
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Step-growth titanium-catalysed dehydropolymerisation of amine–boranes†
Titel
Jurca
,
Theresa
Dellermann
 ,
Naomi E.
Stubbs
,
Diego A.
Resendiz-Lara
,
George R.
Whittell
* and
Ian
Manners
*
,
Naomi E.
Stubbs
,
Diego A.
Resendiz-Lara
,
George R.
Whittell
* and
Ian
Manners
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: Ian.Manners@bristol.ac.uk
First published on 6th March 2018
Abstract
Precatalysts active for the dehydropolymerisation of primary amine–boranes are generally based on mid or late transition metal. We have found that the activity of the precatalyst system formed from CpR2TiCl2 and 2nBuLi towards the dehydrogenation of the secondary amine–borane Me2NH·BH3, to yield the cyclic diborazane [Me2N–BH2]2, increases dramatically with increasing electron-donating character of the cyclopentadienyl rings (CpR). Application of the most active precatalyst system (CpR = η-C5Me5) to the primary amine–borane MeNH2·BH3 enabled the first synthesis of high molar mass poly(N-methylaminoborane), [MeNH–BH2]n, the BN analogue of polypropylene, by an early transition metal such as catalyst. Significantly, unlike other dehydropolymerization precatalysts for MeNH2·BH3 such as [Ir(POCOP)H2], skeletal nickel, and [Rh(COD)Cl]2, the Ti precatalyst system was also active towards a range of substrates including BzNH2·BH3 (Bz = benzyl) yielding high molar mass polymer. Moreover, in contrast to the late transition metal catalysed dehydropolymerisation of MeNH2·BH3 and also the Ziegler–Natta polymerisation of olefins, studies indicate that the Ti-catalyzed dehydropolymerization reactions proceed by a step-growth rather than a chain-growth mechanism.
Introduction
Catalysis plays a pivotal role in molecular and macromolecular C–C bond forming chemistry. The development of comparable reactions for the catenation of other p-block elements, however, has proceeded at a markedly slower pace. Nonetheless, the identification of useful target main group molecules and materials over the past decade has prompted significant progress in the field.1 For example, catalytic dehydrocoupling/dehydrogenation of amine–boranes has become an area of widespread interest, largely motivated by potential applications in hydrogen storage2 and transfer,3 and the formation of novel ceramic thin films and polymeric materials.1,4 The latter can be regarded as BN analogues of polyolefins, but with distinct properties and possible applications, for example as piezoelectrics and precursors to boron-based solid state materials.4 Consequently, a wide variety of catalyst systems have been developed to promote the dehydrogenation of amine–boranes in general, with the vast majority based on mid to late transition metals (e.g. Re,5 Fe,6 Ru,7 Rh,4e,8 Ir4c,9 and Ni10).11 With regards to the dehydropolymerisation of primary amine–boranes using Brookhart's catalyst, [IrH2(POCOP)] (POCOP = 2,6-bis(di-tert-butylphosphinito)benzene),12 our group has reported the formation of high molar mass (Mn > 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) [MeNH–BH2]n (5) from MeNH2·BH3 (4).4a,c Other middle to late metal catalysts, such as [CpFe(CO)2]2,6b [Rh(Ph2P(CH2)4PPh2)]+,4i and [Rh(κ2-P,P-xantphos){η2-H2B(CH2CH2tBu)·NMe3}]+,4e,8e have also been shown to be effective in this role, and in certain cases key mechanistic information has been elucidated. These polymerisations thus appear to proceed by a chain-growth coordination-insertion mechanism.1e,4c,e Metal-free routes involving free, transient aminoborane monomers have also been recently reported, but remain mechanistically unclear.4g,13
000 g mol−1) [MeNH–BH2]n (5) from MeNH2·BH3 (4).4a,c Other middle to late metal catalysts, such as [CpFe(CO)2]2,6b [Rh(Ph2P(CH2)4PPh2)]+,4i and [Rh(κ2-P,P-xantphos){η2-H2B(CH2CH2tBu)·NMe3}]+,4e,8e have also been shown to be effective in this role, and in certain cases key mechanistic information has been elucidated. These polymerisations thus appear to proceed by a chain-growth coordination-insertion mechanism.1e,4c,e Metal-free routes involving free, transient aminoborane monomers have also been recently reported, but remain mechanistically unclear.4g,13
In addition to our report of [CpFe(CO)2]2 (ref. 6b) as an example of an earth abundant transition metal catalyst, we also described the use of the group 4 metallocene precatalysts Cp2TiCl2 (6avide infra)/two equiv. of nBuLi or Cp2Ti(PMe3)2 as reasonably efficient dehydrocoupling catalysts for the secondary amine–borane Me2NH·BH3 (1), yielding the cyclodiborazane [Me2N–BH2]2 (3) (Scheme 1).14 Others11b,15,16 have also reported the use of neutral TiII and ZrII, and also cationic ZrIV precatalysts for the dehydrocoupling of 1. From these studies, two general reaction mechanisms have been proposed. Compound 1 may react with the active catalyst to form Me2N=BH2 as the intermediate, which then dimerizes to afford 3 in an off-metal process,15,17 as shown for late transition metal catalyst systems.1c,2a,10b,18 Alternatively, 1 may be dehydrocoupled to form the linear diborazane Me2NH–BH2–NMe2–BH3 (2) as the intermediate, which then yields 3 in a subsequent on-metal, ring-closing dehydrogenation step and indicates a rather different mechanism.14,19 Our group has also reported the preparation of paramagnetic TiIII species related to the catalytic reaction,20 and identified the TiIII–amido–borane complex [Cp2Ti(NMe2BH3)] (6b, vide infra) as being more active than either [6a + 2nBuLi], or Cp2Ti(PMe3)2 for the dehydrocoupling of 1 to give 3 (via2).21 To date, however, the polymerisation of the primary amine–borane MeNH2·BH3 using a catalyst system based on an early transition metals such as Ti or Zr has not be observed.
 | ||
| Scheme 1 Titanocene-catalysed dehydrocoupling of Me2NH·BH3 (1) to give [Me2N–BH2]2 (3). | ||
Herein, we report structure-correlated kinetic studies of different titanium based precatalyst systems for the dehydrogenation of the secondary amine–borane Me2NH·BH3 (1), and based on these results, the first successful dehydropolymerisation of primary amine–boranes, yielding high molecular weight polyaminoboranes, that proceeds by a step-growth rather than a chain-growth mechanism.
Results
Dehydrogenation of N,N-dimethyl amine–borane
Our initial investigations were based on the influence of cyclopentadienyl ligand substitution on the activity of a series of two-component precatalysts, which were formed by CpR2TiCl2 and 2nBuLi. We therefore explored the dehydrocoupling of amine–borane 1 (1 M in toluene) mediated by 2 mol% of [6c–e + 2nBuLi] at 22 °C in toluene. Previously reported precatalysts [6a + 2nBuLi]11 and Ti(III) species 6b (ref. 21) were also investigated under identical conditions for comparative purposes (Chart 1), as well as the reaction of [6e + 2nBuLi] in THF. All reactions were conducted in sealed J. Young NMR tubes, and monitored by 11B NMR spectroscopy.22a Rapid initial conversion of 1 (δ11B −13.8 ppm) to linear diborazane 2 (δ11B 1.6 ppm (internal BH2), −13.8 ppm (terminal BH3)) was detected, followed by slower subsequent conversion of 2 to cyclodiborazane 3 (δ11B 4.9 ppm), presumably with concomitant release of H2. The compounds (Me2N)2BH (δ11B 28.4 ppm) and Me2N=BH2 (δ11B 37.4 ppm) were also identified in the reaction mixture, but in very minor amounts (Fig. S1–S6†). All chemical shifts and coupling constants for the products were consistent with those reported in the literature.5b,15 | ||
| Chart 1 Ti-based amine–borane dehydrocoupling/dehydropolymerisation precatalyst components 6a–e. | ||
Precatalyst [6a + 2nBuLi] resulted in the slowest conversion to 3, only reaching high (>90%) conversion after 690 min.22b Switching to precatalysts 6b20 and [6c + 2nBuLi] resulted in an increased reaction rate, with reaction completion at 390 and 420 min, respectively. Most significantly, reactions with precatalysts [6d/6e + 2nBuLi] proceeded at a substantially faster rate, reaching complete conversion to 3 after 180 min for 6d, and remarkably, in under 30 min in the case of 6e (Fig. 1 and S6†). A change in solvent from toluene to THF for 6e results in nearly no conversion of 1 after 12 h, despite the latter being a better solvent for 1. This reduction in activity is therefore most probably caused by coordination of the solvent to the active site of the catalyst (Fig. S7†). The observed difference between 6d and 6e is particularly informative, as these ligands are effectively isosteric as indicated by the similar coordination gap aperture (cga) values of ca. 58 and 55°, respectively.23 In addition to influencing the rate and strength of substrate bonding, this feature would also be expected to similarly affect the existence of any off-cycle dimerization, or the formation of an “tucked-in complex”.24 They do, however, exhibit different electronic properties, as shown through IR spectroscopy of the corresponding [CpRFe(CO)2]2 complexes (ν(CO) for [CpRFe(CO)2]2 = 1762, 1938 and 1755, 1922 cm−1 for CpR = tBuC5H4 and C5Me5, respectively).25 This result strongly suggests that the trend of increasing reaction rate from 6a–e is most probably a consequence of the increasing electron-donating character of the CpR ligands rather than any steric factor.
 | ||
| Fig. 1 Reaction profiles for the formation of 3 from the catalytic dehydrocoupling of 1 with precatalysts [2 mol% 6a,c–f + 2nBuLi], and 6b as monitored by 11B{1H} (96 MHz, toluene-d8) NMR spectroscopy. | ||
For the most active precatalyst [6e + 2nBuLi], this translated to a turnover frequency (TOF) of 141 h−1 (based on 45% conversion to 3 after 5 min, see Table S1†) and this value is in the range (35–420 h−1) reported for the conversion of 1 to 3 by isolable CpR2Ti precatalysts.15 Increased reaction rates were also reported for these species on incorporation of electron donating SiMe3 groups on CpR, however, the disambiguation of the role of steric and electronic effects was not possible. Nonetheless, dehydrocoupling with precatalysts [6a,c–e + 2nBuLi] and 6b proceeded via linear diborazane 2 rather than Me2N=BH2 as the major intermediate, which differs from that reported for the isolable TiII precatalysts (Scheme 1).
Dehydropolymerisation of primary amine–boranes
Prompted by the high activity of precatalysts [6d/6e + 2nBuLi] towards 1 we endeavoured to test them towards the dehydropolymerisation of primary amine–borane 4. Preliminary kinetic studies were conducted with ca. 2 mol% catalyst in toluene solution at 22 °C in sealed J. Young NMR tubes, and the reactions were monitored by 11B NMR spectroscopy (Fig. S8, S9†). For the reaction of [6d + 2nBuLi] with 4 the spectra show the instant formation of polyaminoborane 5 (δ11B −6.1 ppm, and −18 ppm assigned to the end-group) as well as the presence of (MeNH)2BH, 9 (δ11B 27.7 ppm), which formed presumably via redistribution of amine–borane 4. Simultaneously, [MeNH–BH2]3, 7 (δ11B −5.8 ppm) could be detected, which was further dehydrogenated forming [MeN–BH]38 (δ11B 32.5 ppm) after ca. 5 h (product assignment based on the literature, see Fig. S8†).4a,c,6b Unreacted amine–borane 4 was still present in the reaction mixture even after 23 h. On the other hand [6e + 2nBuLi] led to a complete consumption of 4 after ca. 8 h and formation of predominantly polymer 5 and byproducts 7 (95% combined for 5 and 7, as the peaks were unresolvable in the 11B NMR spectrum), 8 (4%) and 9 (minimal amounts) (Fig. S9 and S10†).26 It is noteworthy that in this case 7 and 8 are the only species observed after ca. 1 h. Based on these promising results we focused the remainder of our dehydropolymerisation studies on precatalyst [6e + 2nBuLi] (Scheme 2). | ||
| Scheme 2 Catalytic dehydropolymerisation of 4 with precatalyst [6e + 2nBuLi] to give 5 and byproducts 7–9. | ||
Catalytic dehydropolymerisation reactions of 4 were focused on the isolation and characterisation of polymer 5 with precatalyst [6e + 2nBuLi] and conducted in toluene (1.5 M in substrate) at 22 °C. To optimize the conditions for the formation of the high molecular weight polyaminoborane 5, variable catalyst loadings from 0.4–7 mol% were screened initially at both 8 h and 16 h (see Scheme 2 and Fig. S11–S18†). After precipitation of the reaction mixture into cold hexanes and removal of both the soluble catalyst and byproducts, all reactions led to the isolation of white polymeric 5 (with yields of 53–72% limited by the above-mentioned side reactions), which was characterised by 11B NMR spectroscopy and Gel Permeation Chromatography (GPC). A steady increase in molar mass (Mn) and a concomitant decrease in polydispersity index (PDI = Mw/Mn) was observed with increasing catalyst loading. Consistent with the former was the decreasing intensity of the end-group resonance (δ11B ca. −18 ppm) with respect to that of the main-chain (δ11B ca. −6 ppm) in the 11B NMR spectra (Fig. S11–S18†). This observation served to confirm the original assignment, and in combination with the absence of any well-resolved coupling in the corresponding proton-coupled spectra, suggests the lack of significant amounts of unreacted 4. Increased reaction times (from 8 h to 16 h) only served to afford polyaminoboranes with lower Mn values (see Fig. 2, and Table S2†). The optimal conditions for the formation of 5 involved 7 mol% [6e + 2nBuLi] and 8 h reaction time, yielding polymer with Mn = 54000 g mol−1 (PDI = 1.6).
 | ||
| Fig. 2 Graphical representation of molar mass (Mn and Mw in g mol−1) of 5 obtained from the reactions of 4 with precatalyst [6e + 2nBuLi] as a function of catalyst loading (0.4–7 mol%) and reaction time (8 h and 16 h) at 22 °C. | ||
To extend the substrate scope of the dehydropolymerisation reaction, the N-benzyl (Bz) substituted amine–borane BzNH2·BH3 (10a) was reacted under previously optimised conditions, yielding a white, sparingly soluble precipitate (Scheme 3).27 GPC analysis of the THF-soluble fraction indicated the presence of high molar mass polymer 11a with Mn = 101700 g mol−1 (PDI = 1.15) (Fig. S28†). Further studies carried out on the dehydropolymerisation reaction showed the formation of byproducts 12a, 13a and 14a after approximately 1 h, which is analogous to the results for the dehydropolymerisation of 4 (Fig. S20†). Surprisingly, previous attempts to dehydropolymerise this substrate using the well-established Ir catalyst [IrH2(POCOP)], skeletal nickel or [Rh(COD)Cl]2 have been unsuccessful and showed no reaction. These results encouraged us to perform similar dehydropolymerisation reactions using the N-4-phenylbutyl (10b) and the N-thiophenylmethyl amine–borane (10c) as substrates as well as an equimolar mixture of BzNH2·BH3 (10a) and nBuNH2·BH3 (10d) (Scheme 3). This yielded the homopolymers 11b and 11c and the copolymer 11d, respectively. All reactions yielded high molar mass polymers with Mn = 349100 g mol−1 (PDI = 1.30, 11b), 95600 g mol−1 (PDI = 1.29, 11c) and 131900 g mol−1 (PDI = 1.33, 11d) (Table 1, Fig. S28 and S32†). In contrast to poly(N-benzylaminoborane) 11a, the latter polymers (11b–d) were completely soluble and could be further characterized by 1H, 13C and 11B NMR spectroscopy and mass spectrometry (11d) (Fig. S21–S27, S29–S31 and S33†).
 | ||
| Scheme 3 Catalytic dehydropolymerisation of 10a–c (I) and of a mixture of 10a and 10d (II) with precatalyst [6e + 2nBuLi] to give polymers 11a–d and the respective byproducts 12a–c, 13a–c and 14a–c. The ratio of monomers 10a and 10b of the copolymer 11d was determined by 1H NMR spectroscopy giving a n/m ratio of 2. | ||
| Yield (%) | Molecular weight Mn (g mol−1) | Molecular weight Mw (g mol−1) | PDI | |
|---|---|---|---|---|
| 11a | 31 | 101700 |
116700 |
1.15 |
| 11b | 61 | 349100 |
453700 |
1.30 |
| 11c | 44 | 95600 |
124400 |
1.29 |
| 11d | 44 | 131900 |
175400 |
1.33 |
Mechanistic studies
Further mechanistic studies were carried out on the dehydropolymerisation of MeNH2·BH3 (4) using 7 mol% of [6e + 2nBuLi]. We studied the effect of reaction time in more detail by isolating polyaminoborane 5 after 0.5, 1, 2, and 4 h (see Fig. S34–S37†), in addition to the 8 and 16 h time points already recorded. A steady increase in Mn and a concomitant decrease in PDI of 5 with increasing reaction time up to the 8 h time point was observed (see Fig. 3, S38,† and Table 2). The observation of a decrease in molar mass and increased PDI at prolonged (8–16 h) reaction times was attributed to depolymerisation and dehydrogenation to afford 8 and 9 (Fig. S39†). Similar observations have been reported with [CpFe(CO)2]2 (ref. 6b) as a precatalyst, whilst this effect was much less significant in the case of [IrH2(POCOP)].4c We also found that cyclotriborazane 7, which is likely formed as an intermediate during the depolymerisation of 5, was rapidly dehydrogenated by [6e + 2nBuLi] (1 h, toluene, 22 °C) to yield borazine 8 (see Fig. S40†). Interestingly, both the Ti- and Ir-catalysed dehydropolymerisations showed an increase in Mn with catalyst loading. In the case of the Ir precatalyst, this observation was tentatively interpreted in terms of a chain-growth mechanism that involved an initial, rate-determining dehydrogenation step to form transient MeNH=BH2, followed by coordination polymerisation to form 5.4c As for the Ir-catalysed reaction,28 attempts to trap the highly reactive MeNH=BH2 using cyclohexene,29 to form MeNH=BCy2, were unsuccessful in the case of the Ti precatalyst (see Fig. S41†). This suggests that if the primary aminoborane is indeed formed as an intermediate, it either remains coordinated or is consumed more rapidly than it undergoes hydroboration with the cyclic olefin.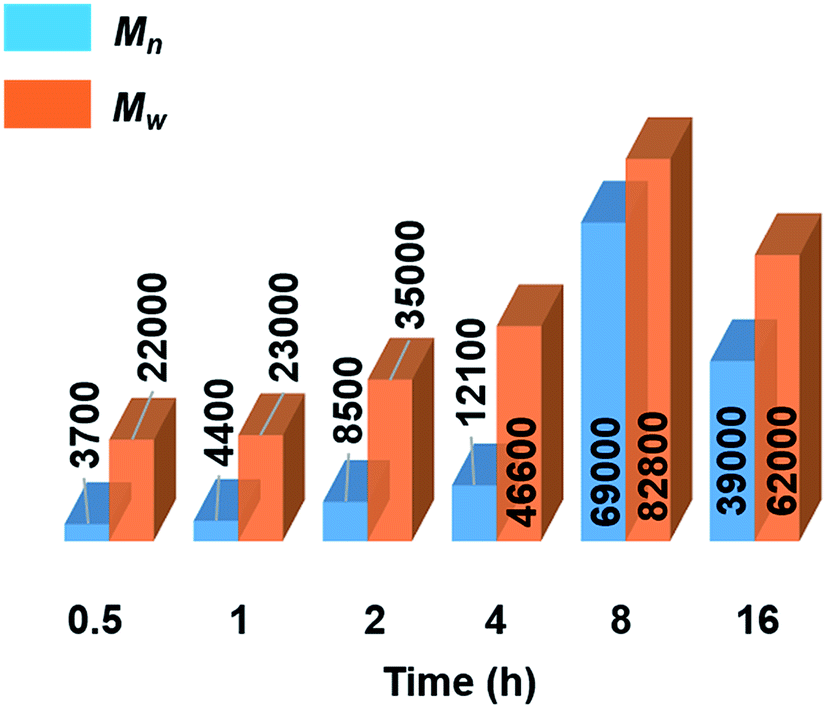 | ||
| Fig. 3 Graphical representation of molecular weights (Mn and Mw in g mol−1) from GPC analysis of isolated polyaminoborane 5 from the reactions of 4 with precatalyst [6e + 2nBuLi] (7 mol%, 0.5–16 h, 22 °C) (see Table 2). | ||
Significantly, in the Ti-catalysed polymerisation a steady increase in molar mass was observed from 0.5 h (conversion of 4 = 80%, Mn = 3700 g mol−1, PDI = 6.2) up to 8 h (conversion = 97%, Mn = 54000 g mol−1, PDI = 1.6) before depolymerisation and dehydrogenation of the polyaminoborane 5 were detected (Table 2) .30 This is indicative of a step-growth polycondensation process and contrasts with the behavior found for the dehydropolymerisation of 4 with [IrH2(POCOP)] as precatalyst. In the latter case high molar mass 5 was detected even at low conversions of 4, as befits a chain-growth mechanism.4c
The existence of a step-growth polymerisation mechanism for the Ti-catalysed dehydropolymerisation of 4 is consistent with the intermediacy of linear diborazane 2 in the dehydrogenation of 1. It is also supported by several further experiments. For example, treatment of isolated, low molar mass 5 (Mn = 2600 g mol−1, PDI = 4.3) with a further quantity of 7 mol% of [6e + 2nBuLi] in toluene for 7.5 h afforded higher molar mass 5 (Mn = 18000 g mol−1, PDI = 1.8), which demonstrates that monomer is not required to form high molar mass polymer (see Fig. S42 and S43†).32 Consistent with the hypothesis that the Ti- and Ir-catalysed polymerisations proceed via fundamentally different mechanisms, the molar mass of 5 (Mn = 3100 g mol−1, PDI = 2.7) only increased marginally (Mn = 6700 g mol−1, PDI = 2.5) upon treatment with [IrH2(POCOP)], (see Fig. S44 and S45†), whereas under these conditions, the Ir precatalyst converts 4 to 5 with a Mn of 262600 g mol−1 (PDI = 1.7) (Fig. S47†).
Conclusions
In summary, we have successfully optimised the precatalyst system for secondary amine–boranes based on CpR2TiCl2/2nBuLi by systematic variation of the cyclopentadienyl ligand steric and electronic properties. Based on these results and with an extension to primary amine–boranes, we report the first example of an early transition metal-mediated synthesis of high molar mass polyaminoboranes via dehydropolymerisation of N-methyl and N-benzyl (and related) substituted amine–boranes. The presented precatalyst system, based on earth abundant titanium, was shown to augment the amine–borane substrate scope exhibited by state-of-the art catalysts, e.g. Brookhart's iridium catalyst, skeletal nickel or [Rh(COD)Cl]2. Further investigations into the mechanistic pathway for the dehydropolymerisation of MeNH2·BH3 suggested that it proceeds by a step-growth rather than a chain-growth mechanism.Previously, the catalytic dehydropolymerisation of intrinsically polar primary amine–borane substrates has required mid to late transition metal centers. It is interesting to note that, in the case of olefins, the analogous developments occurred historically in the reverse order, starting with early metals before the more recent successful development of late transition metal catalysts.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
T. J. thanks the EU for a Marie Curie postdoctoral fellowship, T. D. thanks the Humboldt Foundation for a Feodor-Lynen fellowship, D. R. L. thanks CONACyT and I. M. and N. E. S. thank EPSRC for support. We thank Owen Metters and Lena Stoll for supplying various substrates/catalysts and help with data acquisition.Notes and references
- (a) E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed; (b) R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC; (c) R. J. Less, R. L. Melen and D. S. Wright, RSC Adv., 2012, 2, 2191–2199 RSC; (d) A. M. Priegert, B. W. Rawe, S. C. Serin and D. P. Gates, Chem. Soc. Rev., 2016, 45, 922–953 RSC; (e) H. C. Johnson, T. N. Hooper and A. S. Weller, Top. Organomet. Chem., 2015, 49, 153–220 CrossRef CAS.
- (a) A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed; (b) B. Peng and J. Chen, Energy Environ. Sci., 2008, 1, 479–483 CAS; (c) F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613–2626 RSC; (d) N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 509–521 CrossRef CAS; (e) D. W. Himmelberger, C. W. Yoon, M. E. Bluhm, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2009, 131, 14101–14110 CrossRef CAS PubMed.
- (a) X. Yang, L. Zhao, T. Fox, Z.-X. Wang and H. Berke, Angew. Chem., Int. Ed., 2010, 49, 2058–2062 CrossRef CAS PubMed; (b) A. P. M. Robertson, E. M. Leitao and I. Manners, J. Am. Chem. Soc., 2011, 133, 19322–19325 CrossRef CAS PubMed; (c) E. M. Leitao, N. E. Stubbs, A. P. M. Robertson, H. Helten, R. J. Cox, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2012, 134, 16805–16816 CrossRef CAS PubMed; (d) M. E. Sloan, A. Staubitz, K. Lee and I. Manners, Eur. J. Org. Chem., 2011, 672–675 CrossRef CAS; (e) X. Yang, T. Fox and H. Berke, Chem. Commun., 2011, 47, 2053–2055 RSC; (f) C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 3342–3346 CrossRef CAS PubMed; (g) S. Li, G. Li, W. Meng and H. Du, J. Am. Chem. Soc., 2016, 138, 12956–12962 CrossRef CAS PubMed.
- (a) A. Staubitz, A. Presa Soto and I. Manners, Angew. Chem., Int. Ed., 2008, 47, 6212–6215 CrossRef CAS PubMed; (b) B. L. Dietrich, K. I. Goldberg, D. M. Heinekey, T. Autrey and J. C. Linehan, Inorg. Chem., 2008, 47, 8583–8585 CrossRef CAS PubMed; (c) A. Staubitz, M. E. Sloan, A. P. M. Robertson, A. Friedrich, S. Schneider, P. J. Gates, J. Schmedt auf der Günne and I. Manners, J. Am. Chem. Soc., 2010, 132, 13332–13345 CrossRef CAS PubMed; (d) A. N. Marziale, A. Friedrich, I. Klopsch, M. Drees, V. R. Celinski, J. Schmedt auf der Günne and S. Schneider, J. Am. Chem. Soc., 2013, 135, 13342–13355 CrossRef CAS PubMed; (e) H. C. Johnson, E. M. Leitao, G. R. Whittell, I. Manners, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2014, 136, 9078–9093 CrossRef CAS PubMed; (f) N. E. Stubbs, T. Jurca, E. M. Leitao, C. H. Woodall and I. Manners, Chem. Commun., 2013, 49, 9098–9100 RSC; (g) O. J. Metters, A. M. Chapman, A. P. M. Robertson, C. H. Woodall, P. J. Gates, D. F. Wass and I. Manners, Chem. Commun., 2014, 50, 12146–12149 RSC; (h) V. A. Du, T. Jurca, G. R. Whittell and I. Manners, Dalton Trans., 2016, 45, 1055–1062 RSC; (i) R. Dallanegra, A. P. M. Robertson, A. B. Chaplin, I. Manners and A. S. Weller, Chem. Commun., 2011, 47, 3763–3765 RSC; (j) C. Lichtenberg, M. Adelhardt, T. L. Gianetti, K. Meyer, B. de Bruin and H. Grützmacher, ACS Catal., 2015, 5, 6230–6240 CrossRef CAS; (k) M. W. Lui, N. R. Paisley, R. McDonald, M. J. Ferguson and E. Rivard, Chem.–Eur. J., 2016, 22, 2134–2145 CrossRef CAS PubMed; (l) X. Wang, T. N. Hooper, A. Kumar, I. K. Priest, Y. Sheng, T. O. M. Samuels, A. W. Robertson, M. Pacios, H. Bhaskaran, A. S. Weller and J. H. Warner, CrystEngComm, 2017, 19, 285–294 RSC.
- (a) Y. Jiang and H. Berke, Chem. Commun., 2007, 3571–3573 RSC; (b) Y. Jiang, O. Blacque, T. Fox, C. M. Frech and H. Berke, Organometallics, 2009, 28, 5493–5504 CrossRef CAS.
- (a) R. T. Baker, J. C. Gordon, C. W. Hamilton, N. J. Henson, P.-H. Lin, S. Maguire, M. Murugesu, B. L. Scott and N. C. Smythe, J. Am. Chem. Soc., 2012, 134, 5598–5609 CrossRef CAS PubMed; (b) J. R. Vance, A. P. M. Robertson, K. Lee and I. Manners, Chem.–Eur. J., 2011, 17, 4099–4103 CrossRef CAS PubMed; (c) J. F. Sonnenberg and R. H. Morris, ACS Catal., 2013, 3, 1092–1102 CrossRef CAS; (d) J. R. Vance, A. Schäfer, A. P. M. Robertson, K. Lee, J. Turner, G. R. Whittell and I. Manners, J. Am. Chem. Soc., 2014, 136, 3048–3064 CrossRef CAS PubMed; (e) C. Lichtenberg, L. Viciu, M. Adelhardt, J. Sutter, K. Meyer, B. de Bruin and H. Grützmacher, Angew. Chem., Int. Ed., 2015, 54, 5766–5771 CrossRef CAS PubMed; (f) A. Glüer, M. Förster, V. R. Celinski, J. Schmedt auf der Günne, M. C. Holthausen and S. Schneider, ACS Catal., 2015, 5, 7214–7217 CrossRef; (g) P. Bhattacharya, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 11153–11161 CrossRef CAS PubMed; (h) F. Anke, D. Han, M. Klahn, A. Spannenberg and T. Beweries, Dalton Trans., 2017, 46, 6843–6847 RSC; (i) N. T. Coles, M. F. Mahaon and R. L. Webster, Organometallics, 2017, 36, 2262–2268 CrossRef CAS.
- (a) M. Käβ, A. Friedrich, M. Drees and S. Schneider, Angew. Chem., Int. Ed., 2009, 48, 905–907 CrossRef PubMed; (b) A. Friedrich, M. Drees and S. Schneider, Chem.–Eur. J., 2009, 15, 10339–10342 CrossRef CAS PubMed; (c) D. F. Schreiber, C. O'Connor, C. Grave, Y. Ortin, H. Müller-Bunz and A. D. Phillips, ACS Catal., 2012, 2, 2505–2511 CrossRef CAS; (d) N. Blaquiere, S. Diallo-Garcia, S. I. Gorelsky, D. A. Black and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 14034–14035 CrossRef CAS PubMed.
- (a) T. M. Douglas, A. B. Chaplin and A. S. Weller, J. Am. Chem. Soc., 2008, 130, 14432–14433 CrossRef CAS PubMed; (b) R. Dallanegra, A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2009, 48, 6875–6878 CrossRef CAS PubMed; (c) M. E. Sloan, T. J. Clark and I. Manners, Inorg. Chem., 2009, 48, 2429–2435 CrossRef CAS PubMed; (d) L. J. Sewell, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2012, 134, 3598–3610 CrossRef CAS PubMed; (e) H. C. Johnson and A. S. Weller, Angew. Chem., Int. Ed., 2015, 54, 10173–10177 CrossRef CAS PubMed; (f) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424–9434 CrossRef CAS PubMed.
- (a) M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048–12049 CrossRef CAS PubMed; (b) T. J. Hebden, M. C. Denney, V. Pons, P. M. B. Piccoli, T. G. Koetzle, A. J. Schultz, W. Kaminsky, K. I. Goldberg and D. M. Heinekey, J. Am. Chem. Soc., 2008, 130, 10812–10820 CrossRef CAS PubMed.
- (a) R. J. Keaton, J. M. Balcquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844–1845 CrossRef CAS PubMed; (b) A. P. M. Robertson, R. Suter, L. Chabanne, G. R. Whittell and I. Manners, Inorg. Chem., 2011, 50, 12680–12691 CrossRef CAS PubMed; (c) M. Vogt, B. de Bruin, H. Berke, M. Trincado and H. Grützmacher, Chem. Sci., 2011, 2, 723–727 RSC.
- For examples of early and early-to-mid transition metal catalysts for the dehydrogenation of amine–borane adducts: see, for example; (a) Y. Kawano, M. Uruichi, M. Shimoi, S. Taki, T. Kawaguchi, T. Kakizawa and H. Ogino, J. Am. Chem. Soc., 2009, 131, 14946–14957 CrossRef CAS PubMed; (b) A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 8826–8829 CrossRef CAS PubMed.
- I. Göttker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804–1811 CrossRef PubMed.
- (a) C. Marquardt, T. Jurca, K.-C. Schwan, A. Stauber, A. V. Virovets, G. R. Whittell, I. Manners and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13782–13786 CrossRef CAS PubMed; (b) C. A. De Albuquerque Pinheiro, C. Roiland, P. Jehan and G. Alcaraz, Angew. Chem., Int. Ed., 2018, 57, 1519–1522 CrossRef CAS PubMed.
- M. E. Sloan, A. Staubitz, T. J. Clark, C. A. Russell, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2010, 132, 3831–3841 CrossRef CAS PubMed.
- D. Pun, E. Lobkovsky and P. J. Chirik, Chem. Commun., 2007, 3297–3299 RSC.
- T. Beweries, S. Hansen, M. Kessler, M. Klahn and U. Rosenthal, Dalton Trans., 2011, 40, 7689–7692 RSC.
- Y. Luo and K. Ohno, Organometallics, 2007, 26, 3597–3600 CrossRef CAS.
- C. J. Stevens, R. Dallanegra, A. B. Chaplin, A. S. Weller, S. A. Macgregor, B. Ward, D. McKay, G. Alcaraz and S. Sabo-Etienne, Chem.–Eur. J., 2011, 17, 3011–3020 CrossRef CAS PubMed.
- A similar TiII–TiIV cycle based on theoretical work with 2 as an intermediate: see J. Tao and Y. Qi, J. Organomet. Chem., 2013, 745–746, 479–486 CrossRef CAS.
- (a) The potential presence of TiIII species under catalytic dehydrogenation conditions was initially suggested by the isolation of a TiIII–amido–borane complex [Cp2Ti(NH2BH3)] from the reaction of Cp2TiCl2 with Li[NH2BH3]: see D. J. Wolstenholme, K. T. Traboulsee, A. Decken and G. S. McGrady, Organometallics, 2010, 29, 5769–5772 CrossRef CAS; (b) Recently, TiIII-phosphinoaryloxide species have been shown to catalytically dehydrogenate 1: see M. Klahn, D. Hollmann, A. Spannenberg, A. Brückner and T. Beweries, Dalton Trans., 2015, 44, 12103–12111 RSC.
- H. Helten, B. Dutta, J. R. Vance, M. E. Sloan, M. F. Haddow, S. Sproules, D. Collison, G. R. Whittell, G. C. Lloyd-Jones and I. Manners, Angew. Chem., Int. Ed., 2013, 52, 437–440 CrossRef CAS PubMed.
- (a) Reactions conducted in sealed vessels proceeded at a slower rate than open systems, due to the build-up of H2 pressure; (b) For comparison, in ref. 14 conversion of 1 to 3, by 6a + 2nBuLi (2 mol% in toluene) was complete in 240 min in a system which was periodically opened to draw aliquots for NMR spectroscopy, and thereby releasing H2 gas build up.
- For cga values for analogous CpR2ZrCl2 complexes see: P. C. Möhring and N. J. Coville, Coord. Chem. Rev., 2006, 250, 18–35 CrossRef.
- (a) I. F. Urazowski, V. I. Ponomaryov, O. G. Ellert, I. E. Nifani'ev and D. A. Lemenovskii, J. Organomet. Chem., 1988, 356, 181–193 CrossRef CAS; (b) J. E. Bercaw, R. H. Marvich, L. G. Bell and H. H. Brintzinger, J. Am. Chem. Soc., 1972, 94, 1219–1238 CrossRef CAS.
- IR data (tBuC5H4) see: M. A. El-Hinnawi, M. Y. El-Khateeb, I. Jibril and S. T. Abu-Orabi, Synth. React. Inorg. Met.-Org. Chem., 1989, 19, 809–826 CrossRef CAS . For (C5Me5) see: K. R. Pope and M. S. Wrighton, J. Am. Chem. Soc., 1987, 109, 4545–4552 CrossRef.
- This NMR experiment was performed in THF to provide complete solubility of all substrates in the reaction mixture. The reaction was halted after 8 h by removal of the solvent (see Fig. S10†).
- The lack of solubility prevented purification of the polymers by means of the method described in the ESI.†.
- A. P. M. Robertson, E. M. Leitao, T. Jurca, M. F. Haddow, H. Helten, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2013, 135, 12670–12683 CrossRef CAS PubMed.
- V. Pons, R. T. Baker, N. K. Szymczak, D. J. Heldebrant, J. C. Linehan, M. H. Matus, D. J. Grant and D. A. Dixon, Chem. Commun., 2008, 6597–6599 RSC.
- Due to the optimisation of the reaction for maximised molecular weight and minimised depolymerisation PDIs less than 2 are observed (for an ideal step growth mechanism the Mw/Mn ratio should be 2 after complete monomer conversion).
- These numbers are based on the amount of 4 in solution, and therefore may overestimate conversion due to limited solubility in toluene. In the first 1 h of reaction, there is a observable amount of 4 that is not solubilised, but this is consumed as the reaction progresses.
- The molar mass of the polymer obtained from further treatment of low molar mass 5 with precatalyst is nevertheless significantly lower than that from amine–borane 4 over similar time periods (see Fig. 2). This is consistent with 4 participating as a more reactive substrate in the polycondensation process.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05395a |
| This journal is © The Royal Society of Chemistry 2018 |