
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of the reported structure of ceanothine D via a novel macrocyclization strategy†
Jisun
Lee
 and
Madeleine M.
Joullié
*
and
Madeleine M.
Joullié
*
Department of Chemistry, University of Pennsylvania, 231 S. 34th St. Philadelphia, PA 19104-6323, USA. E-mail: mjoullie@sas.upenn.edu
First published on 31st January 2018
Abstract
The first total synthesis of the reported structure of ceanothine D, a cyclopeptide alkaloid found in red root, was achieved using a highly convergent synthetic strategy. Highlights of the synthesis include the first concomitant macrocyclization and formation of the unique chiral tertiary alkyl-aryl ether bond with complete regio- and stereo-control in the presence of a sensitive Z-enamide moiety to access the strained para-cyclophane present in its structure. This synthetic strategy may be broadly applicable in the generation of other structurally similar cyclopeptide alkaloids, enabling further biological and chemical investigations.
Introduction
Cyclopeptide alkaloids are the most abundant family of natural products isolated from the leaves, stem bark, root bark, and seeds of a wide variety of plant species.1 Their role in plants has not been fully elucidated due to lack of availability as yields either from isolation or synthesis are very low. Therefore, development of new synthetic approaches has been an important endeavour since their discovery.1,2 The structural similarity of cyclopeptide alkaloids is categorized by the size of the macrocycle that can be 13-, 14-, or 15-membered (Fig. 1). The 14-membered group is the most prevalent, yet most challenging to synthesize because of the enhanced rigidity of the molecule, which results in lack of conjugation between the aromatic ring and the double bond of the enamide moiety.1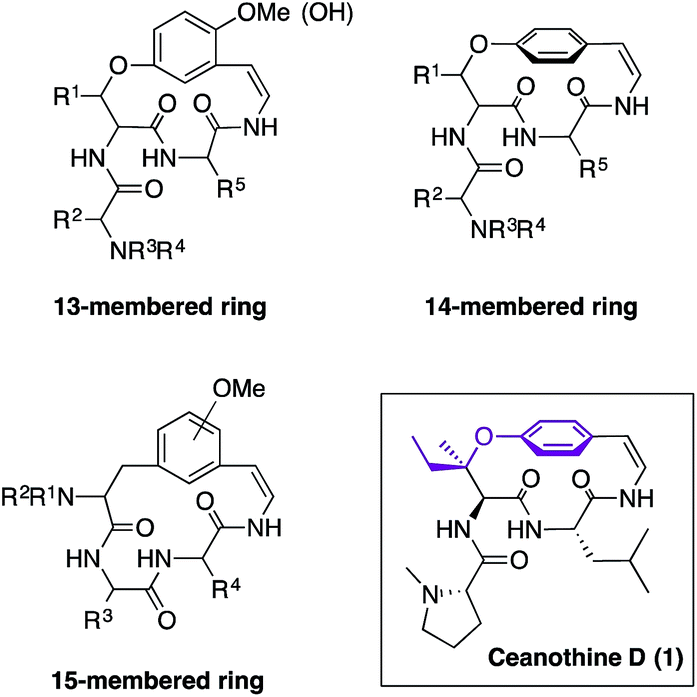 | ||
| Fig. 1 13-, 14-, and 15-Cyclopeptide alkaloids and reported structure of ceanothine D. | ||
Ceanothine D (1) was first reported by Servis from the root bark of Ceanothus americanus (also known as red root or the New Jersey tea plant) along with at least eighteen other cyclopeptide alkaloids that displayed close structural resemblances (Fig. 1).3 The structure of 1 was proposed based on degradation studies, mass spectrometry, and 1H NMR using a 60 MHz spectrometer.3 Interestingly, ceanothine D is the only cyclopeptide alkaloid reported to date to contain the unique chiral tertiary alkyl-aryl ether linkage derived from β-hydroxyisoleucine. The stereochemistry was assigned as L, since it is the most common stereochemical configuration found in nature, and the alkyl-aryl ether stereochemistry was presumed to be R, because most β-hydroxy amino acids in cyclopeptide alkaloids have this stereochemical assignment.1f,4 To the best of our knowledge, there is no report of the structural elucidation of the naturally occurring form of ceanothine D or total synthesis of the reported molecule to date.
The use of Ceanothus americanus to treat a wide variety of ailments including blood coagulation and pressure, spleen pain, and even cancer has been supported by its rich history in folk medicine.3,5 In fact, it has been used as a tea substitute during the American Revolutionary and Civil War,5 and it can be accessed as an over-the-counter dietary supplement even today. Although the metabolites of Ceanothus americanus have been of interest to chemists and biologists for many years,6 isolation of individual compounds remains a laborious and difficult challenge since structurally similar cyclopeptide alkaloids are present in varied amounts as complex mixtures.3,7 Furthermore, extraction yields vary from 0.0002 to 1% depending on the plant species, season, maturity of the plant, geographical location, and isolation method.1f,8 Due to these limitations, surprisingly few cyclopeptide alkaloids have been pharmacologically investigated, and biological activities of this class of compounds remain under investigation.1i,9 Efficient synthetic strategies to generate these compounds will not only facilitate structural elucidation, but also allow rapid biological profiling of the metabolites and related non-natural analogues.
To this end, research in our group10 along with that in other synthetic groups (e.g., Rapoport,11 Schmidt,12 Lipshutz,13 Han,14 Zhu,15 Evano8,16) focused on the development of new synthetic strategies to access cyclopeptide alkaloids. Synthetic challenges include stereocontrolled construction of the alkyl-aryl ether bond under mild reaction conditions and formation of the sensitive Z-enamide moiety. In particular, diverse bond disconnections have been examined for the non-trivial macrocyclization step. For the 13- and 15-membered cyclopeptides, the most widely employed approach is via intramolecular copper(I)-mediated amidation,8,16 while as macrolactamization,17 followed by late-stage elaboration of the Z-enamide10–12,14 is the most common strategy for the 14-membered compounds. Despite the steady progress in this area, synthesis of 14-membered cyclopeptides still remains a relevant synthetic task1i,10–15,18,19 especially in comparison to 13- or 15-membered counterparts because of the intrinsic ring strain12e,20 associated with the 14-membered macrocycle.21 Synthesis of ceanothine D poses an additional unique challenge due to the presence of a chiral tertiary alkyl-aryl ether vicinal to a stereogenic centre in the molecule. Previous research efforts in our group led to the development of a new methodology to generate such motifs via stereocontrolled regioselective ring opening of a trisubstituted aziridine by a variety of phenol nucleophiles.22 This intermolecular reaction was successfully implemented on the syntheses of natural products and analogues containing chiral tertiary alkyl-aryl ethers.23 Ceanothine D presented an excellent opportunity to further highlight the utility and versatility of this robust methodology, utilizing an intramolecular variant of the reaction, which addresses two major synthetic challenges associated with the molecule: the formation of the chiral tertiary alkyl-aryl ether fragment and macrocyclization in a single transformation.
Results and discussion
From the retrosynthetic perspective, access to ceanothine D (1) was envisioned through macrocyclization via intramolecular stereocontrolled regioselective aziridine ring opening by the phenol group of 2 (Scheme 1). Peptide coupling of acid 5 and free amine of L-leucineamide followed by subsequent intramolecular Mitsunobu reaction of the resultant amino alcohol would afford 3. Acid 5 could be easily derived from commercially available N-Boc-D-serine. The Z-vinyl iodide moiety of 4 would be constructed by stereoselective olefination of the silyl protected 4-hydroxybenzaldehyde using the Stork–Zhao reagent.24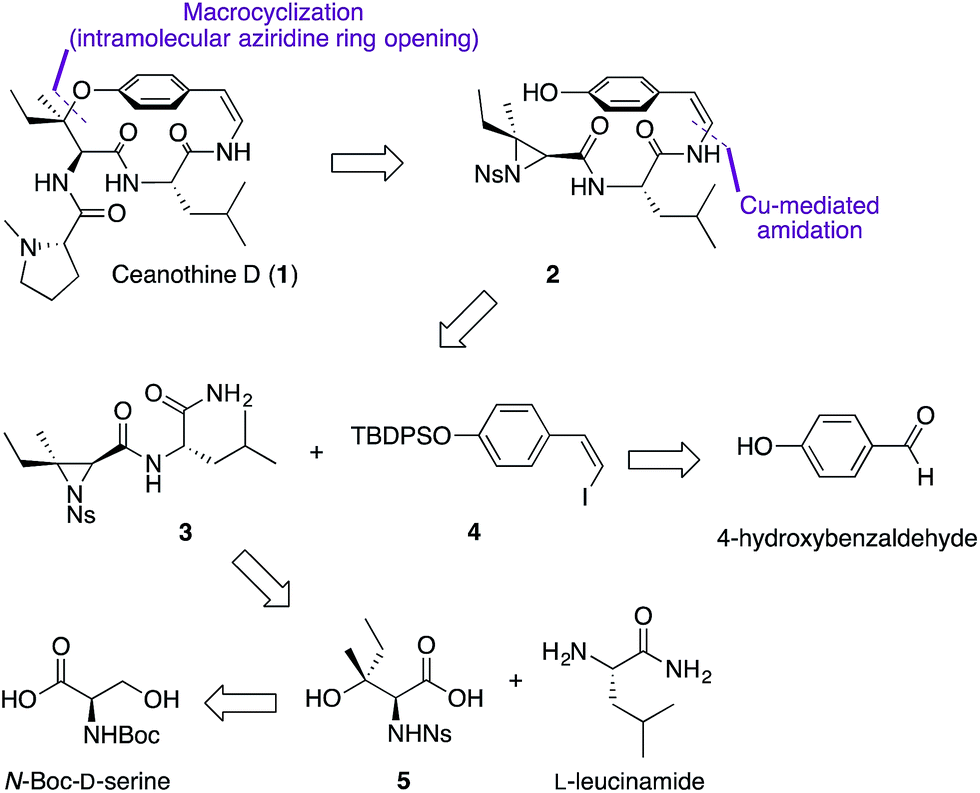 | ||
| Scheme 1 Retrosynthetic analysis. | ||
Starting from commercially available N-Boc-D-serine, known precursor 6 was prepared in five steps (Scheme 2).22b,25 Pinnick oxidation22,23d to the corresponding acid 5, followed by EDCI-mediated coupling with free amine of L-leucineamide afforded the amino alcohol 7 in good yield. Mitsunobu cyclization22,23a,b,d furnished the desired trisubstituted aziridine (3), and its structure was secured by X-ray crystallographic analysis. Treatment of the TBDPS protected commercially available 4-hydroxybenzaldehyde with the Stork–Zhao reagent16,24 gave the corresponding Z-vinyl iodide 4 as the major product (see ESI†). With requisite 3 and 4 in hand, the copper-mediated amidation was examined next. Surveying of current literature revealed the possibility of using catalytic Cu(I)-systems for the formation of Z-enamides especially in the presence of sensitive functional groups.8,16,26 In particular, utilization of diamine ligands with copper(I) iodide was the most prevalent approach in synthesis of enamide containing complex molecules.26d–f Indeed, initial synthetic efforts employing CuI with various diamine ligands in our system led to synthetically significant yields. Size of the diamine ligand27 was crucial in optimizing the yield of the reaction. Switching from sterically less bulky N,N′-dimethylethylenediamine to N,N′-dimethyl-1,2-diphenyl-1,2-ethylenediamine27a,b resulted in significantly increased yields,‡ presumably due to suppression of intermolecular aziridine ring opening side reaction(s) of 3 and/or 8 by the diamine ligand.28 The optimized procedure afforded desired Z-enamide 8 in good yield without any observable epimerization at stereocenters or isomerization of the Z-vinyl iodide (4). Next, rapid removal of the silyl protecting group furnished linear precursor 2, which was suitably positioned for the key macrocyclization step. Gratifyingly, our methodology22 in regio- and stereoselective ring opening of a trisubstituted aziridine translated well into the present intramolecular system despite concerns of creating a strained para-cyclophane (Scheme 2). To the best of our knowledge, this is the first effective macrocyclization of a 14-membered cyclopeptide precursor with the chemically sensitive Z-enamide intact. This successful outcome could be attributed (in part) to comparably large release of ring strain associated with ring-opening of the aziridine that compensates for the formation of the strained macrocycle. Furthermore, previous work by Païs29 suggests that the selection of suitable reagents (i.e. 1,5,7-triazabicyclo[4.4.0]dec-5-ene, TBD) is critical, since a macrocyclization event using a similar, albeit less strained, dihydro version of the linear precursor failed in the presence of Lewis acid promoters. As hypothesized from our past studies, the use of TBD as a bifunctional hydrogen-bonding guanidine base that activates the aziridine ring towards nucleophilic attack by the phenol,22b,c dictated success in the current system. Taken together, the synthetic utility and versatility of our methodology was successfully showcased, resulting in a synthetically significant yield of the desired macrocycle (9) (Scheme 2). Extensive NMR studies further confirmed the structure of 9 (see ESI†). Finally, removal of the nosyl protecting group with thiophenol afforded the free amine 10, which was subsequently coupled to N-methyl-L-proline completing the synthesis of the reported structure of ceanothine D (1) in good yields (Scheme 2). The full structure of 1 was elucidated on the basis of quantum-chemical calculations30 and experimental NMR analysis (see ESI†).
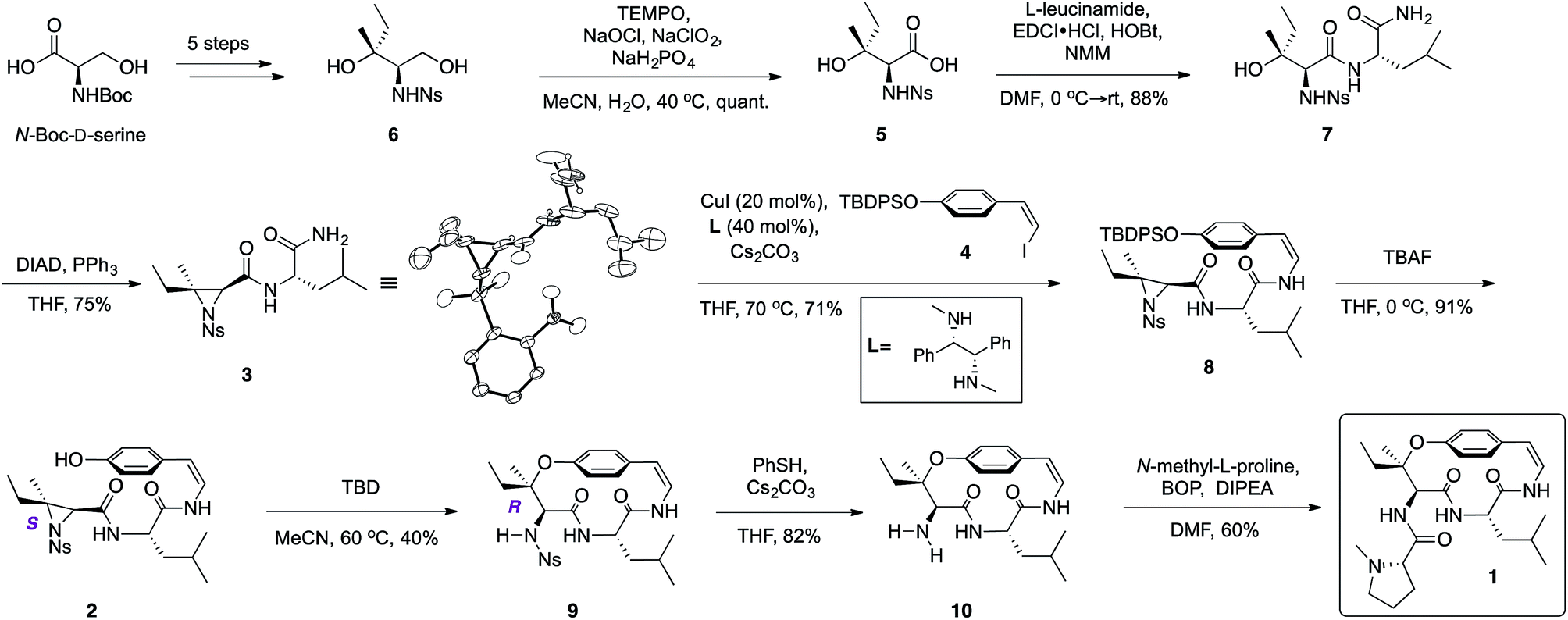 | ||
| Scheme 2 Total synthesis of the reported structure of ceanothine D. | ||
Since the first report of ceanothine D (1) by Servis in 1969,3 the structural elucidation of 1 has been incomplete. This occurrence is not unusual, since the relative or absolute configuration of cyclopeptide alkaloids that were originally reported before 2006 are only recently being investigated.1i Other than the low-resolution 1H NMR spectrum3b of ceanothine D provided by Servis, the only other comparable piece of information was optical rotation. Interestingly, the optical rotation of the synthetic version of 1 did not match that of the reported value of the natural product,§ suggesting the revision of the reported structure of ceanothine D. At this stage, the absolute structure of ceanothine D remains unknown due to the inaccessibility of natural samples for further analysis and full structural elucidation. Isolation and extraction from Ceanothus americanus is non-trivial, since extraction methods suffer from low yields of complex mixtures.1,8 Despite this limitation, usage of Ceanothus americanus is prevalent and widely supported by herbal home remedy enthusiasts.1,3,5 Further chemical and biological investigations enabled by an efficient macrocyclization strategy will expand the knowledge and highlight the relevance of this interesting class of natural products. In particular, the presence of the unique chiral tertiary alkyl-aryl ether linkage in 1 might induce structural changes and result in distinct biological activities relative to other 14-membered cyclopeptide alkaloids.
Conclusions
In conclusion, the first total synthesis of the reported structure of ceanothine D has been achieved with a novel macrocyclization method in eight steps from a known intermediate 6 (ref. 22b and 25) in overall 8.4% yield. Highlights of the synthesis include the first concomitant, sterecontrolled macrocyclization and formation of the chiral tertiary alkyl-aryl ether bond, particularly in the presence of a chemically sensitive Z-enamide moiety to afford the notoriously strained1,19 14-membered cyclopeptide alkaloid. The developed strategy may be useful in related research fields as a contribution to the synthetic methods available for the generation of a variety of macrocycles, and will be the goal of future investigations.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
In memory of Dr George Furst. Financial support was provided by the National Science Foundation (CHE-0951394) and the University of Pennsylvania. We thank Drs George Furst and Jun Gu (University of Pennsylvania) for NMR assistance, Dr Rakesh Kohli for high-resolution mass spectra, and Dr Patrick Carroll for X-ray data. We also thank Dr Simon Berritt for helpful discussions and Drs Erin Skoda (Forbeck) and Brandon T. Kelley whose work led to a better understanding of cyclopeptide alkaloids.Notes and references
- Selected references: (a) E. W. Warnhoff, Fortschr. Chem. Org. Naturst., 1970, 28, 162 CrossRef CAS PubMed; (b) R. Tschesche and E. U. Kaussmann, Cyclopeptide alkaloids, in Alkaloids, ed. R. H. F. Manske, Academic Press, New York, 1975, vol. 15, p. 165 Search PubMed; (c) M. M. Joullié and R. F. Nutt, Cyclopeptide Alkaloids, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, John Wiley & Sons, Inc., New York, 1985, vol. 3, p. 113 Search PubMed; (d) D. C. Gournelis, G. G. Laskaris and R. Verpoorte, Nat. Prod. Rep., 1997, 14, 75 RSC; (e) M. M. Joullié and D. J. Richard, Chem. Commun., 2004, 2011 RSC; (f) N. H. Tan and J. Zhou, Chem. Rev., 2006, 106, 840 CrossRef CAS PubMed; (g) H. R. El-Seedi, M. H. Zahra, U. Goransson and R. Verpoorte, Phytochem. Rev., 2007, 6, 143 CrossRef CAS; (h) A. F. Morel, G. Maldaner and V. Ilha, Cyclopeptide alkaloids from higher plants, in The alkaloids: chemical and biological perspectives, ed. G. Cordell, Wiley, New York, 2009, vol. 67, p. 79 Search PubMed; (i) E. Tuenter, V. Exarchou, S. Apers and L. Pieters, Phytochem. Rev., 2017, 16, 623 CrossRef CAS.
- M. Païs, F. X. Jarreau, X. Lusinchi and R. Goutarel, Ann. Chim., 1966, 11, 83 Search PubMed.
- (a) R. E. Servis, A. I. Kosak, R. Tschesche, E. Frohberg and H.-W. Fehlhabe, J. Am. Chem. Soc., 1969, 91, 5619 CrossRef CAS; (b) R. E. Servis, PhD thesis, New York University, 1969.
- (a) J. Marchand, F. Rocchiccioli, M. Païs and F. X. Jarreau, Bull. Soc. Chim. Fr., 1972, 4699 CAS; (b) J. Marchand, M. Païs and F. X. Jarreau, Bull. Soc. Chim. Fr., 1971, 3742 CAS.
- (a) J. T. Groot, J. Pharmacol. Exp. Ther., 1927, 30, 275 CAS; (b) C. E. Tharaldsen and J. Krawetz, Am. J. Physiol., 1927, 79, 445 CAS; (c) C. E. Tharaldsen, J. Am. Inst. Homeopathy, 1929, 22, 428 CAS.
- (a) J. H. M. Clinch, Am. J. Pharm., 1884, 56, 131 Search PubMed; (b) A. H. Clark, Am. J. Pharm., 1926, 98, 147 CAS; (c) A. H. Clark, Am. J. Pharm., 1928, 100, 240 CAS.
- H. Saltzman, PhD thesis, New York University, 1965.
- M. Toumi, V. Rincheval, A. Young, D. Gergeres, E. Turos, F. Couty, B. Mignotte and G. A. Evano, Eur. J. Org. Chem., 2009, 20, 3368 CrossRef.
- Selected references: (a) E. Tuenter, K. Segers, K. B. Kang, J. Viaene, S. H. Sung, P. Cos, L. Maes, Y. V. Heyden and L. Pieters, Molecules, 2017, 22, 224 CrossRef PubMed; (b) K. B. Kang, G. Ming, G. J. Kim, T.-K.-Q. Ha, H. Choi, W. K. Oh and S. H. Sung, Phytochemistry, 2015, 119, 90 CrossRef CAS PubMed; (c) W. A. Kaleem, N. Muhammad, M. Qayum, H. Khan, A. Khan, L. Aliberti and V. De Feo, Fitoterapia, 2013, 91, 154 CrossRef CAS PubMed; (d) P. Panseeta, K. Lomchoey, S. Prabpai, P. Kongsaeree, A. Suksamrarn, S. Ruchirawat and S. Suksamrarn, Phytochemistry, 2011, 72, 909 CrossRef CAS PubMed.
- (a) R. F. Nutt, K.-M. Chen and M. M. Joullié, J. Org. Chem., 1984, 49, 1013 CrossRef CAS; (b) D. M. Flanagan and M. M. Joullié, Synth. Commun., 1989, 19, 1 CrossRef CAS; (c) M. M. Bowers, P. Carroll and M. M. Joullié, J. Chem. Soc., Perkin Trans. 1, 1989, 857 RSC; (d) R. J. Heffner and M. M. Joullié, Tetrahedron Lett., 1989, 30, 7021 CrossRef CAS; (e) R. J. Heffner, J. Jiang and M. M. Joullié, J. Am. Chem. Soc., 1992, 114, 10181 CrossRef CAS; (f) J. Jiang, W.-R. Li, R. M. Przeslawski and M. M. Joullié, Tetrahedron Lett., 1993, 34, 6705 CrossRef CAS; (g) L. Williams, Z. Zhang, F. Shao, P. J. Carroll and M. M. Joullié, Tetrahedron, 1996, 52, 11673 CrossRef CAS; (h) S. P. East, F. Shao, L. Williams and M. M. Joullié, Tetrahedron, 1998, 54, 13371 CrossRef CAS; (i) S. P. East and M. M. Joullié, Tetrahedron Lett., 1998, 39, 9631 CrossRef; (j) D. M. Flanagan and M. M. Joullié, Synth. Commun., 1990, 20, 459 CrossRef.
- (a) J. C. Lagarias, R. A. Houghten and H. Rapoport, J. Am. Chem. Soc., 1978, 100, 8202 CrossRef CAS; (b) D. G. Goff, J. C. Lagarias, W. C. Shih, M. P. Klein and H. Rapoport, J. Org. Chem., 1980, 45, 4813 CrossRef CAS; (c) J. C. Lagarias, W. H. Yokoyama, J. Bordner, W. C. Shih, M. P. Klein and H. Rapoport, J. Am. Chem. Soc., 1983, 105, 1031 CrossRef CAS.
- (a) U. Schmidt, H. Griesser, A. Lieberknecht and J. Talbiersky, Angew. Chem., 1981, 93, 271 ( Angew. Chem., Int. Ed. Engl. , 1981 , 20 , 280 ) CrossRef CAS; (b) U. Schmidt, A. Lieberknecht, H. Griesser and J. Haüsler, Angew. Chem., 1981, 93, 272 ( Angew. Chem., Int. Ed. Engl. , 1981 , 20 , 181 ) CrossRef CAS; (c) U. Schmidt, A. Lieberknecht, H. Bökens and H. Griesser, Angew. Chem., 1981, 93, 1121 ( Angew. Chem., Int. Ed. Engl. , 1981 , 20 , 1026 ) CrossRef CAS; (d) U. Schmidt, A. Lieberknecht, H. Griesser and J. Talbiersky, J. Org. Chem., 1982, 47, 3261 CrossRef CAS; (e) U. Schmidt, A. Lieberknecht, H. Bökens and H. Griesser, J. Org. Chem., 1983, 48, 2680 CrossRef CAS; (f) U. Schmidt and U. Schanbacher, Angew. Chem., 1983, 95, 150 ( Angew. Chem., Int. Ed. Engl. , 1983 , 22 , 152 ) CrossRef CAS; (g) U. Schmidt and U. Schanbacher, Liebigs Ann. Chem., 1984, 1205 CrossRef CAS; (h) U. Schmidt, M. Zäh and A. Lieberknecht, J. Chem. Soc., Chem. Commun., 1991, 1002 RSC.
- (a) B. H. Lipshutz, R. W. Hungate and K. E. McCarthy, Tetrahedron Lett., 1983, 24, 5155 CrossRef CAS; (b) B. H. Lipshutz, R. W. Hungate and K. E. McCarthy, J. Am. Chem. Soc., 1983, 105, 7703 CrossRef CAS; (c) B. H. Lipshutz, B. E. Huff, K. E. McCarthy, T. A. Miller, S. M. J. Mukarram, T. J. Siahaan, W. D. Vaccaro, H. Webb and A. M. Falick, J. Am. Chem. Soc., 1990, 112, 7032 CrossRef CAS.
- Y.-A. Kim, H.-N. Shin, M.-S. Park, S.-H. Cho and S.-Y. Han, Tetrahedron Lett., 2003, 44, 2557 CrossRef CAS.
- (a) J. Zhu, T. Laïb, J. Chastanet and R. Beugelmans, Angew. Chem., 1996, 108, 2664 ( Angew. Chem., Int. Ed. Engl. , 1996 , 35 , 2517 ) CrossRef; (b) T. Laïb and J. Zhu, Tetrahedron Lett., 1998, 39, 283 CrossRef; (c) J. Zhu, Synlett, 1997, 133 CrossRef CAS; (d) T. Laïb and J. Zhu, Tetrahedron Lett., 1990, 40, 83 CrossRef; (e) T. Temal-Laïb, J. Chastanet and J. Zhu, J. Am. Chem. Soc., 2002, 124, 583 CrossRef; (f) P. Cristau, T. Temal-Laïb, M. Bois-Choussy, M.-T. Martin, J.-P. Vors and J. Zhu, Chem.–Eur. J., 2005, 11, 2668 CrossRef CAS PubMed.
- (a) M. Toumi, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2007, 46, 572 CrossRef CAS PubMed; (b) M. Toumi, F. Couty and G. Evano, Synlett, 2008, 29 CAS; (c) M. Toumi, F. Couty and G. Evano, J. Org. Chem., 2008, 73, 1270 CrossRef CAS PubMed.
- G. He, J. Wang and D. Ma, Org. Lett., 2007, 9, 1367 CrossRef CAS PubMed.
- M. de Greef, S. Abeln, K. Belkasmi, A. Dömling, R. V. A. Orru and L. A. Wessjohann, Synthesis, 2006, 3997 CAS.
- Selected recent review: T. Gulder and P. S. Baran, Nat. Prod. Rep., 2012, 29, 899 RSC.
- (a) A. Kirfel and G. Z. Will, Kristallografiya, 1976, 142, 368 CAS; (b) A. Kirfel, G. Will, R. Tschesche and H. Wilhelm, Z. Naturforsch., B: J. Chem. Sci., 1976, 31, 279 Search PubMed.
- Selected recent references on strained para-cyclophanes: (a) J.-P. Krieger, G. Ricci, D. Lesuisse, C. Meyer and J. Cossy, Angew. Chem., Int. Ed., 2014, 53, 8705 CrossRef CAS PubMed; (b) S. Jung, Y. Kitajima, Y. Ueda, K. Suzuki and K. Ohmori, Synlett, 2016, 27, 1521 CrossRef CAS.
- (a) P. Li, E. M. Forbeck, C. D. Evans and M. M. Joullié, Org. Lett., 2006, 8, 5105 CrossRef CAS PubMed; (b) E. M. Forbeck, C. D. Evans, J. A. Gilleran, P. Li and M. M. Joullié, J. Am. Chem. Soc., 2007, 129, 14463 CrossRef CAS PubMed; (c) E. M. Forbeck, PhD thesis, University of Pennsylvania, 2009; (d) B. T. Kelley, P. Carroll and M. M. Joullié, J. Org. Chem., 2014, 79, 5121 CrossRef CAS PubMed.
- (a) P. Li, C. D. Evans and M. M. Joullié, Org. Lett., 2005, 7, 5325 CrossRef CAS PubMed; (b) P. Li, C. D. Evans, Y. Wu, B. Cao, E. Hamel and M. M. Joullié, J. Am. Chem. Soc., 2008, 130, 2351 CrossRef CAS PubMed; (c) J. S. Grimley, A. M. Sawayama, H. Tanaka, M. M. Stohlmeyer, T. F. Woiwode and T. J. Wandless, Angew. Chem., Int. Ed., 2007, 46, 8157 CrossRef CAS PubMed; (d) M. M. Joullié, S. Berritt and E. Hamel, Tetrahedron Lett., 2011, 52, 2136 CrossRef PubMed.
- G. Stork and K. Zhao, Tetrahedron Lett., 1989, 30, 2173 CrossRef CAS.
- G. Ageno, L. Banfi, G. Cascio, G. Guanti, E. Manghisi, R. Riva and V. Rocca, Tetrahedron, 1995, 51, 8121 CrossRef CAS.
- Selected references: (a) L. Jiang, G. E. Job, A. Klapars and S. L. Buchwald, Org. Lett., 2003, 5, 3667 CrossRef CAS PubMed; (b) R. Shen, C. T. Lin, E. J. Bowman, B. J. Bowman and J. A. Porco Jr, J. Am. Chem. Soc., 2003, 125, 7889 CrossRef CAS PubMed; (c) X. Pan, Q. Cai and D. Ma, Org. Lett., 2004, 6, 1809 CrossRef CAS PubMed; (d) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed; (e) G. Evano, C. Theunissen and A. Pradal, Nat. Prod. Rep., 2013, 30, 1467 RSC; (f) T. Kuranaga, Y. Sesoko, K. Sakata, N. Maeda, A. Hayata and M. Inoue, J. Am. Chem. Soc., 2013, 135, 5467 CrossRef CAS PubMed; (g) T. Kuranaga, Y. Sesoko and M. Inoue, Nat. Prod. Rep., 2014, 31, 514 RSC.
- (a) K. Okamoto, M. Sakagami, F. Feng, H. Togame, H. Takemoto, S. Ichikawa and A. Matsuda, Org. Lett., 2011, 13, 5240 CrossRef CAS PubMed; (b) K. Okamoto, M. Sakagami, F. Feng, H. Togame, H. Takemoto, S. Ichikawa and A. Matsuda, J. Org. Chem., 2012, 77, 1367 CrossRef CAS PubMed; (c) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13 RSC.
- B. T. Kelley and M. M. Joullié, Org. Lett., 2010, 12, 4244 CrossRef CAS PubMed.
- F. Rocchiccioli, F.-X. Jarreau and M. Païs, Tetrahedron, 1978, 34, 2917 CrossRef CAS.
- (a) M. W. Lodewyk, C. Soldi, P. B. Jones, M. M. Olmstead, J. Rita, J. T. Shaw and D. J. Tantillo, J. Am. Chem. Soc., 2012, 134, 18550 CrossRef CAS PubMed; (b) G. K. Pierens, J. Comput. Chem., 2014, 35, 1388 CrossRef CAS PubMed; (c) M. G. Chini, C. R. Jones, A. Zampella, M. V. D'Auria, B. Renga, S. Fiorucci, C. P. Butts and C. G. Bifulco, J. Org. Chem., 2012, 77, 1489 CrossRef CAS PubMed; (d) M. S. B. Caro, L. H. de Oliveira, V. Ilha, R. A. Burrow, I. I. Dalcol and A. F. Morel, J. Nat. Prod., 2012, 75, 1220 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data are provided. CCDC 1588828. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00234g |
| ‡ No preference for either (S,S)- or (R,R)-N,N′-dimethyl-1,2-diphenyl-1,2-ethylenediamine enantiomer was observed in the reaction. |
| § The observed optical rotation of synthetic 1 ([α]22D = +128.6) was found to be different from the reported value3a ([α]D = −347). The reason for this discrepancy is unclear at the present as it could be attributed to a variety of factors. |
| This journal is © The Royal Society of Chemistry 2018 |