
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of quaternary centres by copper catalysed asymmetric conjugate addition to β-substituted cyclopentenones with the aid of a quantitative structure–selectivity relationship†
Ruchuta
Ardkhean
 a,
Mike
Mortimore
b,
Robert S.
Paton
*a and
Stephen P.
Fletcher
*a
a,
Mike
Mortimore
b,
Robert S.
Paton
*a and
Stephen P.
Fletcher
*a
aDepartment of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA. E-mail: robert.paton@colostate.edu
bVertex Pharmaceuticals (Europe) Ltd, 86-88 Jubilee Avenue, Milton Park, Abingdon, Oxfordshire OX14 4RW, UK
First published on 5th February 2018
Abstract
A new asymmetric conjugate addition method was developed for β-substituted cyclopentenones to form quaternary centres using alkylzirconocene nucleophiles giving up to 97% yield and 92% ee. Key to the reaction's success was the design of suitable phosphoramidite ligands which was aided by a linear quantitative structure–selectivity relationship (QSSR). QSSR models were created from the ligand screening data (a total of 36 ligands) which revealed important electronic and steric requirements and led to the synthesis of more enantioselective ligands. DFT calculations of competing transition structures enable the interpretation of the electronic and steric terms present in the QSSR models.
Quaternary centres are important in chemistry and pharmaceuticals1 but, the enantioselective synthesis of these centres is still a challenge.2–4 Five-membered substrates are relatively flat and are recognised as particularly challenging substrates for asymmetric conjugate addition (ACA).3,5 Generally, methods optimised for ACAs to six-membered rings are not suitable or are more limited when applied to 5-membered rings.6–17 Despite the presence of quaternary centres in cyclopentanone-containing in natural products, drugs and related compounds18–23 (Scheme 1), only a handful of methods exist to allow the formation of quaternary centres using this substrate.24,25
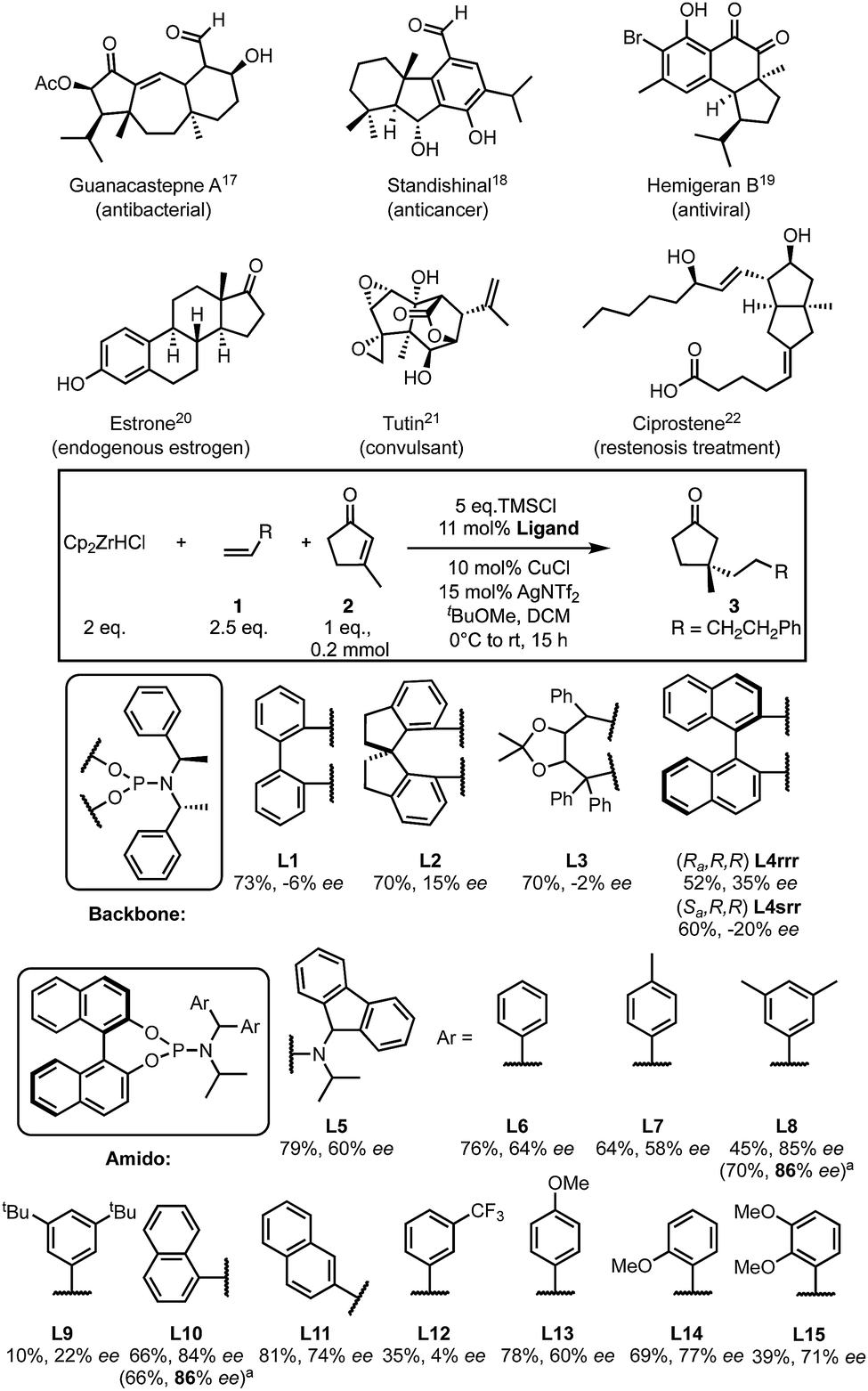 | ||
| Scheme 1 Screening condition and results. Yield based on 1H NMR spectroscopy with MeNO2 as internal standard. Ee were determined by chiral HPLC. aResults from the reaction which give the highest ee out of all repeats. | ||
Conjugate additions are commonly used to generate stereocentres from prochiral starting materials26–29 using various metals including Cu,5,27,30,31 Rh,32 and Pd25,33,34 or by organocatalysis.9,35 We have developed copper catalysed ACAs on β-di-substituted cyclic36 and linear substrates37 to form quaternary centres using alkylzirconium nucleophiles. However, we were unable to develop effective asymmetric additions to the relevant 5-membered ring substrates.38
There are few methods to form quaternary centres that are bound to four sp3-hybridised carbons in cyclopentanone. Alexakis reported the first example of ACA to β-substituted cyclopentenones in 2007 using two trialkylaluminium nucleophiles (AlR3, R = Me and Et).39 In 2008, Hoveyda disclosed, to our knowledge, the only other example of ACA onto unactivated β-substituted cyclopentenones, demonstrating good substrate scope but yet was limited to addition of simple alkyl nucleophiles (10 examples using three trialkylaluminium nucleophiles).24 Here we report a method for quaternary all Csp3 centre formation to β-substituted cyclopentenones with a variety of alkyl nucleophiles.
We started by examining a variety of phosphoramidite ligands using the conditions shown in Scheme 1. Results are reported as average values where repeats were carried out (see ESI†). A preliminary screen of the ligand backbone (for additional backbones, see ESI†) showed that BINOL is most suitable, with a higher enantiomeric excess (ee) being observed. Retaining the BINOL moiety we then examined ligands with variations on the amido group (see ESI†) and found that diaryl ligands gave the most promising results. Chirality of the amido group is not essential for enantioselectivity (Scheme 1). The ee drastically varies with the identity of the aryl from nearly racemic (4% ee with L12) to 86% ee observed with L8 and L10.
Based on these data, some preliminary qualitative conclusions can be drawn; an electron-withdrawing group is detrimental to ee (L12), while electron donating groups have little effect on the enantioselectivity compared to unsubstituted phenyl rings (60–77% ee, L13–15). Additionally, naphthyl groups provide good ee values depending on their substitution pattern (L10, L11).
However, some results were difficult to rationalise. Aryl groups bearing weakly electron-donating alkyl substituents perform variably; p-methyl giving 58% ee (L7), di-meta-methyl substitution gave much better results (86% ee, L8), while increasing the size of the meta-substituents to t-butyl markedly decreases the ee (22% ee, L9). Such non-intuitive trends in ligand structure/product ee relationships are frequently observed in asymmetric catalysis, and particularly in transition-metal catalysed reactions.40 From a qualitative perspective, these data only offer limited additional guidance in ligand design except to avoid electron-withdrawing groups. With the goal of designing better ligands to improve the ee of this transformation we set out to use these results in a more quantitative way.
We recently reported experimental and computational investigations that offer insights into enantioinduction of ACAs to cyclohexenone,41 using QSSR inspired by Sigman's leading work using multidimensional parametric descriptors.42–45 Therefore, to help us design better ligands, we constructed a QSSR model in the form of a linear regression between observed and calculated enantioselectivity (in kJ mol−1) as a function of ligand descriptors. We computed 28 chemical descriptors to describe the ligands as a whole, as well as just the aromatic substituents (see ESI†). We found that the key electronic parameter of the aryl rings is the DFT-computed HOMO energy of the truncated ring with a methyl group. The steric component of the amido-aryl substituents is described by first generation Sterimol parameters:46,47L, B1 and B3, each quantify a different dimension of the aryl rings (Fig. 1).
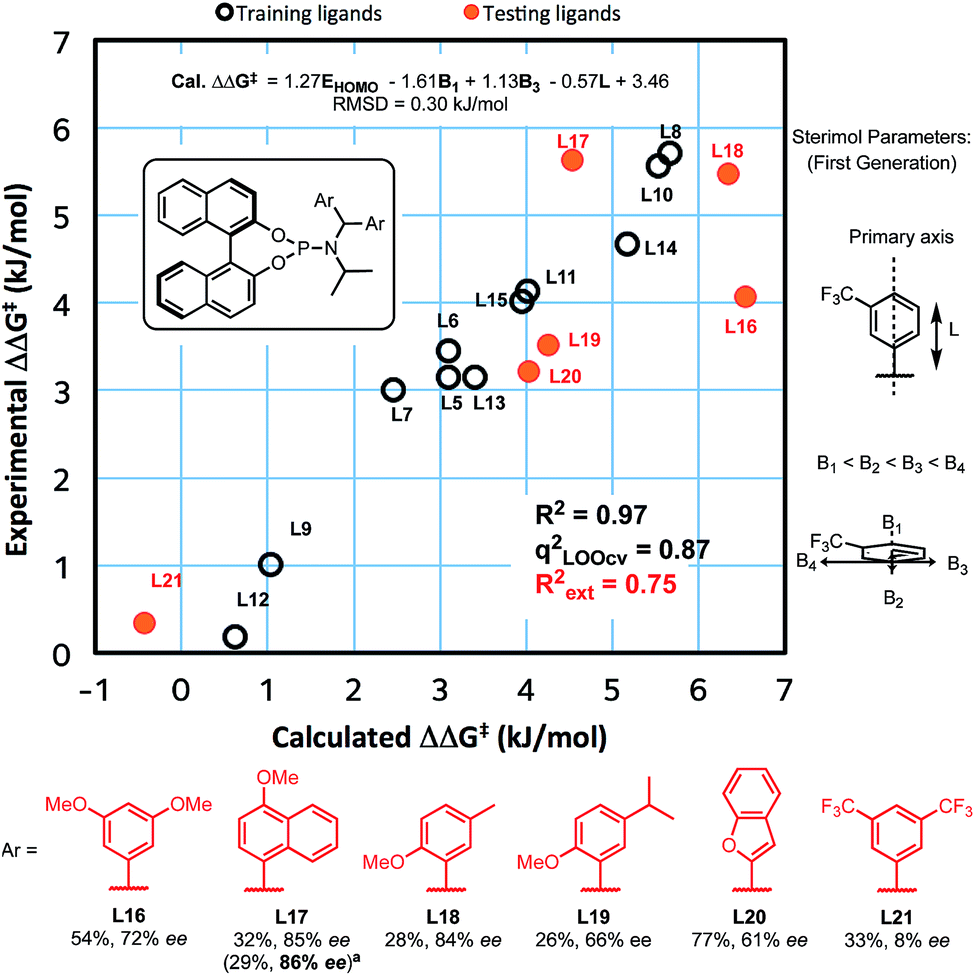 | ||
| Fig. 1 Correlation of experimentally observed enantioselectivity and calculated enantioselectivity (Model I) and results from new testing ligands. Yield based on 1H NMR spectra with MeNO2 as internal standard. Ee's were determined by chiral HPLC. aResults from the reaction which give the highest ee out of all repeats. | ||
The L parameter describes the length of the ring along the bond that connects the substituent to the rest of the ligand. In cases of flat aryl rings, B1 represents the thickness of the ring (minimum width) and B3 represents the width of the ring (width perpendicular to B1). To select the most significant variables out of the 28 collected, we used ‘forward selection’ implemented in the R package (see ESI for details†). From linear regression, Model I was obtained in terms of the normalised parameters:
| ΔΔG‡ (kJ mol−1) = 1.27EHOMO − 1.61B1 + 1.13B3 − 0.57L + 3.46 |
This model shows a good fit (R2 = 0.97) with root mean squared deviation (RMSD) of 0.30 kJ mol−1. Leave-one-out cross-validation (LOOCV) was carried out, giving a q2 of 0.87. To validate the model's predictive power, a testing set of six new ligands was synthesised (shown in red, Fig. 1). The model also performed acceptably for these testing ligands, with an Rext2 = 0.75. One testing set ligand with two CF3 substituents (L21) gave a low ee of 8%. This was captured by the model with a calculated ee close to zero. Analysis of variance (ANOVA) showed that the model and each parameter are statistically significant at 5% level of confidence (see ESI†).
This model contains four ligand descriptors; the HOMO energy of the aryl rings, and three steric parameters: L, B1, B3. The enantioselectivity is positively correlated with the aromatic HOMO energy i.e. more electron rich substituents. To interpret this electronic term, we performed DFT calculations of competing transition structures (TSs) of the stereodetermining carbocupration step (see ESI for details†). The three lowest energy TSs are shown in Fig. 2.
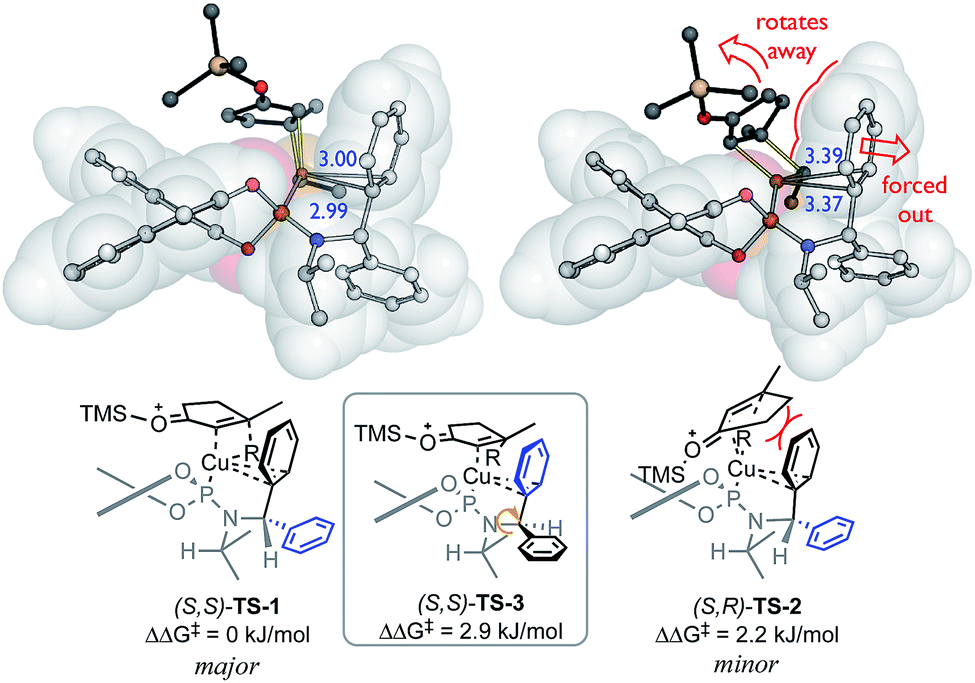 | ||
| Fig. 2 Competing transition structures, SMD(CH2Cl2)-B97D/def2-TZVPP//B97D/6-31G(d)&LANL2DZ level of theory. Distances between Cu and CAr (in blue, Å). | ||
The calculations suggest a favourable non-covalent interaction between Cu and one of the aryl rings – as judged from the interatomic separation and computational (e.g. NCI and NBO) analyses.41,48 This Cu–arene interaction is more favourable in TS-1 leading to the major enantiomer. The minor pathway thought to be viaTS-2, where the bulk of the enone ring disrupts this interaction since it is oriented to force the aromatic substituent away from the Cu centre. This interaction is enhanced by aromatic electron-donating ability, and favours the major enantiomer: a positive correlation between enantioselectivity and EHOMO is therefore expected.
Initially, enantioselectivity data was obtained from ligands with two identical aryl rings. However, the two aryl rings are diastereotopic and so distinct TSs are possible depending on which one coordinates to Cu, as shown in Fig. 2. TS-3 is an alternative conformation leading to the major enantiomer, 3 kJ mol−1 higher in energy than the lowest energy TS-1. Model I shows a correlation of three steric terms, all belong to aryl rings but with both aryls of each ligand being identical, the current training ligands offers no information to distinguish which steric terms associate to which ring (black ring next to copper or the blue ring). To further understand the meaning underlying the steric and electronic terms in QSSR, a set of unsymmetrical diaryl ligand were designed. We synthesised 19 new ligands bearing (R)-BINOL and a chiral amido group with two distinct aryl substituents with two goals in mind; (1) to obtain ligands that are predicted to improve ee as suggested by Model I, and (2) to have a set of ligand that spans suitable chemical space (Scheme 2).
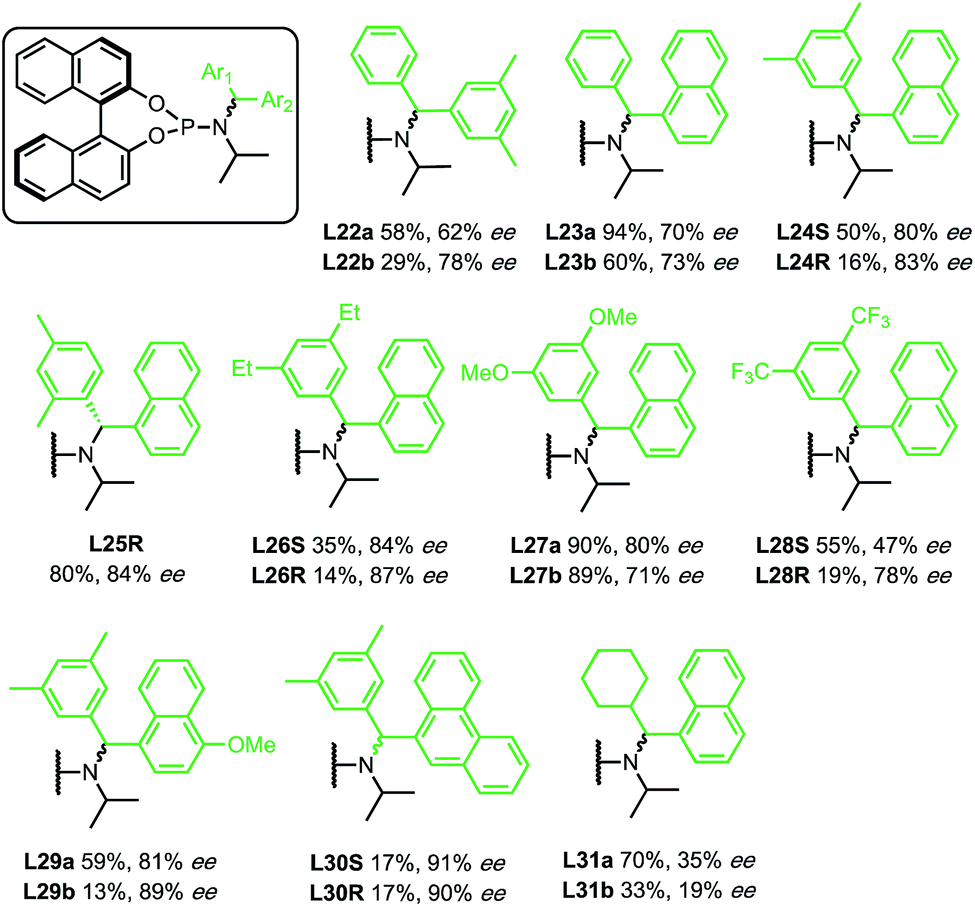 | ||
| Scheme 2 Results from new chiral ligands. Yield based on 1H NMR with MeNO2 as internal standard. Ee's were determined by chiral HPLC. The ligand series ‘a’ corresponds to the ligands that were synthesised using the enantiomer of the amine precursor which have negative αD, and the ligand series ‘b’ corresponds to the ligands that were synthesised using the enantiomer of the amine precursor which have positive αD. | ||
In contrast to the previous set of ligands, these new ligands contain a stereogenic centre at the benzylic carbon. Where possible, the absolute stereochemistry of the ligands or the corresponding amines were identified by X-ray crystallography, otherwise we assume that the diastereomer of the ligand that yields higher ee corresponds to the stereochemistry that will place the aryl with higher HOMO energy next to the copper (see ESI for details†). With data from new ligands in combination with the previous training set, we derived a new QSSR model for all 30 ligands (once again based on a forward selection from a total of 12 normalized electronic and steric parameters), Model II (Fig. 3):
| ΔΔG‡ (kJ mol−1) = 0.74EHOMOR1 + 0.59EHOMOR2 − 1.10B1R1 + 0.65B3R1 + 4.17 |
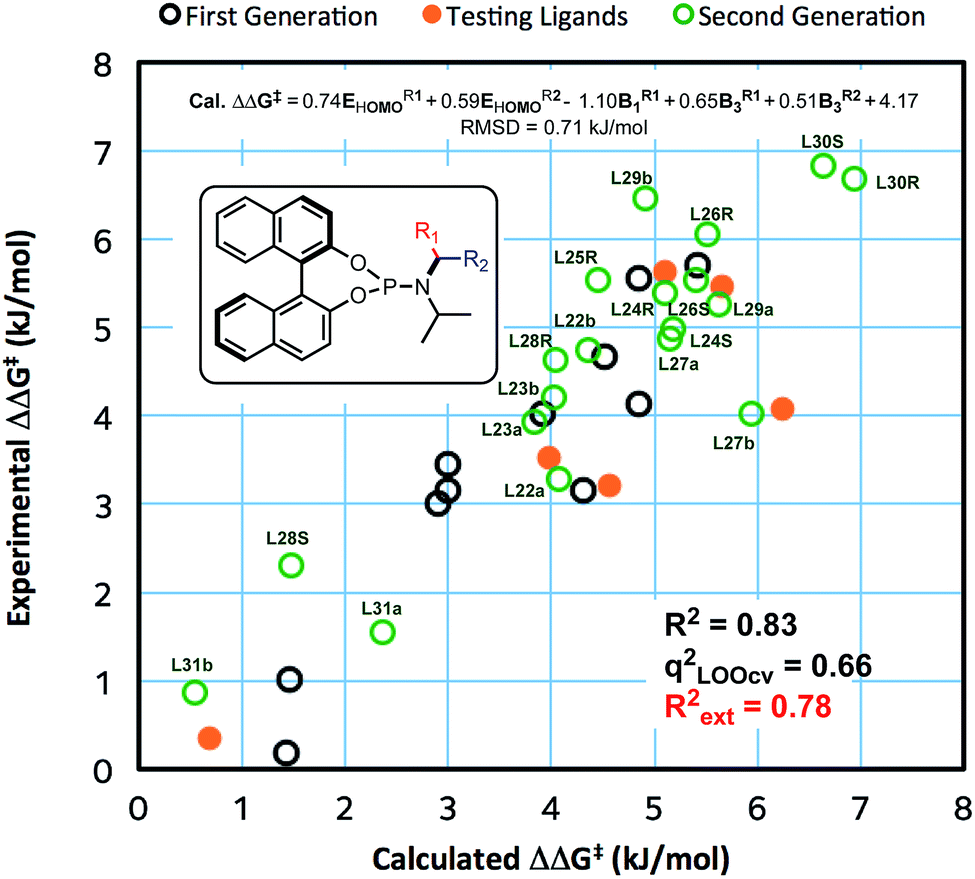 | ||
| Fig. 3 Correlation of experimentally observed and calculated enantioselectivity (Model II). | ||
The previous set of ligands (L5–L15) is shown as black circles and the new ligands (L22–L31) are shown in green. The model has good fit, R2 = 0.83 with RMSD = 0.71 kJ mol−1. Internal validation with LOOCV gave q2 = 0.66. Using the same testing set ligands as the previous models, the model performs well, giving Rext2 = 0.78. ANOVA showed that the model and each parameter are statistically significant at 5% level of confidence except for the term B3R2 which lies at the borderline (see ESI†). This model is balanced between a good fit and using few parameters. Inclusion of the steric parameter LR2 to this model resulted in a new model with a slightly better fit but with fewer degrees of freedom, while exclusion of B3R2 gave lower quality of fit (see ESI†).
Reassuringly, the model shows a positive correlation of enantioselectivity with the aromatic HOMO energy and with the B3 parameter, and a negative correlation with B1 parameter as obtained in Model I. The presence of HOMO terms for both aromatic substituents (R1 and R2) can be taken as an indication that both rings may coordinate to copper, and that transition structures corresponding to both conformations are populated and contribute to the observed enantioselectivity.
Among the new generation of ligands, five give higher ee values (87–91% ee, Scheme 2) than the previous set (86% ee in for L8, L10 and L17, Scheme 1). Unfortunately, these new ligands gave relatively lower yields in the screening conditions. Compromising between product yield and ee, we choose L10 as a ligand to further explore the substrate scope of this reaction under optimised conditions where the solvent is switched to Et2O at 0.40 mmol scale (Scheme 3). Under these conditions, the yield and ee for the model substrate were improved (3; 89%, 90% ee). The optimised conditions were applied to other simple aliphatic alkenes (4–8) giving up to 97% yield and 92% ee. The method is also compatible with functionalised nucleophiles such as silyl, bromide, benzyl-protected alcohol and internal alkynyl groups (9–13; 57–82%, 85–90% ee). Other β-substituted cyclopentenones were also tested giving moderate yield and good ee's (14–16; 54–66%, 82–87% ee).
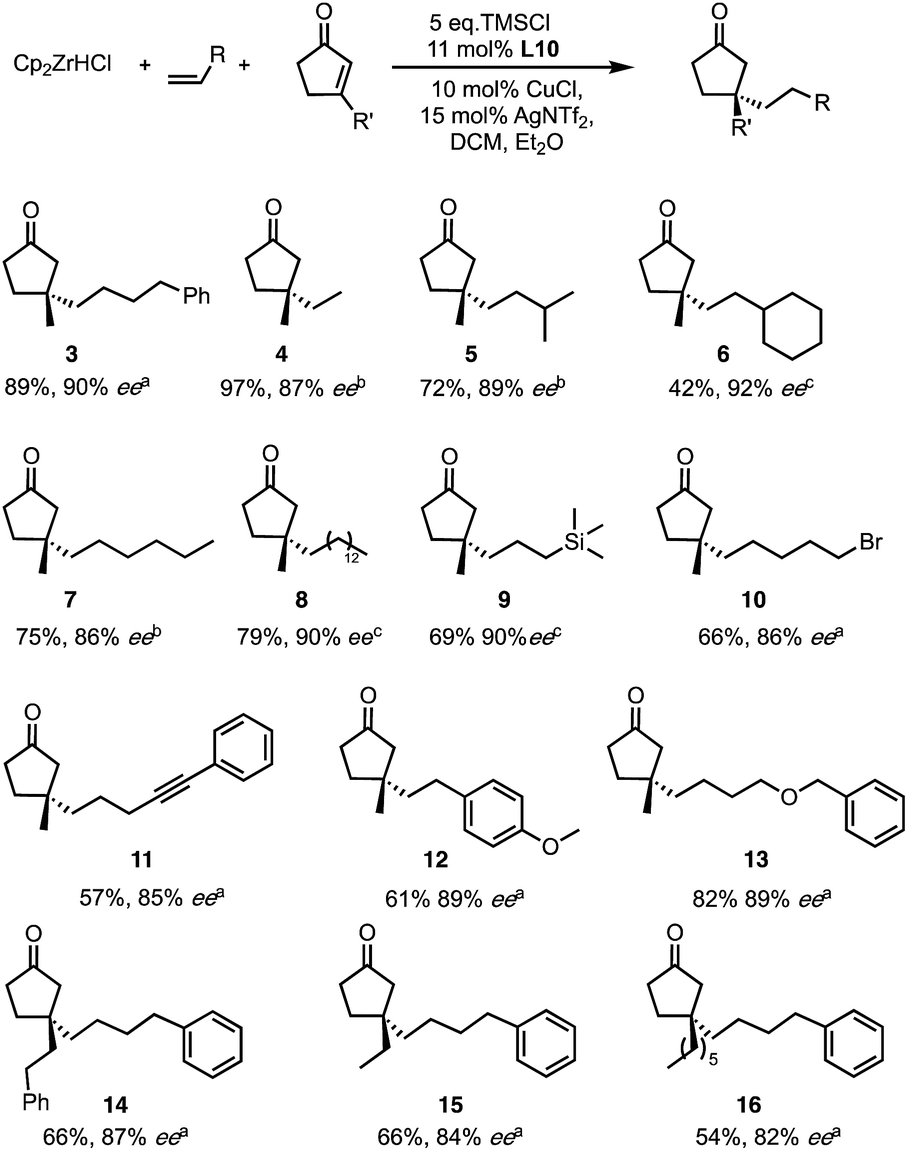 | ||
| Scheme 3 Optimised condition and substrate scope for both reaction partners. Ee determined by aChiral HPLC or SFC, bChiral GC, cDerivatisation followed by HPLC. | ||
Conclusions
In summary, we have developed a method for asymmetric conjugate addition to β-substituted cyclopentenones that works well with functionalised nucleophiles. This method circumvents the handling of highly reactive organometallic species. The nucleophiles are generated in situ from Schwartz reagents and readily available alkenes allowing addition of a much broader range of nucleophiles than previously possible. The design of more enantioselective chiral ligands was initiated by ligand screening in which the data was later used to construct QSSR models to aid ligand design. The key non-covalent interaction between copper and the aryl moiety was established in QSSR models which was structurally substantiated by DFT calculations. The application of this knowledge led to the development of a new set of phosphoramidite ligands that give higher enantioselectivities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Development and Promotion of Science and Technology Talents Project (DPST), Royal Thai Government and the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for a studentship to RA, generously supported by AstraZeneca, Diamond Light Source, Defense Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. We thank Thomas Staroske and Stefan Richardson for chiral separation of the racemic amines, precusors of the phosphoramidite ligands. We thank Tim Claridge for his help in the determination of the diastereomeric ratio of the ligands by NMR spectrometry. Special thanks to Steven Mansfield for his efficient execution of X-ray structure elucidation of the amines and the crystallography team, chemistry Oxford for X-ray structures of the ligands.References
- T. Ling and F. Rivas, Tetrahedron, 2016, 72, 6729–6777 CrossRef CAS.
- C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295–7306 RSC.
- K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed.
- J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564–12580 CrossRef CAS PubMed.
- A. Alexakis, J. E. Bäckvall, N. Krause, O. Pàmies and M. Diéguez, Chem. Rev., 2008, 108, 2796–2823 CrossRef CAS PubMed.
- M. Vuagnoux-d'Augustin and A. Alexakis, Chem.–Eur. J., 2007, 13, 9647–9662 CrossRef PubMed.
- C. Hawner, K. Li, V. Cirriez and A. Alexakis, Angew. Chem., Int. Ed., 2008, 47, 8211–8214 CrossRef CAS PubMed.
- L. Palais and A. Alexakis, Chem.–Eur. J., 2009, 15, 10473–10485 CrossRef CAS PubMed.
- X. Gu, Y. Dai, T. Guo, A. Franchino, D. J. Dixon and J. Ye, Org. Lett., 2015, 17, 1505–1508 CrossRef CAS PubMed.
- K. Kikushima, J. C. Holder, M. Gatti and B. M. Stoltz, J. Am. Chem. Soc., 2011, 133, 6902–6905 CrossRef CAS PubMed.
- D. Müller, C. Hawner, M. Tissot, L. Palais and A. Alexakis, Synlett, 2010, 2010, 1694–1698 CrossRef.
- C. Hawner, D. Müller, L. Gremaud, A. Felouat, S. Woodward and A. Alexakis, Angew. Chem., Int. Ed., 2010, 49, 7769–7772 CrossRef CAS PubMed.
- S. Kehrli, D. Martin, D. Rix, M. Mauduit and A. Alexakis, Chem.–Eur. J., 2010, 16, 9890–9904 CrossRef CAS PubMed.
- L. Palais and A. Alexakis, Chem.–Eur. J., 2009, 15, 10473–10485 CrossRef CAS PubMed.
- M. W. Paixão, N. Holub, C. Vila, M. Nielsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 7338–7342 CrossRef PubMed.
- S. Liu, Q. Wang, L. Ye, Z. Shi, Z. Zhao, X. Yang, K. Ding and X. Li, Tetrahedron, 2016, 72, 5115–5120 CrossRef CAS.
- D. Martin, S. Kehrli, M. d'Augustin, H. Clavier, M. Mauduit and A. Alexakis, J. Am. Chem. Soc., 2006, 128, 8416–8417 CrossRef CAS PubMed.
- S. F. Brady, M. P. Singh, J. E. Janso and J. Clardy, J. Am. Chem. Soc., 2000, 122, 2116–2117 CrossRef CAS.
- G. Majetich and J. M. Shimkus, J. Nat. Prod., 2010, 73, 284–298 CrossRef CAS PubMed.
- K. D. Wellington, R. C. Cambie, P. S. Rutledge and P. R. Bergquist, J. Nat. Prod., 2000, 63, 79–85 CrossRef CAS.
- H. Kuhl, Climacteric, 2005, 8, 3–63 CrossRef CAS PubMed.
- H. Zhou, Y.-H. Tang and Y. Zheng, Brain Res., 2006, 1092, 207–213 CrossRef CAS PubMed.
- W.-K. P. Cho, Methods for Inhibiting Sterol Absorption, US Pat., 7612058, 2009.
- T. L. May, M. K. Brown and A. H. Hoveyda, Angew. Chem., Int. Ed., 2008, 47, 7358–7362 CrossRef CAS PubMed.
- J. Buter, R. Moezelaar and A. J. Minnaard, Org. Biomol. Chem., 2014, 12, 5883–5890 CAS.
- K. Soai and S. Niwa, Chem. Rev., 1992, 92, 833–856 CrossRef CAS.
- A. H. M. de Vries, A. Meetsma and B. L. Feringa, Angew. Chem., Int. Ed. Engl., 1996, 35, 2374–2376 CrossRef CAS.
- N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 2001, 0171–0196 CrossRef.
- M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033–8061 CrossRef CAS.
- T. Jerphagnon, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2009, 38, 1039–1075 RSC.
- B. Maciá, in Progress in Enantioselective Cu(I)-catalyzed Formation of Stereogenic Centers, Springer, Cham, 2015, pp. 41–98 Search PubMed.
- T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed.
- S. E. Shockley, J. C. Holder and B. M. Stoltz, Org. Process Res. Dev., 2015, 19, 974–981 CrossRef CAS PubMed.
- J. C. Holder, L. Zou, A. N. Marziale, P. Liu, Y. Lan, M. Gatti, K. Kikushima, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 14996–15007 CrossRef CAS PubMed.
- S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 2007, 1701–1716 CrossRef.
- P. M. C. Roth, M. Sidera, R. M. Maksymowicz and S. P. Fletcher, Nat. Protoc., 2014, 9, 104–111 CrossRef CAS PubMed.
- Z. Gao and S. P. Fletcher, Chem. Sci., 2016, 8, 641–646 RSC.
- M. Sidera, P. M. C. Roth, R. M. Maksymowicz and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 7995–7999 CrossRef CAS PubMed.
- M. Vuagnoux-d'Augustin, S. Kehrli and A. Alexakis, Synlett, 2007, 2007, 2057–2060 CrossRef.
- R. L. Reyes, T. Harada, T. Taniguchi, K. Monde, T. Iwai and M. Sawamura, Chem. Lett., 2017, 46, 1747–1750 CrossRef CAS.
- R. Ardkhean, P. M. C. Roth, R. M. Maksymowicz, A. Curran, Q. Peng, R. S. Paton and S. P. Fletcher, ACS Catal., 2017, 7, 6729–6737 CrossRef CAS.
- K. C. Harper, E. N. Bess and M. S. Sigman, Nat. Chem., 2012, 4, 366–374 CrossRef CAS PubMed.
- M. Orlandi, M. J. Hilton, E. Yamamoto, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 12688–12695 CrossRef CAS PubMed.
- J.-Y. Guo, Y. Minko, C. B. Santiago and M. S. Sigman, ACS Catal., 2017, 7, 4144–4151 CrossRef CAS.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292–1301 CrossRef CAS PubMed.
- A. Verloop, W. Hoogenstraaten and J. Tipker, in Drug Design, ed. E. J. Ariens, 1976, vol. 7, p. 165 Search PubMed.
- T. Piou, F. Romanov-Michailidis, M. Romanova-Michaelides, K. E. Jackson, N. Semakul, T. D. Taggart, B. S. Newell, C. D. Rithner, R. S. Paton and T. Rovis, J. Am. Chem. Soc., 2017, 139, 1296–1310 CrossRef CAS PubMed.
- R. N. Straker, Q. Peng, A. Mekareeya, R. S. Paton and E. A. Anderson, Nat. Commun., 2016, 7, 10109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1811310–1811312, 1818574 and 1818575. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05304e |
| This journal is © The Royal Society of Chemistry 2018 |