
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct evidence of charge separation in a metal–organic framework: efficient and selective photocatalytic oxidative coupling of amines via charge and energy transfer†
Caiyun
Xu‡
a,
Hang
Liu‡
a,
Dandan
Li
a,
Ji-Hu
Su
b and
Hai-Long
Jiang
 *a
*a
aHefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Soft Matter Chemistry, Collaborative Innovation Center of Suzhou Nano Science and Technology, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: jianglab@ustc.edu.cn; Web: http://staff.ustc.edu.cn/∼jianglab/
bCAS Key Laboratory of Microscale Magnetic Resonance, Synergetic Innovation Center of Quantum Information and Quantum Physics, Department of Modern Physics, University of Science and Technology of China, Hefei 230026, P. R. China
First published on 20th February 2018
Abstract
The selective aerobic oxidative coupling of amines under mild conditions is an important laboratory and commercial procedure yet a great challenge. In this work, a porphyrinic metal–organic framework, PCN-222, was employed to catalyze the reaction. Upon visible light irradiation, the semiconductor-like behavior of PCN-222 initiates charge separation, evidently generating oxygen-centered active sites in Zr-oxo clusters indicated by enhanced porphyrin π-cation radical signals. The photogenerated electrons and holes further activate oxygen and amines, respectively, to give the corresponding redox products, both of which have been detected for the first time. The porphyrin motifs generate singlet oxygen based on energy transfer to further promote the reaction. As a result, PCN-222 exhibits excellent photocatalytic activity, selectivity and recyclability, far superior to its organic counterpart, for the reaction under ambient conditions via combined energy and charge transfer.
Introduction
Efficient and selective oxidation reactions based on molecular oxygen (O2) or even ambient air as the oxidant have received tremendous attention from the view of green chemistry. In particular, the selective oxidative coupling of amines to imine derivatives as important building blocks for the synthesis of fine chemicals and pharmaceuticals is highly desired.1 Diverse metal complex catalysts have been reported toward the aerobic oxidative coupling of amines. Unfortunately, relatively high temperature (∼373 K) and/or oxygen pressure (∼5 atm) are required to proceed.1,2 Alternatively, photo-induced aerobic oxidation emerges to be an effective and sustainable approach under moderate conditions. Photosensitizer molecules absorb sunlight to generate singlet oxygen (1O2) that can promote amine oxidative coupling. However, with 1O2 as a sole oxidant, the reaction efficiency needs to be improved and homogeneous systems are associated with intrinsic difficulty of catalyst separation and/or intermolecular deactivation.3 In contrast, heterogeneous semiconductor photocatalysts possess excellent catalytic performance for the aerobic oxidative coupling of amines, in which photogenerated electrons transferring to O2 to give superoxide radicals (O2˙−) and amines activated by the remaining holes are proposed. Though O2˙− is detectable via the electron spin resonance (ESR) technique, actually, the oxidation product has never been evidenced and the reaction mechanism has remained disputable in heterogeneous catalytic systems thus far.4 Moreover, given the strong oxidation ability of holes, semiconductor photocatalysts usually produce aldehyde byproducts, lowering the selectivity to imines.5 Therefore, the exploration of a specific mechanism regarding this reaction over semiconductor-like photocatalysts for excellent activity and selectivity is imperative.Metal–organic frameworks (MOFs) are a class of crystalline porous materials and exhibit potential applications in diverse fields.6–8 The well-defined structures constructed from metal (oxide) nodes and organic linkers endow MOFs with semiconductor-like behavior for photocatalysis.7,8 Upon light irradiation, different processes might occur in MOFs, such as ligand-to-metal charge transfer (LMCT), metal-to-ligand charge transfer (MLCT), π–π* transition of the delocalized linker, etc.8 However, only a half-reaction radical intermediate, usually electron-reduced metal ions, was detected in previous reports, while the radical cation (e.g., excited ligand) cannot be recognized, possibly due to its very short lifetime. Solid evidence collection for both electron- and hole-involved intermediates in MOF photocatalysts remains a highly desired goal to understand charge separation in MOFs.
In this work, a representative porphyrinic MOF, PCN-222 (also known as MOF-545 or MMPF-6),9 was chosen for the aerobic oxidative coupling of amines under visible light and ambient conditions. The porphyrin linkers in PCN-222 can be excited by visible light and the photogenerated electrons transfer to Zr-oxo clusters to form oxygen-centered active sites, leaving enhanced porphyrin π-cation radical signals by hole oxidation, both of which have been detected via ESR. The results clearly manifest the linker-to-cluster charge transfer (LCCT) procedure, accounting for the semiconductor-like behavior of PCN-222. The electrons and holes further transfer and produce O2˙− and benzylamine radicals (PhCH˙NH2 and PhCH2NH˙), respectively, which have been directly evidenced in heterogeneous photocatalytic systems for the first time, synergistically promoting the selective formation of target imines. In addition, the energy transfer (ET) gives rise to 1O2 generation over porphyrin motifs by light irradiation. The combination of charge and energy transfer processes in PCN-222 leads to its excellent catalytic activity and selectivity under ambient conditions (Scheme 1).
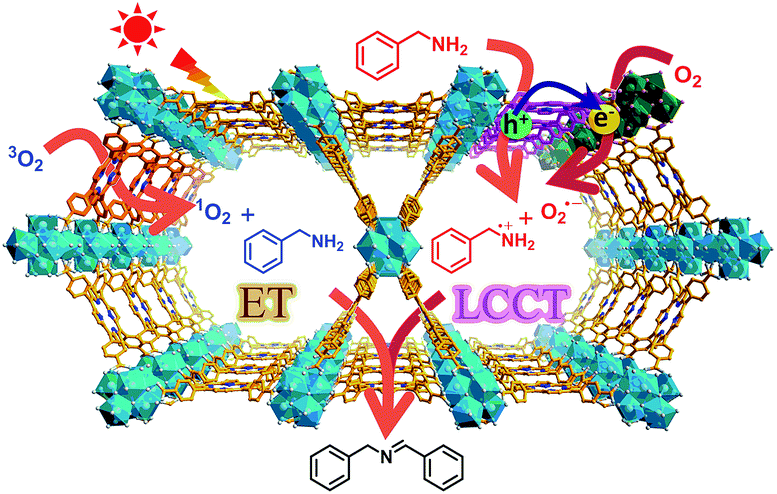 | ||
| Scheme 1 Schematic illustration of LCCT and ET processes involved in PCN-222 for the selective oxidative coupling of amines under visible light irradiation. | ||
Results and discussion
The PCN-222, Zr6(μ3-OH)8(OH)8(TCPP)2 (TCPP = tetrakis(4-carboxyphenyl)porphyrin), was selected as it features a 3D structure with large channels larger than 3 nm and a high BET surface area of 1659 m2 g−1 (Fig. S1†). Importantly, it exhibits a strong UV-Vis response in the 300–800 nm range, inheriting the Soret band (∼420 nm) and four Q bands (500–700 nm) from the porphyrin ligand (Fig. S2†). The Soret band and Q bands are deemed to originate from two different transitions from the HOMOs (a1u and a2u) to the LUMO (eg),10 indicating the similar electronic structure of PCN-222 and the porphyrin molecule TPPCOOMe. This hypothesis is supported by density functional theory (DFT) calculations (Fig. S3 and S4†) and cyclic voltammetry measurements (Fig. S5†). Therefore, behaving like a semiconductor, PCN-222 can be excited by visible light and the photogenerated electrons can transfer from the porphyrin ligand to Zr-oxo clusters, where the HOMO and LUMO are located, respectively.7g Based on the UV-Vis spectra and electrochemical characterization of PCN-222 and TPPCOOMe, their energy diagrams of the HOMO−n, HOMO and LUMO can be clearly given (Fig. S6†).11 Given the more negative potential of the LUMO level (−0.39 V vs. NHE) in PCN-222 than the reduction potential of O2 to related reactive oxygen species (ROS, E(O2/O2˙−) = −0.33 V vs. NHE),10a it is theoretically feasible for photocatalytic O2˙− generation. The porphyrin ligand would act as a visible-light-harvesting unit and contribute to the HOMO and HOMO−n at +1.43 V and +2.20 V vs. NHE, respectively, calculated by using the Tauc plot (Fig. S6†).11 The relatively lower occupied molecular orbital level guarantees a high oxidation ability for benzylamine (E1/2(M+/M) of ∼+1.47 V vs. NHE).12The above results encourage us to investigate the oxidative coupling of benzylamines over PCN-222 in air under visible light irradiation (Table 1). Acetonitrile was found to be the most suitable medium among different solvents examined (Table S1†). Benzylamine can be completely converted to the corresponding target product, N-benzylidenebenzylamine, in 1 h over PCN-222, where light irradiation and air atmosphere are necessary (entries 1–4). A higher activity can be achieved under an O2 atmosphere (entry 5). Moreover, this catalyst exhibits excellent activity under simulated solar irradiation (entry 6), extending its potential to practical application. Upon gradually increasing the light intensity, the conversion increases accordingly, reflecting the photo-driven process (Fig. S7†). In contrast, the active motif, 5,10,15,20-tetrakis(4-methoxycarbonyl phenyl)porphyrin (TPPCOOMe), gives only a 26.7% yield and a much lower activity than PCN-222 with a fixed porphyrin amount (entry 7; Fig. S8†), which indicates that the porphyrin ligand itself cannot give the high efficiency of PCN-222. The stability and reusability of PCN-222 were examined and its activity was maintained very well in five consecutive runs (Fig. S9†). After that, the powder X-ray diffraction pattern of PCN-222 revealed its structural integrity and stability (Fig. S10†). After 30 min of reaction, the catalyst was filtered out and the results showed that no further product was formed even after 1.5 h (Fig. S11†), indicating that the process should be truly heterogeneous. Therefore, PCN-222 possesses truly heterogeneous nature with excellent activity, selectivity, stability and recyclability toward the photocatalytic oxidative coupling of benzylamine.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Air | Light | t [h] | Conv.b [%] |
| a Reaction conditions: 0.1 mmol benzylamine, 5 mg PCN-222, 100 mW cm−2 Xe lamp cutoff below 420 nm, 3 mL CH3CN, and air atmosphere. b Determined by GC analysis. c Under a 1 atm N2 atmosphere. d Under a 1 atm O2 atmosphere. e Under an AM 1.5 illumination of 100 mW cm−2. f 3.6 mg TPPCOOMe. | |||||
| 1 | PCN-222 | + | + | 1 | 100 |
| 2 | — | + | + | 2 | 1 |
| 3 | PCN-222 | + | − | 2 | — |
| 4c | PCN-222 | − | + | 1 | 44.4 |
| 5d | PCN-222 | − | + | 0.75 | 100 |
| 6e | PCN-222 | + | + | 1.5 | 95.6 |
| 7f | TPPCOOMe | + | + | 1 | 26.7 |
Upon the optimization of reaction conditions by using benzylamine as a probe substrate, to demonstrate the general applicability of PCN-222, different amines have been examined (Table 2). Primary benzylamine derivatives substituted with various functional groups were converted to corresponding imines with excellent conversions, in which the oxidation of the benzylamines with electron-donating groups proceeded slightly faster than those with electron-withdrawing groups (entries 1–8). The reaction rate of the regioisomers decreased in the para > meta > ortho order, indicating the presence of the steric effect (entries 2–4).4a Heterocyclic amines containing N, O and S atoms, which usually poison metal catalysts,4a can be well tolerated and displayed excellent yields (entries 9–11). Moreover, both secondary and cyclic amines were amenable to the procedure, giving the corresponding imines in satisfactory yields (entries 12 and 13). However, the oxidative coupling of aniline without a hydrogen atom at its α-carbon did not proceed (entry 14), suggesting that reaction might occur via hydrogen abstraction.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | t [min] | Conv.b [%] | Sel.b [%] |
| a Reaction conditions: 0.1 mmol amine, 5 mg PCN-222, 100 mW cm−2 Xe lamp cutoff below 420 nm, 3 mL CH3CN, and air atmosphere. b Determined by GC-MS. | |||||
| 1 | X = H | X = H | 60 | 100 | 100 |
| 2 | X = o-OCH3 | X = o-OCH3 | 65 | 100 | 100 |
| 3 | X = m-OCH3 | X = m-OCH3 | 55 | 100 | 100 |
| 4 | X = p-OCH3 | X = p-OCH3 | 45 | 100 | 100 |
| 5 | X = p-CH3 | X = p-CH3 | 60 | 100 | 100 |
| 6 | X = p-F | X = p-F | 70 | 100 | 100 |
| 7 | X = p-Cl | X = p-Cl | 65 | 100 | 100 |
| 8 | X = p-CF3 | X = p-CF3 | 75 | 100 | 100 |
| 9 |
|
|
80 | 100 | 100 |
| 10 |
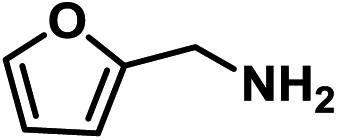
|
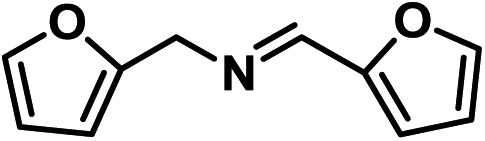
|
65 | 100 | 100 |
| 11 |
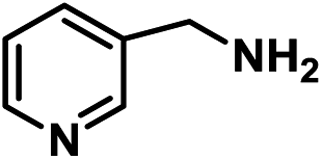
|
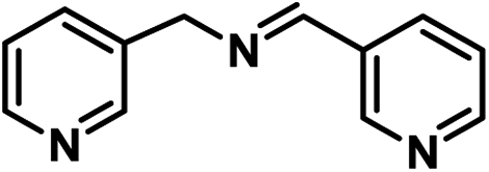
|
60 | 100 | 100 |
| 12 |
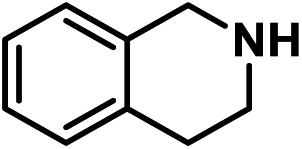
|

|
70 | 100 | 100 |
| 13 |
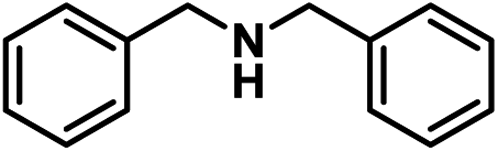
|
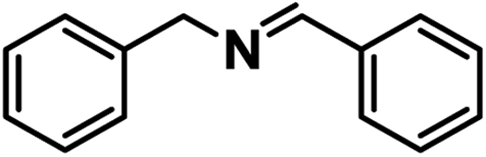
|
60 | 100 | 100 |
| 14 |
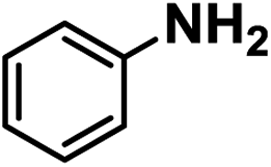
|
|
120 | — | — |
It is necessary to elucidate the reaction mechanism behind the above excellent photocatalytic performance of PCN-222. The light-induced porphyrin π-cation radical was monitored in situ by using the ESR technique (Fig. S12–S14†). Interestingly, the porphyrin ligand (TPPCOOMe) shows an unchanged ESR signal while PCN-222 gives a much enhanced porphyrin π-cation radical signal upon light irradiation, as shown by the ESR light–dark difference spectra (Fig. S13 and S14†), indicating the generation of photogenerated radicals (i.e., e–h pairs) in PCN-222, compared to porphyrin molecules.10a Hence, the effective electron transfer in PCN-222 results in the remaining holes contributing to the enhanced ESR signal of cation radicals. The electron transfer destination from the ligand in PCN-222 can be deduced by using the ESR technique as well. A new ESR signal at giso = 2.0021 (close to the value of free electrons, ge-2.0023) emerged and its signal intensity strengthened with a prolonged irradiation time (Fig. 1a), which might originate from oxygen-centered active sites in Zr-oxo clusters.13 The above results demonstrate that photo-generated electrons transfer from the porphyrin ligand to Zr-oxo clusters, generating oxygen-centered active sites in Zr-oxo clusters and porphyrin π-cation radicals in the ligand, which, unprecedentedly provide direct evidence for the LCCT procedure in a MOF.
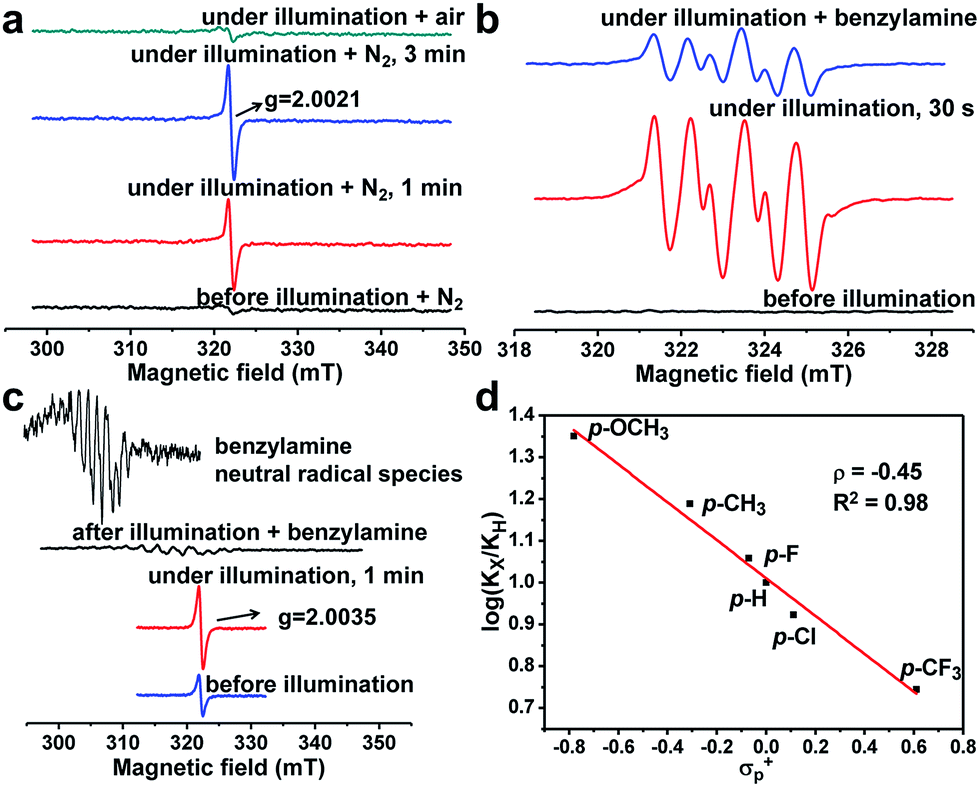 | ||
| Fig. 1 (a) ESR detection of the electron transfer process in PCN-222. (b) ESR detection of O2˙− generation over PCN-222 trapped by DMPO. (c) The detection of hole-involved intermediates in PCN-222. (d) Hammett plot for the oxidation of benzylamine and its para-substituted derivatives photocatalyzed by PCN-222. All ESR tests were described in detail in the Experimental section. | ||
More evidence has been gleaned to track the transfer pathway of photo-generated carriers occurred in PCN-222 and reactants. Upon exposure to O2/air, the signal at g = 2.0021 was eliminated (Fig. 1a), indicating that the formed active sites possess reducibility and are able to reduce O2 to O2˙− species (shown below further). It has been well established that t-butanol and 1,4-benzoquinone (BQ, an known electron inhibitor) can effectively inhibit the generation of OH˙ and O2˙−, respectively,14 in which only BQ can effectively inhibit the reaction (Fig. S15†), possibly due to the high oxidation potential of E(–OH/˙OH) (+2.80 V vs. NHE) that is insufficient for the HOMO−n of PCN-222.10 No target product was detected by introducing H2O2 (Table S2†), indicating that it is not the ROS. In addition, 5,5-dimethyl-pyrroline-N-oxide (DMPO) was employed as the O2˙−-sensitive trapping agent, further manifesting the O2˙− formation over PCN-222 (Fig. 1b).4a Upon the addition of benzylamine, the signal intensity of DMPO-O2˙− evidently decreases, suggesting the strong interaction between benzylamine and O2˙−. These results jointly point out that electron transfer occurs between Zr-oxo clusters and O2 to generate O2˙− as the ROS for the subsequent oxidative coupling of amines. In addition, the photocatalytic oxidation rates of benzylamine and its para-substituted derivatives (X = OMe, Me, H, F, Cl, and CF3 groups) were examined over PCN-222 to obtain the Hammett plot (Fig. 1d). A reasonable linearity between log(kx/kh) values and Brown–Okamoto constant (σp+) parameters15 was obtained with a negative slope, indicating the hole transfer from porphyrin π-cation radicals in PCN-222 to benzylamine to give a positively charged intermediate, possibly PhCH2NH2˙+.4a Once benzylamine was injected, the porphyrin π-cation radical ESR signal was quenched immediately; a set of weak signals simultaneously emerged that can be assigned to the combined signals of benzyl-type carbon-centered (PhCH˙NH2) and nitrogen-centered (PhCH2NH˙) radicals (Fig. 1c, S16 and S17†), generated from the hydrogen abstraction from PhCH2NH2˙+,16 indicative of hole transfer from porphyrin π-cation radicals to benzylamine.
The specific catalytic sites in the charge transfer process were further determined by the analysis of the energy diagram of PCN-222. The steady-state photoluminescence (PL) spectra show that the excitation of the Soret band (λ = 420 nm) leads to much lower PL intensity in PCN-222, which indicates the rapid and effective charge transfer occurred in this process (Fig. S2 and S18†).8e In contrast, the PL is stronger than that of TPPCOOMe upon the excitation of Q bands (λ = 520 nm). The results clearly support the previous argument that the transition from the HOMO−n to the LUMO in PCN-222 mainly contributes to the benzylamine oxidation. Further photocurrent tests also prove this point, as the photocurrent under a single excitation wavelength (i.e., λ = 420 nm) accounts for a greater portion of the total photocurrent (Fig. S19b†).
With the above data in hand, the charge transfer mechanism in PCN-222 for the photocatalytic oxidative coupling of benzylamine has been proposed (Fig. 2a). Upon irradiation, the LCCT process mainly from the HOMO−n to the LUMO in PCN-222 gives rise to effective e–h separation, which reduces O2 to O2˙− and oxidizes benzylamine to PhCH2NH2˙+, respectively. Thanks to the synergistic effect of electron–hole pairs, proton transfer from PhCH2NH2˙+ to O2˙− takes place under mild conditions, generating hydrogen abstracted benzylamine radicals (PhCH˙NH2 and PhCH2NH˙) and hydroperoxyl radicals (HO2˙), which are converted to benzylideneamine (PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH) and H2O2. The H2O2 byproduct has been detected in the reaction system via a well-established method (Fig. S20†). The PhCHNH further reacts with benzylamine to produce the target product.16
NH) and H2O2. The H2O2 byproduct has been detected in the reaction system via a well-established method (Fig. S20†). The PhCHNH further reacts with benzylamine to produce the target product.16
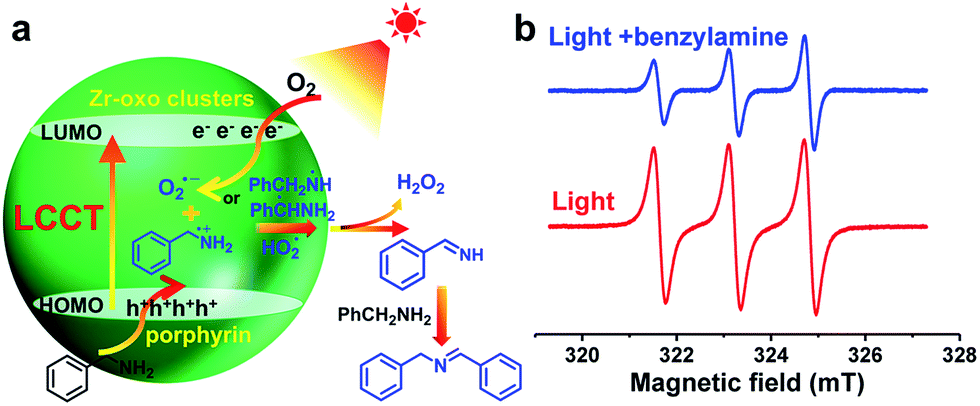 | ||
| Fig. 2 (a) Proposed charge transfer process for the photocatalytic oxidative coupling of benzylamine over PCN-222, in which the complex HOMO constitution is simplified. (b) ESR spectra (light–dark difference) of PCN-222 for the 1O2 detection in the presence of 4-oxo-TMP. | ||
In addition to the O2˙− species generated during the charge transfer process, photosensitized porphyrin motifs can react with ground state oxygen (3O2) to generate 1O2via triplet–triplet annihilation (TTA) in PCN-222.14b To evidence 1O2 generation, 4-oxo-TMP, the classical 1O2 probe, was employed to display intense characteristic signals (g = 2.0055, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet) of 4-oxo-TEMPO under light irradiation for PCN-222 (Fig. 2b and S21†). The signals evidently decreased upon the addition of benzylamine, manifesting that 1O2 indeed participates in the reaction. The addition of an 1O2 scavenger remarkably impaired the conversion (Fig. S15†), again supporting the 1O2 production. The generated 1O2 based on the energy transfer process featuring high electrophilicity would directly oxidize amines to imines.3,12 Therefore, the combination of O2˙− generation via charge transfer and 1O2 production via energy transfer contributes to the ultimate great activity of PCN-222 (Scheme 1 and Fig. S22†).
:1:1 triplet) of 4-oxo-TEMPO under light irradiation for PCN-222 (Fig. 2b and S21†). The signals evidently decreased upon the addition of benzylamine, manifesting that 1O2 indeed participates in the reaction. The addition of an 1O2 scavenger remarkably impaired the conversion (Fig. S15†), again supporting the 1O2 production. The generated 1O2 based on the energy transfer process featuring high electrophilicity would directly oxidize amines to imines.3,12 Therefore, the combination of O2˙− generation via charge transfer and 1O2 production via energy transfer contributes to the ultimate great activity of PCN-222 (Scheme 1 and Fig. S22†).
To identify the ROS that are responsible for the activity of TPPCOOMe, scavenging experiments were conducted on the ligand. Among different ROS scavengers, the conversion can be completely inhibited in the presence of an 1O2 scavenger (Fig. S23†). In sharp contrast to that of PCN-222, the reaction over TPPCOOMe was almost insusceptible to the introduction of ˙OH and O2˙− scavengers and less affected by hole scavengers (Fig. S24†). The characteristic 1O2 ESR signal intensity for TPPCOOMe is similar to that for PCN-222 (Fig. S25†), while the signal intensity of O2˙− radicals in TPPCOOMe is much weaker (Fig. S26†), due to the much lower charge separation efficiency in the organic entity (Fig. S18a and S19a†). Given the above results, compared with the cooperative effect of O2˙− and 1O2 in the reaction over PCN-222, 1O2 being the nearly sole ROS accounts for the poor activity of TPPCOOMe (Table 1, entry 7). In addition, there is no obvious linear correlation between log(kx/kh) and σp+ in the Hammett plot (Fig. S27†), indicating the lack of positively charged intermediates produced by hole activation for the process over TPPCOOMe.
Experimental section
Synthesis of the porphyrin ligand (H2TCPP)
The tetrakis(4-carboxyphenyl)porphyrin (H2TCPP) ligand was synthesized according to the previous report with minor modifications.17 The typical synthetic procedures are described below.Synthesis of PCN-222
The MOF was synthesized based on the previous report with some modifications.17 Typically, ZrOCl2·8H2O (108.6 mg), H2TCPP (30 mg) and CF3COOH (0.45 mL) were ultrasonically dissolved in 10 mL of DMF in a 20 mL Pyrex vial. The mixture was heated at 120 °C in an oven for at least 16 h. Purple needle shaped crystals were harvested by filtration and washed with DMF and acetone, respectively. Prior to use, the solid was soaked in 100 mL acetone for 48 h to exchange out DMF, and then filtered and dried in a vacuum.Activity evaluation on photocatalytic oxidative coupling of amines
In a typical procedure, 5 mg of PCN-222 or 3.6 mg of TPPCOOMe as a photocatalyst was dispersed in 3 mL of solvent, followed by the addition of 0.1 mmol of amine substrate in a 5 mL Pyrex vial equipped with a rubber septum and a tiny syringe needle connected the reaction system with the surrounding air atmosphere outside. The sample was stirred vigorously and irradiated with a 300 W xenon lamp equipped with an ultraviolet-cutoff filter (cutting out light below 420 nm) at a fixed light intensity of 100 mW cm−2 unless otherwise stated. Then, the conversion was quantified by gas chromatography and the products were confirmed by GC-MS after centrifugation. The mass of TPPCOOMe was calculated based on the equivalent amount of porphyrin motifs in PCN-222. The optimized reaction conditions for the photocatalytic oxidative coupling reaction of amines were determined, which consist of 5 mg PCN-222, 0.1 mmol amine, 3 mL CH3CN, under a Xe lamp cutoff below 420 nm at a light intensity of 100 mW cm−2 and ambient conditions (in the surrounding air atmosphere).000 rpm for 2 min after each cycle and washed with CH3CN 2 times. Then the catalyst was reused for the subsequent run with fresh benzylamine (0.1 mmol) under the optimized reaction conditions.
ESR measurements
ESR was recorded on a JES-FA200 electron paramagnetic resonance spectrometer under visible-light irradiation (λ > 400 nm). The in situ ESR tests were conducted with a homemade tube with a special setup (Fig. S12†).Detection of hydrogen peroxide (H2O2)
The production of H2O2 was detected by a well-established N,N-diethyl-p-phenylenediamine (DPD)/horseradish peroxidase (POD) method.19 Typically, 100 mg of DPD was dissolved in 10 mL of H2SO4 (0.05 M) solution and 100 mg of POD was dissolved in 10 mL of distilled water, both of which were stored in the dark at below 278 K before use. The filtrate obtained after the photocatalytic oxidative coupling reaction of benzylamine over PCN-222 was added to 20 mL of H2O, and the mixture was further extracted by using ethyl acetate (EtOAc, 20 mL) three times to remove the organic compounds. Then the mixture was diluted to 400 mL with distilled H2O. 9 mL of the above aqueous solution was mixed with 1 mL of PBS buffer (pH = 7.4) and used as a test sample. After adding 20 μL of DPD and 20 μL of POD solution, the UV-Vis spectra of the sample were collected.Electrochemical characterization
Photoluminescence (PL) spectra
Typically, 2 mg PCN-222 (or 1.44 mg TPPCOOMe, a fixed content of porphyrin) was dispersed in 6 mL CH3CN. Steady-state PL spectra were recorded by using a PerkinElmer LS 55 Fluorescence Spectrometer.DFT calculations
Density functional theory (DFT) calculations were carried out in vacuo for a better understanding of charge transfer. Optimizations were carried out with B3LYP[LANL2DZ] without any symmetry restraint. All calculations were performed using G03 software. The geometry optimization of the singlet ground state was performed with a basis set composed of 6-31 G* for C, H, N, and O atoms.Conclusions
In summary, the visible-light photocatalytic oxidative coupling of diverse amines has been promoted by using a porphyrinic MOF (PCN-222) under ambient conditions. In addition to the direct oxidative coupling of amines via1O2 that is generated by the porphyrin ligand based on energy transfer, the synergistic effect between charge transfer-induced electrons and holes greatly contributes to the reaction. As a result, PCN-222 exhibits excellent photocatalytic activity, selectivity and recyclability toward this reaction, far superior to its corresponding TPPCOOMe counterpart that involves an energy transfer process only. The photoexcited PCN-222 produces oxygen-centered active sites in Zr-oxo clusters and porphyrin π-cation radicals in the reduction and oxidation ends, respectively, verified by ESR, which, for the first time, afford direct evidence for the charge separation and semiconductor-like behavior of MOFs. Moreover, the electron- and hole-engaged reaction intermediates have been clearly identified by ESR, which has remained unprecedented thus far, to understand the mechanism based on charge transfer for this reaction. This work not only provides an insightful understanding for charge transfer/separation in MOFs but would also stimulate further studies on photocatalytic organic transformation over MOFs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the NSFC (21725101, 21673213, 21701160, 21521001 and 11227901), the National Key Basic Research Program of China (2014CB931803, 2013CB921800), the General Financial Grant from the China Postdoctoral Science Foundation (2016M602018) and the Recruitment Program of Global Youth Experts.Notes and references
- M. T. Schümperli, C. Hammond and I. Hermans, ACS Catal., 2012, 2, 1108–1117 CrossRef.
- (a) H. Huang, J. Huang, Y.-M. Liu, H.-Y. He, Y. Cao and K.-N. Fan, Green Chem., 2012, 14, 930–934 RSC; (b) S. Ahmad, K. Gopalaiah, S. N. Chandrudu and R. Nagarajan, Inorg. Chem., 2014, 53, 2030–2039 CrossRef CAS PubMed; (c) S. Biswas, B. Dutta, K. Mullick, C.-H. Kuo, A. S. Poyraz and S. L. Suib, ACS Catal., 2015, 5, 4394–4403 CrossRef CAS.
- (a) R. Kumar, E. H. Gleißner, E. G. V. Tiu and Y. Yamakoshi, Org. Lett., 2016, 18, 184–187 CrossRef CAS PubMed; (b) A. Berlicka and B. König, Photochem. Photobiol. Sci., 2010, 9, 1359–1366 RSC; (c) L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS PubMed.
- (a) F. Su, S. C. Mathew, L. Möhlmann, M. Antonietti, X. Wang and S. Blechert, Angew. Chem., Int. Ed., 2011, 50, 657–660 CrossRef CAS PubMed; (b) C. Su, M. Acik, K. Takai, J. Lu, S.-J. Hao, Y. Zheng, P. Wu, Q. Bao, T. Enoki, Y. J. Chabal and K. P. Loh, Nat. Commun., 2012, 3, 1298–1306 CrossRef PubMed; (c) F. Raza, J. H. Park, H.-R. Lee, H.-I. Kim, S.-J. Jeon and J.-H. Kim, ACS Catal., 2016, 6, 2754–2759 CrossRef CAS; (d) D. Sun, L. Ye and Z. Li, Appl. Catal. B Environ., 2015, 164, 428–432 CrossRef CAS.
- (a) S. Furukawa, Y. Ohno, T. Shishido, K. Teramura and T. Tanaka, ACS Catal., 2011, 1, 1150–1153 CrossRef CAS; (b) X. Lang, H. Ji, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934–3937 CrossRef CAS PubMed; (c) Z. Zhang, F. Wang, M. Wang, S. Xu, H. Chen, C. Zhang and J. Xu, Green Chem., 2014, 16, 2523–2527 RSC.
- (a) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed; (b) H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC; (c) T. Islamoglu, S. Goswami, Z. Li, A. J. Howarth, O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2017, 50, 805–813 CrossRef CAS PubMed; (d) B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- (a) T. Zhou, Y. Du, A. Borgna, J. Hong, Y. Wang, J. Han, W. Zhang and R. Xu, Energy Environ. Sci., 2013, 6, 3229–3234 RSC; (b) A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed; (c) S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed; (d) Y. An, Y. Liu, P. An, J. Dong, B. Xu, Y. Dai, X. Qin, X. Zhang, M.-H. Whangbo and B. Huang, Angew. Chem., Int. Ed., 2017, 56, 3036–3040 CrossRef CAS PubMed; (e) S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS PubMed; (f) H. Zhang, G. Liu, L. Shi, H. Liu, T. Wang and J. Ye, Nano Energy, 2016, 22, 149–168 CrossRef CAS; (g) H. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- (a) C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed; (b) M. Wen, K. Mori, T. Kamegawa and H. Yamashita, Chem. Commun., 2014, 50, 11645–11648 RSC; (c) A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed; (d) Y. Horiuchi, T. Toyaok, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CrossRef CAS; (e) J.-D. Xiao, Q. Shang, Y. Xiong, Q. Zhang, Y. Luo, S. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9389–9393 CrossRef CAS PubMed; (f) L. Shen, M. Luo, L. Huang, P. Feng and L. Wu, Inorg. Chem., 2015, 54, 1191–1193 CrossRef CAS PubMed; (g) J.-D. Xiao, L. Han, J. Luo, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 1103–1107 CrossRef CAS PubMed.
- (a) D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed; (b) Y. Chen, T. Hoang and S. Ma, Inorg. Chem., 2012, 51, 12600–12602 CrossRef CAS PubMed; (c) W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- (a) W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS; (b) M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- X. Zhang, H. Wang, H. Wang, Q. Zhang, J. Xie, Y. Tian, J. Wang and Y. Xie, Adv. Mater., 2014, 26, 4438–4443 CrossRef CAS PubMed.
- J. A. Johnson, J. Luo, X. Zhang, Y.-S. Chen, M. D. Morton, E. Echeverría, F. E. Torres and J. Zhang, ACS Catal., 2015, 5, 5283–5291 CrossRef CAS.
- H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS PubMed.
- (a) R. Palominos, J. Freer, M. A. Mondaca and H. D. Mansilla, J. Photochem. Photobiol. Chem., 2008, 193, 139–145 CrossRef CAS; (b) Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS PubMed.
- H. C. Brown and Y. Okamoto, J. Am. Chem. Soc., 1958, 80, 4979–4987 CrossRef CAS.
- (a) K. Ohkubo, T. Nanjo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 1489–1500 CrossRef CAS; (b) R. V. Hoffman and R. Cadena, J. Am. Chem. Soc., 1977, 99, 8226–8232 CrossRef CAS.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- H. Bader, V. Sturzenegger and J. Hoignié, Water Res., 1988, 22, 1109–1115 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and instrumentation, and supplemental figures and tables. See DOI: 10.1039/c7sc05296k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |