq, O. Jarjayes and F. Thomas, ChemMedChem, 2016, 4, 1133–1136 CrossRef PubMed.
- K. J. Davis, C. Richardson, J. L. Beck, B. M. Knowles, A. Guédin, J.-L. Mergny, A. C. Willis and S. F. Ralph, Dalton Trans., 2015, 44, 3136–3150 RSC.
- P. Wu, D.-L. Ma, C.-H. Leung, S.-C. Yan, N. Zhu, R. Abagyan and C. M. Che, Chem.–Eur. J., 2009, 15, 13008–13021 CrossRef CAS PubMed.
- O. Domarco, D. Lötsch, J. Schreiber, C. Dinhof, S. Van Schoonhoven, M. D. García, C. Peinador, B. K. Keppler, W. Berger and A. Terenzi, Dalton Trans., 2017, 46, 329–332 RSC.
- N. H. Campbell, N. H. A. Karim, G. N. Parkinson, M. Gunaratnam, V. Petrucci, A. K. Todd, R. Vilar and S. Neidle, J. Med. Chem., 2012, 55, 209–222 CrossRef CAS PubMed.
- N. M. Smith, B. Corry, K. S. Iyer, M. Norret and C. L. Raston, Lab Chip, 2009, 9, 2021–2025 RSC.
- S. Lin, W. Wang, C. Hu, G. Yang, C.-N. Ko, K. Ren, C.-H. Leung and D.-L. Ma, J. Mater. Chem. B, 2017, 5, 479–484 RSC.
- S. Panich, M. Haj Sleiman, I. Steer, S. Ladame and J. B. Edel, ACS Sens., 2016, 1, 1097–1102 CrossRef CAS.
- M. Srisa-Art, A. J. DeMello and J. B. Edel, Anal. Chem., 2007, 79, 6682–6689 CrossRef CAS PubMed.
- A. De Cian, L. Guittat, M. Kaiser, B. Saccà, S. Amrane, A. Bourdoncle, P. Alberti, M. P. Teulade-Fichou, L. Lacroix and J. L. Mergny, Methods, 2007, 42, 183–195 CrossRef CAS PubMed.
- A. De Rache and J. L. Mergny, Biochimie, 2015, 115, 194–202 CrossRef CAS PubMed.
- N. H. Abd Karim, O. Mendoza, A. Shivalingam, A. J. Thompson, S. Ghosh, M. K. Kuimova and R. Vilar, RSC Adv., 2014, 4, 3355–3363 RSC.
- I. Pietro Oliveri, S. Failla, A. Colombo, C. Dragonetti, S. Righetto and S. Di Bella, Dalton Trans., 2014, 43, 2168–2175 RSC.
- W. H. Pitchford, H. J. Kim, A. P. Ivanov, H. M. Kim, J. S. Yu, R. J. Leatherbarrow, T. Albrecht, K. B. Kim and J. B. Edel, ACS Nano, 2015, 9, 1740–1748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00528a |
| This journal is © The Royal Society of Chemistry 2018 |