
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Competition between Li+ and Na+ in sodium transporters and receptors: Which Na+-Binding sites are “therapeutic” Li+ targets?†
Todor
Dudev
 *a,
Karine
Mazmanian
bc and
Carmay
Lim
*bd
*a,
Karine
Mazmanian
bc and
Carmay
Lim
*bd
aFaculty of Chemistry and Pharmacy, Sofia University, Sofia 1164, Bulgaria. E-mail: todorminkov2013@gmail.com
bInstitute of Biomedical Sciences, Academia Sinica, Taipei 11529, Taiwan. E-mail: carmay@gate.sinica.edu.tw
cChemical Biology and Molecular Biophysics Program, Taiwan International Graduate Program, Academia Sinica, Taipei 11529, Taiwan
dDepartment of Chemistry, National Tsing Hua University, Hsinchu 300, Taiwan
First published on 2nd April 2018
Abstract
Sodium (Na+) acts as an indispensable allosteric regulator of the activities of biologically important neurotransmitter transporters and G-protein coupled receptors (GPCRs), which comprise well-known drug targets for psychiatric disorders and addictive behavior. How selective these allosteric Na+-binding sites are for the cognate cation over abiogenic Li+, a first-line drug to treat bipolar disorder, is unclear. Here, we reveal how properties of the host protein and its binding cavity affect the outcome of the competition between Li+ and Na+ for allosteric binding sites in sodium transporters and receptors. We show that rigid Na+-sites that are crowded with multiple protein ligands are well-protected against Li+ attack, but their flexible counterparts or buried Na+-sites containing only one or two protein ligands are vulnerable to Li+ substitution. These findings suggest a novel possible mode of Li+ therapeutic action: By displacing Na+ bound by ≤2 protein ligands in buried GPCR sites and stabilizing the receptor's inactive state, Li+ could prohibit conformational changes to an active state, leading to lower cytosolic levels of activated guanine nucleotide-binding proteins, which are hyperactive/overexpressed in bipolar disorder patients.
Introduction
Lithium (Li+), a cation with unknown biological function in mammals, has been used (in the form of soluble salts) as a first-line mood stabilizer for people with depressive conditions, particularly those suffering from bipolar disorder.1,2 It also has beneficial effects in ameliorating damages induced by traumatic brain injury,3 demyelinating diseases,4,5 and chronic neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's diseases.1,6 Although the clinical and pharmacodynamical/pharmacokinetic effects of Li+ therapy have been known for decades, its mechanism of therapeutic action remains unclear. Several hypotheses have been put forth, most of which focus on the competition or cooperation between Li+ and native ions such as Mg2+ and Na+ in biological compartments.One of the leading hypotheses posits that Li+ competes with Mg2+ in signal-transducing metalloenzymes that are overexpressed in bipolar patients such as glycogen synthase kinase 3β, inositol monophosphatase, and inositol polyphosphate phosphatase.2,7–10 By displacing weakly-bound Mg2+, Li+ inhibits these enzymes and eventually normalizes cell signaling. Subsequent calculations9 delineated the key factors controlling the outcome of the competition between Mg2+ and free, hydrated Li+ in proteins and elucidated why Li+ can replace Mg2+ only in enzymes known to be targets of Li+ therapy, but not in Mg2+-enzymes essential for cell function: Li+ can replace Mg2+ bound to only a few bulky ligands in sites with a net high positive charge that is reduced upon Li+ substitution. These findings were subsequently confirmed by 7Li magic-angle-spinning solid-state NMR experiments on E. coli SuhB inositol monophosphatase.11
Instead of competing with Mg2+, Li+ can co-bind with the native dication to modulate the properties of metal-loaded nucleotides such as adenosine triphosphate (ATP) and guanosine triphosphate (GTP), which act as activators of signal-transducing proteins.12–14 The bimetallic nucleotide complex can modulate the response of the host receptor and thus, the downstream signal transduction cascade in the cell: the binuclear ATP–Mg–Li complex, when bound to the neuronal P2X receptor (a ligand-gated Ca2+ channel), has been shown to elicit longer P2X activation than the native ATP–Mg cofactor.12 Subsequent calculations revealed that Li+ binding to [ATP–Mg]2− does not alter the native cofactor's overall charge, conformation, and interactions with protein ligands lining the P2X binding site.13,14 However, if the protein matrix prevented ADP from binding Li+ in its preferred solution mode, then Li+ binding to ATP–Mg could reduce the native cofactor's susceptibility to hydrolysis, resulting in prolonged residence of the bimetallic ATP–Mg–Li complex in P2X and thus, prolonged receptor response.14 On the other hand, compared to the native GTP–Mg cofactor, bimetallic GTP–Mg–Li interacts less well with guanine nucleotide-binding proteins (G-proteins), which are hyperactive/overexpressed in bipolar disorder patients. Thus, Li+ binding to [GTP–Mg]2− results in lower cytosolic levels of activated G-proteins and eventually normalizes cell signaling.14
Another hypothesis posits that Li+, in its free hydrated state, can compete with Na+. Bipolar disorder patients have abnormally high Na+ cytosolic concentrations.15,16 Monovalent Li+, by entering the cytosol via sodium channels, accumulates inside the cell lowering the intracellular concentration of Na+, which in turn reduces the Ca2+ cytosolic level. Decreasing the intracellular concentrations of both Na+ and Ca2+ attenuates the cell excitability, eventually normalizing the neuronal activity in bipolar disorder patients.15
However, another possible mode of Li+ therapeutic action has not been considered (to our knowledge); viz., the competition between Li+ and Na+ for specific Na+-binding sites in signal-transducing proteins, notably neurotransmitter transporters and G-protein coupled receptors (GPCRs), which comprise well-known drug targets for psychiatric disorders17,18 and addictive behavior.19 Many transport proteins involved in trafficking of neurotransmitters such as glycine, serotonin, dopamine, glutamate, and γ-aminobutyric acid (GABA) across the cell membrane bind Na+, which acts as an indispensable allosteric regulator of their activities.20–22 Membrane receptors such as glutamate receptors and GPCRs that mediate excitatory transmissions are also subject to Na+ allosteric control.23,24 Several fundamental questions regarding the competition between Li+ and Na+ in these systems have not been addressed:
(1) What are the key determinants controlling the selectivity for Na+ over Li+ in Na+-proteins?
(2) Which Na+-binding sites are more vulnerable to Li+ substitution? If Li+ can displace Na+ from its binding site, how would this affect the host protein function?
Here, we aim to answer these questions. First, we surveyed the Protein Data Bank (PDB)25 for neurotransmitter transporters and GPCRs containing functional Na+-binding sites to determine their characteristic features. Then, we evaluated the thermodynamic outcome of the competition between Na+ and Li+ in various model transporter/receptor Na+-binding sites, as described in Methods. We reveal how this outcome depends on properties of the host protein and its binding cavity and which Na+-sites are prone to Li+ substitution. Our findings suggest a novel possible mode of Li+ therapeutic action.
Methods
PDB survey
The PDB41 was searched for structures of neurotransmitter transporters and GPCRs that contain bound Na+; the resulting PDB entries are listed in ESI Table S1.† Na+-bound ligands were identified by a cutoff of 3 Å. If the same protein has more than one structure, the highest-resolution one with the most number of Na+-bound ligands was chosen. This yielded 16 representative proteins (Table 1) with distinct Na+-sites. For a given structure, we computed the % ratio of the solvent-accessible surface area (SASA) of the residue X in the protein to the accessible surface area of X in the tripeptide –Gly–X–Gly– using the MOLMOL42 program with a solvent probe radius of 1.4 Å. For a given structure, we also computed all hydrogen bonds made by the Na+-ligating residues using the HBPLUS Hydrogen Bond Calculator version 3.15.43 The relative % SASA and hydrogen bonds made by the Na+-ligating residues in the representative PDB structures are listed in ESI Table S2.†| PDB entry | Resoln (Å)a | Transporter proteins | Na+-bound ligandsb |
|---|---|---|---|
| a Resolution of the PDB structure. b Residues with a superscript “bb” bind to Na+via their backbone carbonyl groups; otherwise, they bind Na+via their sidechain O atoms. Atoms in parenthesis after a residue denotes that both atoms binds to Na+. | |||
2A65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 26 |
1.65 | Leucine transporter LeuT | Na1: A22bb N27 T254(OG1, O) N286 Leu (substrate) |
| Na2: G20bb V23bb A351bb T354 S355 | |||
| 3KBC 27 | 3.51 | Glutamate transporter GltPh | Na1: G306bb N310bb N401bb D405(OD1; OD2) |
| Na2: T308 S349bb I350bb T352bb | |||
|
4XP128 |
2.89 | Dopamine transporter DAT | Na1: A44bb N49 S320bb S320 N352 H2O |
| Na2: G42bb V45bb L417bb D420 S421 | |||
| 5E9S 29 | 2.80 | Glutamate transporter GltTk | Na1: G309bb N313bb N405bb D409(OD1 OD2) |
| Na2: T311bb S352bb I353bb T355bb | |||
| Na3: Y91bb T94 S95 N313 D315 | |||
|
5I7130 |
3.15 | Serotonin transporter SERT | Na1: A96bb N101 S336bb S336 N368bb |
| Na2: G94bb V97bb L434bb D437 S438 H2O | |||
|
5LM431 |
3.10 | Glutamate transporter GLAST1/EAAT1 | T376bb S417bb I418bb A420bb |
|
|||
| Receptor proteins | |||
|
3C3223 |
1.72 | Glutamate receptor kainate GluK1 (GluR5) | E96(OE1; O) I99bb D100 H2O |
| 3G3F 32 | 1.38 | Glutamate receptor kainate GluK2 (GluR6) | E97(OE1; O) I100bb D101 H2O |
|
3OM133 |
1.68 | Glutamate receptor kainate GluK5 (KA2) | F162bb S165bb E167bb 2(H2O) |
| 3S9E 34 (chain B) | 1.60 | Glutamate receptor kainate GluK3 (GluR7) | Na1: S24bb R26bb 4(H2O) |
| Na2: N5bb S52 2(H2O) | |||
|
3VW735 |
2.20 | GPCR receptor protease-activated PAR1 | D148 S189 D367 2(H2O) |
| 4BVN 36 | 2.10 | GPCR receptor adrenergic β1AR | Na1: C192bb D195bb C198bb 2(H2O) |
| Na2: D87 S128 3(H2O) | |||
| 4EIY 37 | 1.80 | GPCR receptor adenosine A2AAR | D52 S91 3(H2O) |
| 4LDE 38 | 2.79 | GPCR receptor adrenergic β2AR | N1103 C1184bb E1187bb C1190bb H2O |
| 4N6H 39 | 1.80 | GPCR receptor opioid δ-OR | D95 N131 S135 2(H2O) |
| 5NDD 40 | 2.80 | GPCR receptor protease-activated PAR2 | D121 D340 N158 S162 N33 |
Models used
On the basis of the structure/composition of Na+-sites from the protein crystal structures, the Na+-binding sites (without the substrate) were modeled as mononuclear penta/hexacoordinated complexes, comprising mostly neutral O-containing protein ligands (see Table 1 and Results). N-Methylacetamide (CH3CONHCH3) was used to model the backbone peptide group as well as the Asn/Gln side chain rather than acetamide (CH3CONH2), as replacing a H from the acetamide NH2 with a methyl group does not alter metal binding and N-methylacetamide and acetamide have similar solvation free energies.44,45 The Ser/Thr and Asp−/Glu− side chains were modeled as ethanol (CH3CH2OH) and acetate (CH3COO−), respectively.Geometry optimization
Among various combinations of ab initio methods/density functionals (HF, MP2, SVWN, B3LYP) and basis sets (6-31+G(d,p), 6-31+G(2d,2p), 6-31+G(3d,p), 6-31+G(3d,2p), 6-311++G(d,p) and 6-311++G(3df,3pd)), the B3-LYP/6-31+G(3d,p) method has been found to be the most efficient in reproducing experimental observables: it could reproduce (to within experimental error) the dipole moments of water and protein ligands,46 bond distances between oxygen and various cations (Na+, K+, Li+, Mg2+, Ca2+, Fe3+, and Ga3+) in aqua and crown ether complexes9,46–49 and the free energies of metal (Li+, Na+, K+, Mg2+ and Ca2+) exchange in crown ether, acetate, oxalate and nitrilotriacetic acid complexes.9,46,50 Hence, it was used to optimize the geometry of each metal complex and to compute the electronic energies, Eel, using the Gaussian 09 program.51Initially, we built hexacoordinated complexes with all six protein and/or water ligands bound to Na+. During geometry optimization, some hexacoordinated Na+ complexes isomerized to more stable pentacoordinated structures with a ligand relegated to the second shell. These were retained for further evaluation. The fully optimized Na+ complexes were used as starting structures for optimizing the respective Li+ counterparts. In the fully optimized Li+ complexes, two ligands spontaneously moved to the metal's second shell, leaving four ligands coordinated to Li+, consistent with lithium's known preference for tetrahedral complexes.52,53 Frequency calculations for each optimized construct were performed at the same level of theory – no imaginary frequency was found in any of the fully optimized structures.
Reaction modeled
Based on the fully optimized structures, we computed the free energy ΔGε for eqn (1)| [Li+-aq] + [Na+-protein] → [Li+-protein] + [Na+-aq] | (1) |
| ΔGε = ΔG1 + ΔGsolvε([Li+-protein]) + ΔGsolvε(Na+) − ΔGsolvε([Na+-protein]) − ΔGsolvε(Li+) | (2) |
Computing the gas-phase free energy, ΔG1
This was computed from the differences in the electronic energies (ΔEel), thermal energies (ΔEth) and entropies (ΔS) between the products and reactants in eqn (1) at a temperature T of 298.15 K according to:| ΔG1 = ΔEel + ΔEth − TΔS | (3) |
In evaluating the thermal energies and entropies, the vibrational frequencies were scaled by an empirical factor of 0.9613.54 The basis set superposition error and dispersion correction for eqn (1) were found to be insignificant (<1.5 kcal mol−1),46 thus they were not included.
Computing the solvation free energy, ΔGsolvε
This was computed by solving Poisson's equation with natural bond orbital atomic charges55 using the MEAD (Macroscopic Electrostatics with Atomic Detail) program.56 The effective solute radii were obtained by adjusting the CHARMM57 van der Waals radii to reproduce the experimental hydration free energies of Li+, Na+, and model ligands to within 1.1 kcal mol−1.9,58 The resulting values are (in Å): RLi = 1.38, RNa = 1.70, RC = 1.95, RN = 1.75, RO(–CONH–) = 1.72, RO(–CH2OH) = 1.90, RO(H2O/Na–H2O) = 1.85, RO(Li–H2O) = 1.84, RO(COO) = 1.40, RH = 1.50, RH(Na–H2O) = 1.26, and RH(Li–H2O) = 1.44.Results
Analysis of PDB structures
The results in Table 1 show some interesting differences between sodium transporters and receptors in terms of the number and composition of allosteric Na+-binding sites: sodium transporters generally possess two or more relatively “dry” juxtaposed Na+ sites where each Na+ is bound by mostly protein ligands rather than water molecules. In contrast, sodium receptors exhibit only one Na+-binding site, where the Na+ is generally bound by fewer (≤3) protein ligands and up to four water molecules.We further analyzed the sodium transporters and receptors in Table 1 for the number and type of ligands bound to Na+ as well as the relative burial and flexibility of allosteric Na+-binding sites. A Na+-binding site is deemed to be buried if the average relative SASA of the Na+-ligating amino acid (aa) residues is <20%, partially buried if it is between 20 and 50%, and solvent accessible if it is >50% (see ESI Table S2†).59 A Na+-binding site with a dense hydrogen-bonding network that would presumably prohibit structural reorganization of Na+-site maybe considered to be relatively rigid. The results in Table 2 show the following features common to allosteric Na+-binding sites in both sodium transporters and receptors:
| Na+ CNa | # of H2O ligandsb | # of neutral aa ligandsc | # of anionic ligandsd | PDB entriese | Buriedf | Total # of HBsg |
|---|---|---|---|---|---|---|
| a The number of aa and water atoms bound to Na+. b Number of Na+-bound water molecules. c Number of Na+-bound neutral aa ligands. OH denotes Ser/Thr side chain and “amide” denotes backbone/Asn/Gln amide group. d Number of Na+-bound negatively charged Asp−/Glu− ligands. “Bi” means that both carboxylate oxygen atoms coordinate Na+. e PDB entries in italics have resolution ≥3.0 Å; those in bold correspond to receptor proteins. f The mean % SASA of all Na+-binding aa residues is given if it exceeds 20%.59 g The total number of hydrogen bonds formed by all the Na+-ligating aa residues (excluding Na+-bound water molecules). | ||||||
| 4 | — | 1 OH + 3 amides | 0 | 3KBC (Na2) | Yes | 8 |
| 4 | — | 4 amides | 0 | 5LM4 | Yes | 4 |
| 4 | — | 4 amides | 0 | 5E9S (Na2) | Yes | 8 |
| 5 | 2 | 3 amides | 0 | 4BVN (Na1) | Partially (23%) | 7 |
| 5 | 2 | 3 amides | 0 | 3OM1 | Yes | 4 |
| 5 | — | 2 OH + 3 amides | 0 | 2A65 (Na2) | Yes | 10 |
| 6 | — | 1 OH + 5 amides | 0 | 2A65 (Na1) | Yes | 12 |
| 6 | 1 | 1 OH + 4 amides | 0 | 4XP1 (Na1) | Yes | 8 |
| 5 | — | 1 OH + 4 amides | 0 | 5I71 (Na1) | Yes | 11 |
| 5 | 3 | 1 OH | 1 | 4EIY | Yes | 5 |
| 4BVN (Na2) | Yes | 9 | ||||
| 5 | 2 | 1 OH + 1 amide | 1 | 4N6H | Yes | 15 |
| 5 | — | 3 amides | 1 (Bi) | 3KBC(Na1) | Yes | 9 |
| 5E9S (Na1) | Yes | 12 | ||||
| 6 | 1 | 1 OH + 3 amides | 1 | 5I71(Na2) | Yes | 10 |
| 5 | — | 1 OH + 3 amides | 1 | 4XP1(Na2) | Yes | 10 |
| 5 | — | 2 OH + 2 amides | 1 | 5E9S (Na3) | Yes | 16 |
| 5 | — | 1 OH + 2 amides | 2 | 5NDD | Yes | 14 |
| 5 | 1 | 2 amides | 2 | 3G3F | Yes | 11 |
| 3C32 | Yes | 10 | ||||
| 6 | 2 | 1 OH | 2 | 3VW7 | Yes | 11 |
| 5 | 1 | 4 amides | — | 4LDE | Yes | 6 |
| 6 | 4 | 2 amides | — | 3S9E (Na1) | Partially (40%) | 3 |
| 4 | 2 | 1 OH + 1 amide | — | 3S9E (Na2) | No (56%) | 4 |
(a) Na+-binding sites are mononuclear rather than bi/polynuclear with an average Na+⋯Na+ distance of 7.9 ± 1.1 Å in transporters containing multiple Na+-binding sites.
(b) Na+ is generally penta/hexacoordinated in receptors and transporters, albeit for structures with resolution >2.5 Å, the Na+ coordination number is uncertain since bound water molecule(s) may not be seen in the structure.
(c) Na+, being a “hard” cation, binds to “hard” oxygen atoms from the protein ligands.
(d) Na+ is bound by at least two aa ligands and no more than two negatively charged ones.
(e) Na+ is preferentially bound by neutral protein ligands (backbone carbonyl or small Ser/Thr/Asn side chains) rather than negatively charged ones (Asp−/Glu−), which generally bind Na+via one rather than both carboxylate oxygen atoms.
(f) Na+-binding sites in both transporter and receptor proteins are generally quite buried with multiple (≥3) hydrogen bonds (see also ESI Table S2†).
Assessing the outcome of the Li+vs. Na+ competition in Na+-binding sites
This was assessed by the free energies for replacing Na+ with Li+ in relatively buried (ε = 4) or partially solvent-exposed (ε = 30) protein binding pockets based on the fully optimized structures of Na+ and Li+ complexes shown in Fig. 1–3. As the protein matrix and the multiple hydrogen bonds formed by the Na+-ligating aa ligands may prohibit the incoming Li+ from adopting its favored tetrahedral ligand arrangement, we computed two sets of Na+ → Li+ free energies: those in black characterize cation competition in flexible Na+-sites that permit structural changes upon Li+ binding and those in blue portray the Na+ → Li+ exchange in rigid Na+-sites that prohibit structural reorganization. To model such inflexible binding sites, we fixed the binding cavity's geometry by freezing the protein ligand positions in the fully optimized Na+ complexes and replaced Na+ with Li+, followed by partial optimization, allowing Li+ and the water ligands to find their optimal positions.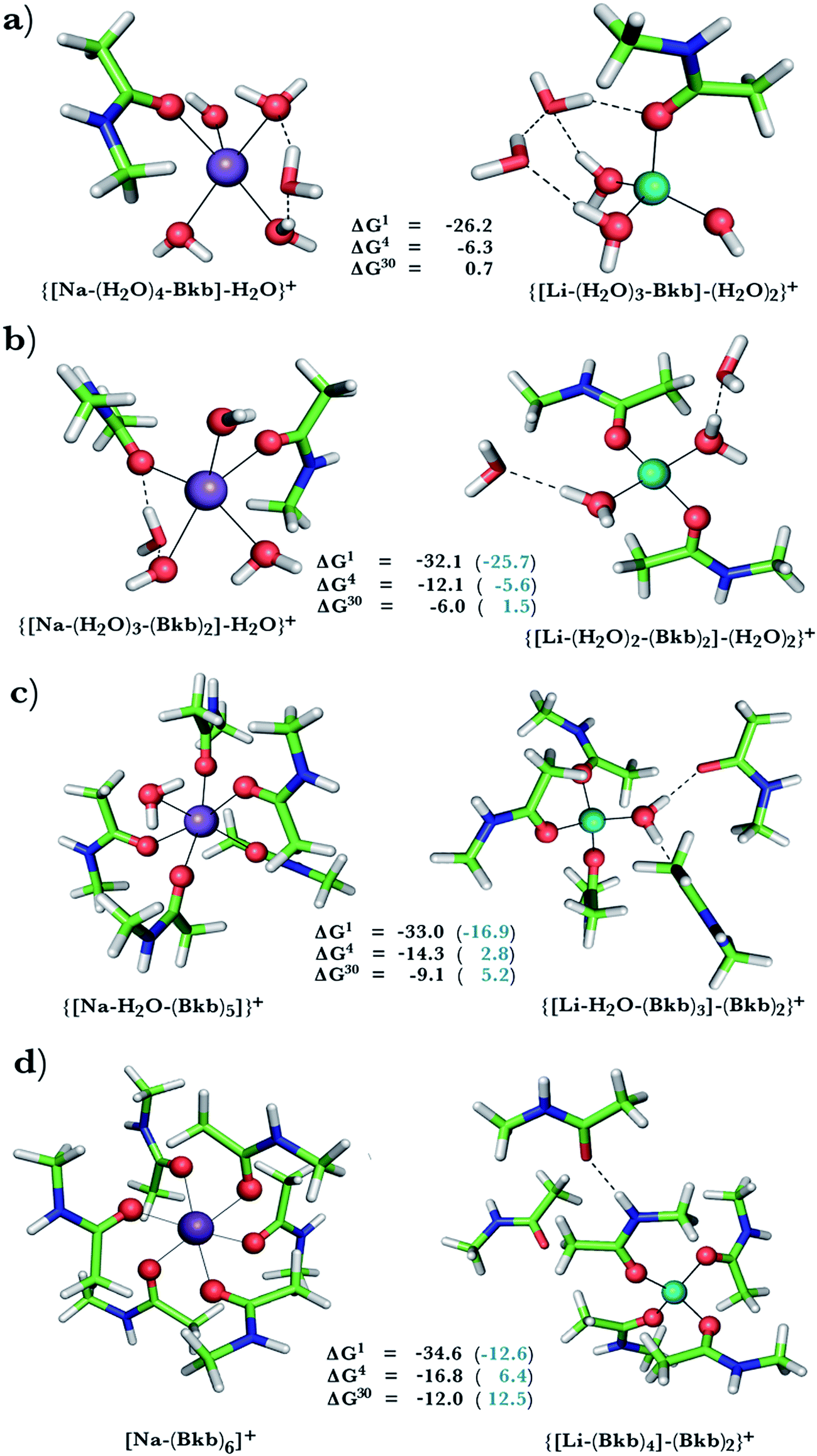 | ||
| Fig. 1 Calculated free energies and optimized structures of Na+ (purple) and Li+ (turquoise) complexes with amides. Metal-coordinating atoms are rendered as red spheres with non-coordinating ones in stick format with oxygen in red, carbon in green, nitrogen in blue, and hydrogen in white. Hydrogen bonds between first-shell and second-shell ligands are indicated by dash lines. | ||
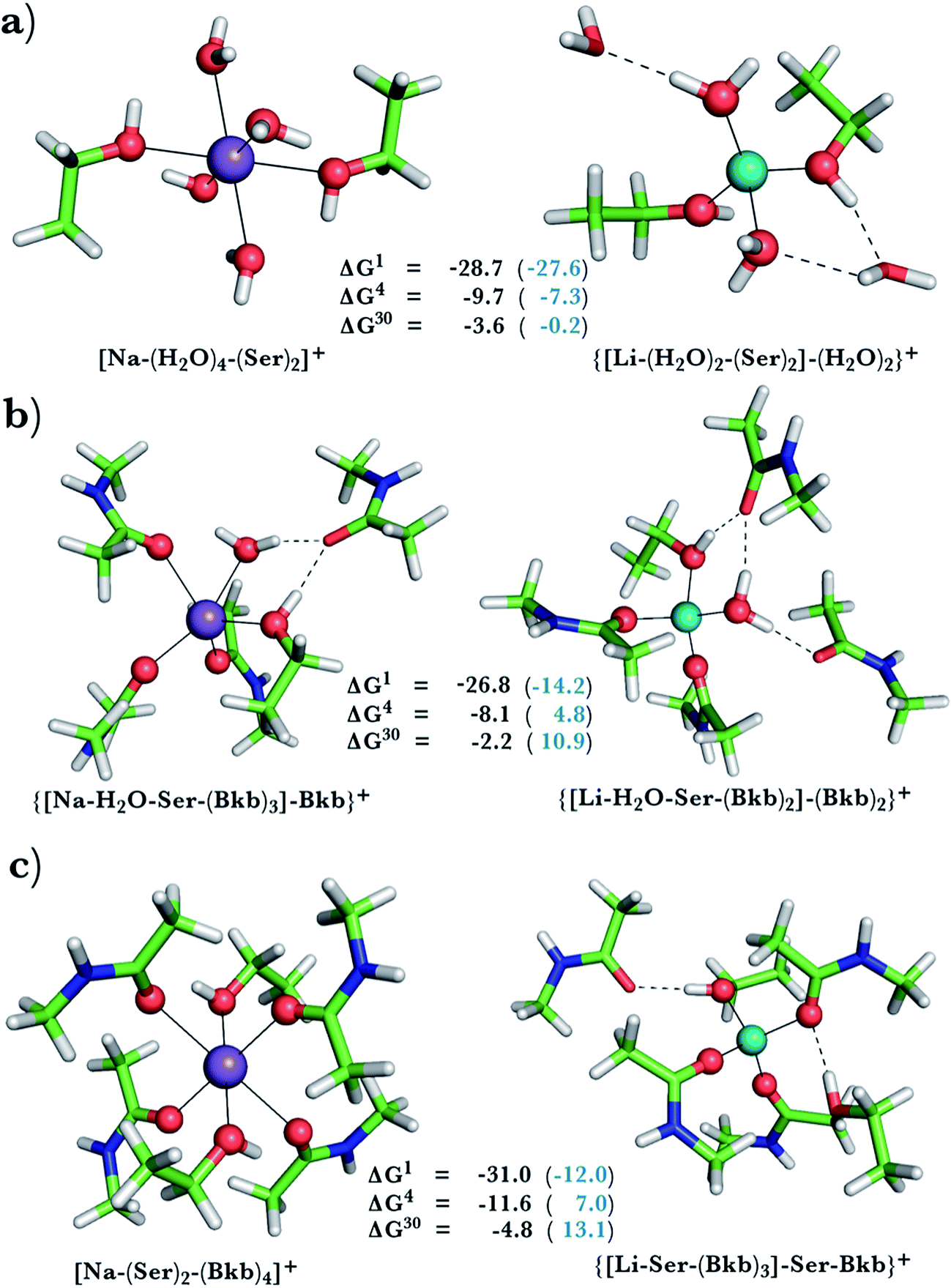 | ||
| Fig. 2 Calculated free energies and optimized structures of Na+ and Li+ complexes containing amide and hydroxyl ligands. Atom colors as in Fig. 1. | ||
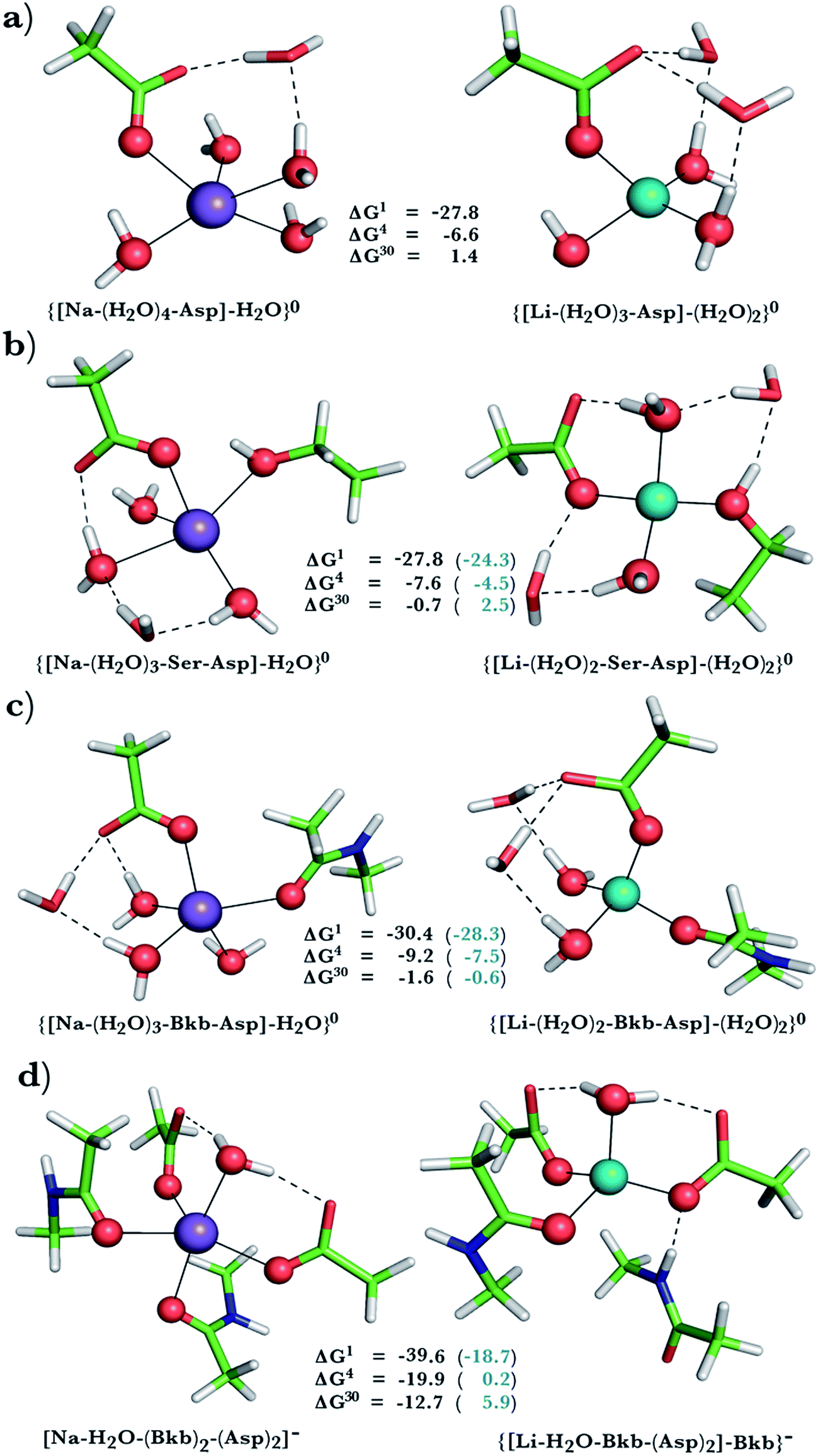 | ||
| Fig. 3 Calculated free energies and optimized structures of Na+ and Li+ complexes containing amide, hydroxyl and acidic ligands. Atom colors as in Fig. 1. | ||
A positive ΔGε in Fig. 1–3 implies a Na+-selective site, whereas a negative value implies a Li+-selective one. Below, we focus on how the Na+ → Li+ free energy changes with varying solvent exposure, rigidity, composition, and net charge of the metal-binding site rather than the absolute free energy. Note that the trends in the free energies computed using the approach herein have been found to be in accord with experimental observations in previous studies.9,47,49,50,58,60,61
Na+ → Li+ exchange in complexes containing neutral ligands
Since Na+ is found coordinated to only neutral ligands in several proteins (Table 2), we optimized complexes of Na+ bound to neutral ligands such as the backbone peptide group (Bkb)/Asn side chain (modeled by N-methylacetamide) or Ser/Thr side chain (modeled by ethanol) and water molecules. Flexible Na+-binding sites containing many neutral amide residues are prone to Li+ substitution, as evidenced by the favorable Na+ → Li+ free energies in Fig. 1b–d. This is mainly because Li+ is a better charge acceptor and forms more favorable interactions with the ligands than the native Na+. Consequently, increasing the number of amides lining the flexible binding pocket enhanced the selectivity for Li+ over Na+: as the number of amide ligands increased from one (Fig. 1a) to two (Fig. 1b) to five (Fig. 1c) to six (Fig. 1d), the ΔGε (ε = 1–30) became more negative. Such electronic effects favoring Li+ over Na+ are attenuated in solvent-exposed sites, where lithium's higher desolvation penalty outweighs sodium's solvation free energy gain upon Na+ → Li+ substitution (ΔG30 less negative than ΔG4). Hence, buried Na+-sites seem to be more susceptible to Li+ attack than solvent-accessible ones.However, if the Na+-binding sites containing many neutral amide residues were rigid, they would be well protected against Li+ attack (Fig. 1c and d, positive ΔG4/ΔG30 in blue). In contrast to flexible sites, the more crowded a rigid binding site, the more Na+-selective it becomes: as the number of amide ligands in a rigid binding site increased from two to six (Fig. 1b–d), the Na+ → Li+ free energy also increased; e.g., ΔG4 (in blue) increased from −5.6 to 2.8 and 6.4 kcal mol−1, respectively. This is because in a rigid site, Li+ is forced to adopt sodium's longer bond distances, resulting in reduced charge transfer, as compared to flexible sites. Furthermore, the backbone/Asn amides often form hydrogen bonds in the Na+-transporter and receptor structures (see ESI Table S2†). Hence, increasing the number of amide ligands lining a rigid binding site might make it harder to restructure the protein cavity in favor of Li+: this is supported by an increase in the free energy penalty for restructuring the Na+-binding site upon Li+ substitution: the ΔG4 difference between rigid and flexible sites increased from 6.5 to 17 to 23 kcal mol−1 in going from Fig. 1b to d. Thus, rigid, crowded Na+-binding sites, in contrast to their flexible counterparts, prefer Na+ over Li+.
When we replaced one or two amide ligands in Fig. 1b–d by ethanol (modeling the Ser/Thr side chain), the above trends do not change. Thus, Na+-binding sites are selective for the cognate cation over non-native Li+ if they are rigid and comprise >2 neutral protein ligands (Fig. 1c/1d and 2b/2c, positive ΔG4/ΔG30 in blue). They become prone to Li+ substitution if they can undergo structural changes to meet lithium's coordination preferences (Fig. 2, negative ΔG4/ΔG30 in black). Compared to an amide however, the hydroxyl group is a poorer charge donor and its interactions with Li+ are attenuated more than those with Na+, making the Na+-site less susceptible to Li+ attack when one or two amide ligands are replaced by hydroxyl groups (less negative ΔG4/ΔG30 in Fig. 2a–c than those in Fig. 1b–d, respectively).
Na+ → Li+ exchange in complexes comprising neutral and negatively charged ligands
Since Na+ is found bound to one or at most two acidic ligands in some proteins in Table 2, we also assessed the Na+vs. Li+ competition in complexes comprising one or two acetates (modeling Asp−/Glu− carboxylates) and a few neutral ligands. As found above, increasing the number of protein ligands lining a flexible binding pocket favored the cation that is the stronger electron acceptor; i.e., Li+ over Na+: as the number of protein ligands increased from one (Fig. 3a) to two (Fig. 3b/3c) to four (Fig. 3d), the ΔG4/ΔG30 became more negative. However, rigidifying the binding site, which forces the incoming Li+ to adopt unfavorable coordination distances rendered the most crowded binding pocket in the series Na+-selective (Fig. 3d, positive “blue” ΔGε, ε > 4). As found above, substitution of an amide in Fig. 3c with OH (a weaker charge donor) in Fig. 3b yielded less negative ΔG4/ΔG30.Discussion
Factors governing the competition between Li+ and Na+ in Na+-binding sites
The results herein reveal how the properties of the host protein and its binding cavity affect the outcome of the competition between Na+ and Li+ in Na+-binding sites:Comparison with experiment
Our findings are in line with available experimental data on sodium transporters/receptors. The calculations predict that rigid Na+-binding sites containing >3 protein ligands would be well-protected against Li+ attack (positive “blue” ΔG4/ΔG30 values for metal complexes comprising >3 neutral protein ligands in Fig. 1c, d, 2b and c). Our PDB analyses show that such rigid Na+-binding sites crowded with >3 protein ligands, which form hydrogen-bonding networks, are found in most of the sodium transporters and receptors listed in Table 1. In accord with our findings, the Na(1)-binding sites in neurotransmitter transporters have been experimentally found to be especially well-protected from Li+ substitution.63–65 Furthermore, experiments on aspartate20 and glutamate66 transporters using Li+ concentrations of 100 and 150 mM, respectively, show that Li+ indeed binds with lower affinity to these allosteric Na+-binding sites than the native metal co-factor.Interestingly, X-ray structures of the glutamate receptor 5 bound to Na+ (PDB entry 3C32) and to Li+ (PDB entry 3C31)23 have been solved to a resolution of 1.72 and 1.49 Å, respectively. The Na+-bound 3C32 structure shows Na+ bound to two neutral (Glu96 O, Ile99 O) and two acidic (Glu96 OE1, Asp100 OD1) aa ligands that altogether form ten hydrogen bonds (see Tables 1 and S2†). Superimposition of the Na+ and Li+-bound structures in Fig. 4 shows a rigid Na+-site that does not change its original structure/size upon accommodating Li+: the three protein ligands (Glu96, Asp100, and Ile99) that coordinate Na+ adopt similar positions/orientations, but Li+ is closer to Glu96 (Li+–OOE1 = 2.0 Å) at the expense of losing contact with Asp100 (Li+–OOD1 = 3.5 Å). Thus, Li+ does not fit favorably in the rigid cavity and binds with lower affinity than the cognate Na+ to the glutamate receptor 5.23 Consistent with this experimental finding, we find that in a rigid cavity, Li+ cannot successfully compete with Na+ bound to two amides and two carboxylates (Fig. 3d, positive “blue” ΔGε, ε > 4).
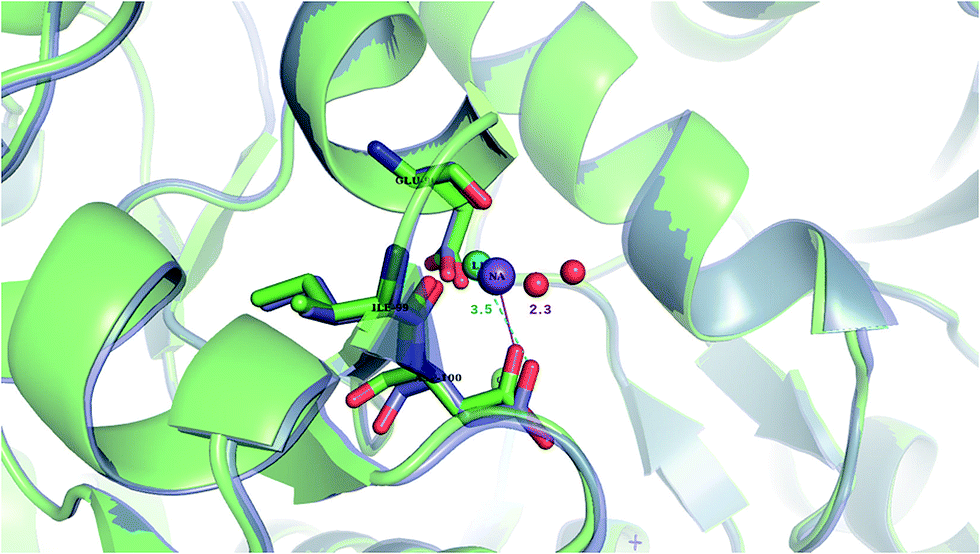 | ||
| Fig. 4 The superimposed X-ray structures of Na+ (3C32) and Li+ (3C31) binding sites in the glutamate kainate receptor (GluR5). The Li+ structure is in purple with Li+ in turquoise and the Na+ structure is in green with Na+ in purple. The respective metal-binding residues are depicted as sticks with nitrogen in blue and oxygen in red. Distances from Asp100 to the cation are indicated. | ||
Biological implications: Na+-binding sites vulnerable to Li+ attack
The calculations reveal that solvent-inaccessible Na+-binding sites lined by only one or two aa residues are susceptible to Li+ attack, regardless of the aa type and relative rigidity of pocket (negative ΔG4 in Fig. 1a, b, 2a, and 3a–c). Notably, they predict that Li+ can displace Na+ bound by Asp− and Ser in a buried Na+-binding site (Fig. 3b, negative ΔGε, ε < 30). Such a deeply buried Na+-binding site comprising an Asp− and a Ser is found in two GPCRs; viz., the A2AAR adenosine receptor (PDB entry 4EIY) and the β1AR adrenergic receptor (PDB entry 4BVN, Na2-site). Bound Na+ allosterically controls the GPCR activity: It stabilizes the inactive/resting receptor conformation, which cannot transmit signal to the G-protein and associated effectors.24,37,67 Agonist binding to the receptor disrupts the allosteric Na+-binding site (by activating transmembrane helix motions) and translocates Na+ to another protein locality,24 causing conformational changes that activate the receptor and trigger G-protein downstream signaling.The aforesaid mechanism of allosteric regulation by Na+ suggests that factors stabilizing the allosteric Na+-binding site would prohibit structural distortions induced by agonist binding, leading to lower cytosolic levels of activated G-proteins, which are hyperactive/overexpressed in bipolar disorder patients. Here, we propose that such a factor is Li+, which can displace Na+ in a buried site (modeled in Fig. 3b). Molecular dynamics simulations (despite limitations of the classical force fields68,69) also suggest that Li+ coordination to such an allosteric binding site is energetically favored over Na+ binding.70 Compared with the cognate Na+ cation, Li+, the better charge acceptor, forms stronger interactions with Asp− and Ser and enhances the stability of the allosteric binding pocket, leading to decreased G-protein activity in the cell. Such effects have indeed been observed in a recent study71 of the GluK2/K5 receptor heterodimer, which showed that Li+ could stabilize the heterodimer and slow its desensitization.
Lithium's replacement of Na+ can not only lock the GPCR protein in an inactive state, but also disrupt conformational transitions in neurotransmitter Na+ symporters, thus compromising substrate transport.72,73 Furthermore, it can alter the selectivity for the co-transported substrate, as found for the neuronal excitatory amino-acid transporter 3 (EAAT3), also known as EAAC1.74 Thus, lithium's replacement of Na+ in sodium transporter/receptor proteins could affect their biological functions. Conversely, Li+ might be scavenged by certain sodium proteins before it can reach its intracellular targets (see Introduction), hence it would be interesting to assess if Li+ could displace Na+ in Na-proteins that are ubiquitous in the blood in future studies.
Conclusion
This is the first study delineating the key factors determining the outcome of the competition between Li+ and Na+ for allosteric Na+-binding sites in biologically important neurotransmitter transporters and GPCRs. We show that abiogenic Li+ can target those Na+-binding sites that are flexible with multiple neutral protein ligands or those that are buried containing only one or two aa ligands. By displacing Na+ bound to an Asp− and a Ser in the solvent-inaccessible metal-binding sites of the A2AAR adenosine and the β1AR adrenergic GPCRs, Li+ could stabilize the receptor's inactive state by prohibiting conformational changes to the active state, thus leading to decreased G-protein activity, which are hyperactive in bipolar patients.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by funds from Academia Sinica and MOST (grant no. 98-2113-M-001-011), Taiwan. T. D. was supported by the project Materials Networking H2020-TWINN-2015.References
- F. Marmol, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2008, 32, 1761–1771 CrossRef CAS PubMed.
- M. Vosahlikova and P. Svoboda, Acta Neurobiol. Exp., 2016, 76, 1–19 Search PubMed.
- P. R. Leeds, F. Yu, Z. Wang, C.-T. Chiu, Y. Zhang, Y. Leng, G. R. Linares and D.-M. Chuang, ACS Chem. Neurosci., 2014, 5, 422–433 CrossRef CAS PubMed.
- S. Levine and A. Saltzman, Immunopharmacology, 1991, 22, 207–213 CrossRef CAS PubMed.
- P. De Sarno, R. C. Axtell, C. Raman, K. A. Roth, D. R. Alessi and R. S. Jope, J. Immunol., 2008, 181, 338–345 CrossRef CAS.
- C.-T. Chiu and D.-M. Chuang, Pharmacol. Ther., 2010, 128, 281–304 CrossRef CAS PubMed.
- D. M. De Freitas, M. M. C. A. Castro and C. F. G. C. Geraldes, Acc. Chem. Res., 2006, 39, 283–291 CrossRef PubMed.
- J. A. Quiroz, R. Machado-Vieira, C. A. Zarate Jr and H. K. Manji, Neuropsychobiology, 2010, 62, 50–60 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2011, 133, 9506–9515 CrossRef CAS PubMed.
- K. M. Brown and D. K. Tracy, Ther. Adv. Psychopharmacol., 2013, 3, 163–176 CrossRef CAS PubMed.
- A. Haimovich, U. Eliav and A. Goldbourt, J. Am. Chem. Soc., 2012, 134, 5647–5651 CrossRef CAS PubMed.
- K. T. Briggs, G. G. Giulian, G. Li, J. P. Y. Kao and J. P. Marino, Biophys. J., 2016, 111, 294–300 CrossRef CAS PubMed.
- T. Dudev, C. Grauffel and C. Lim, Sci. Rep., 2017, 7, 42377 CrossRef CAS PubMed.
- T. Dudev, C. Grauffel, S.-T. D. Hsu and C. Lim, 2017, submitted.
- R. S. El-Mallakh, Clin. Neurosci. Res., 2004, 4, 227–231 CrossRef CAS.
- X. Huang, Z. Lei and R. S. El-Mallakh, Bipolar Disord., 2007, 9, 298–300 CrossRef CAS PubMed.
- L. Malinauskaite, M. Quick, L. Reinhard, J. A. Lyons, H. Yano, J. A. Javitch and P. Nissen, Nat. Struct. Mol. Biol., 2014, 21, 1006–1012 CAS.
- R. R. Neubig and D. P. Siderovski, Nat. Rev. Drug Discov., 2002, 1, 187–197 CrossRef CAS PubMed.
- S. G. Amara and M. S. Sonders, Drug Alcohol Depend., 1998, 51, 87–96 CrossRef CAS PubMed.
- O. Boudker, R. M. Ryan, D. Yernool, K. Shimamoto and E. Gouaux, Nature, 2007, 445, 387–393 CrossRef CAS PubMed.
- H. Krishnamurthy, C. L. Piscitelli and E. Gouaux, Nature, 2009, 459, 347–355 CrossRef CAS PubMed.
- B. Felts, A. B. Pramod, W. Sandtner, N. Burbach, S. Bulling, H. H. Sitte and L. K. Henry, J. Biol. Chem., 2014, 289, 1825–1840 CrossRef CAS PubMed.
- A. J. Plested, R. Vijayan, P. C. Biggin and M. L. Mayer, Neuron, 2008, 58, 720–735 CrossRef CAS PubMed.
- V. Katritch, G. Fenalti, E. E. Abola, B. L. Roth, V. Cherezov and R. C. Stevens, Trends Biochem. Sci., 2014, 39, 233–244 CrossRef CAS PubMed.
- H. M. Berman, T. Battistuz, T. N. Bhat, W. F. Bluhm, P. E. Bourne, K. Burkhardt, Z. Feng, G. L. Gilliland, L. Iype, S. Jain, P. Fagan, J. Marvin, D. Padilla, V. Ravichandran, B. Schneider, N. Thanki, H. Weissig, J. D. Westbrook and C. Zardecki, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 899–907 CrossRef.
- A. Yamashita, S. K. Singh, T. Kawate, Y. Jin and E. Gouaux, Nature, 2005, 437, 215–223 CrossRef CAS PubMed.
- N. Reyes, C. Ginter and O. Boudker, Nature, 2009, 462, 880–885 CrossRef CAS PubMed.
- K. H. Wang, A. Penmatsa and E. Gouaux, Nature, 2015, 521, 322–327 CrossRef CAS PubMed.
- A. Guskov, S. Jensen, I. Faustino, S. J. Marrink and D. J. Slotboom, Nat. Commun., 2016, 13420 CrossRef CAS PubMed.
- J. A. Coleman, E. M. Green and E. Gouaux, Nature, 2016, 532, 334–339 CrossRef CAS PubMed.
- J. C. Canul-Tec, R. Assal, E. Cirri, P. Legrand, S. Brier, J. Chamot-Rooke and N. Reyes, Nature, 2017, 544, 446–451 CrossRef CAS PubMed.
- C. Chaudhry, M. C. Weston, P. Schuck, C. Rosenmund and M. L. Mayer, EMBO J., 2009, 28, 1518–1530 CrossRef CAS PubMed.
- J. Kumar and M. L. Mayer, J. Mol. Biol., 2010, 404, 680–696 CrossRef CAS PubMed.
- R. Venskutonyte, K. Frydenvang, M. Gajhede, L. Bunch, D. S. Pickering and J. S. Kastrup, J. Struct. Biol., 2011, 176, 307–314 CrossRef CAS PubMed.
- C. Zhang, Y. Srinivasan, D. H. Arlow, J. J. Fung, D. Palmer, Y. Zheng, H. F. Green, A. Pandey, R. O. Dror, D. E. Shaw, W. I. Weis, S. R. Coughlin and B. K. Kobilka, Nature, 2012, 492, 387–392 CrossRef CAS PubMed.
- J. L. Miller-Gallacher, R. Nehmé, T. Warne, P. C. Edwards, G. F. Schertler, A. G. Leslie and C. G. Tate, PLoS One, 2014, 9, e92727 Search PubMed.
- W. Liu, E. Chun, A. A. Thompson, P. Chubukov, F. Xu, V. Katritch, G. W. Han, C. B. Roth, L. H. Heitman, A. P. IJzerman, V. Cherezov and R. C. Stevens, Science, 2012, 337, 232–236 CrossRef CAS PubMed.
- A. M. Ring, A. Manglik, A. C. Kruse, M. D. Enos, W. I. Weis, K. C. Garcia and B. K. Kobilka, Nature, 2013, 502, 575–579 CrossRef CAS PubMed.
- G. Fenalti, P. M. Giguere, V. Katritch, X. P. Huang, A. A. Thompson, V. Cherezov, B. L. Roth and R. C. Stevens, Nature, 2014, 506, 191–196 CrossRef CAS PubMed.
- R. K. Y. Cheng, C. Fiez-Vandal, O. Schlenker, K. Edman, B. Aggeler, D. G. Brown, G. A. Brown, R. M. Cooke, C. E. Dumelin, A. S. Dore, S. Geschwindner, C. Grebner, N. O. Hermansson, A. Jazayeri, P. Johansson, L. Leong, R. Prihandoko, M. Rappas, H. Soutter, A. Snijder, L. Sundstrom, B. Tehan, P. Thornton, D. Troast, G. Wiggin, A. Zhukov, F. H. Marshall and N. Dekker, Nature, 2017, 545, 112–115 CrossRef CAS PubMed.
- P. W. Rose, A. Prlić, A. Altunkaya, C. Bi, A. R. Bradley, C. H. Christie, L. D. Costanzo, J. M. Duarte, S. Dutta, Z. Feng, R. K. Green, D. S. Goodsell, B. Hudson, T. Kalro, R. Lowe, E. Peisach, C. Randle, A. S. Rose, C. Shao, Y. P. Tao, Y. Valasatava, M. Voigt, J. D. Westbrook, J. Woo, H. Yang, J. Y. Young, C. Zardecki, H. M. Berman and S. K. Burley, Nucleic Acids Res., 2017, 45, D271–D281 CrossRef CAS PubMed.
- R. Koradi, M. Billeter and K. Wuthrich, J. Mol. Graphics, 1996, 14, 51–55 CrossRef CAS PubMed.
- I. K. McDonald and J. M. Thornton, J. Mol. Biol., 1994, 238, 777–793 CrossRef CAS PubMed.
- R. Wolfenden, Biochemistry, 1978, 17, 201–204 CrossRef CAS PubMed.
- R. Wolfenden, L. Andersson, P. M. Cullis and C. C. B. Southgate, Biochemistry, 1981, 20, 849–855 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2009, 131, 8092–8101 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Phys. Chem. B, 2012, 116, 10703–10714 CrossRef CAS PubMed.
- T. Dudev and C. Lim, Phys. Chem. Chem. Phys., 2012, 14, 12451–12456 RSC.
- V. Nikolova, S. Angelova, N. Markova and T. Dudev, J. Phys. Chem. B, 2016, 120, 2241–2248 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2013, 135, 17200–17208 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- M. Dudev, J. Wang, T. Dudev and C. Lim, J. Phys. Chem. B, 2006, 110, 1889–1895 CrossRef CAS PubMed.
- I. Tunell and C. Lim, Inorg. Chem., 2006, 45, 4811–4819 CrossRef CAS PubMed.
- M. W. Wong, Chem. Phys. Lett., 1996, 256, 391–399 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- D. Bashford, in Scientific Computing in Object-Oriented Parallel Environments, ed. Y. Ishikawa, R. Oldehoeft, J. W. Reynders and M. Tholburn, Springer Berlin, Heidelberg, 1997, Vol. 1343, ch. 30, pp. 233–240 Search PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- T. Dudev, K. Mazmanian and C. Lim, Phys. Chem. Chem. Phys., 2016, 18, 16986–16997 RSC.
- J. Wright and C. Lim, Protein Eng., 2001, 14, 479–486 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2010, 132, 2321–2332 CrossRef CAS PubMed.
- T. Dudev and C. Lim, Sci. Rep., 2015, 5, 7864 CrossRef PubMed.
- S. Y. Noskov and B. Roux, J. Mol. Biol., 2008, 377, 804–818 CrossRef CAS PubMed.
- R. J. Vandenberg and R. M. Ryan, Physiol. Rev., 2013, 93, 1621–1657 CrossRef CAS PubMed.
- L. Borre, T. F. Andreassen, L. Shi, H. Weinstein and U. Gether, J. Biol. Chem., 2014, 289, 25764–25773 CrossRef CAS PubMed.
- Y. Zhou, E. Zomot and B. I. Kanner, J. Biol. Chem., 2006, 281, 22092–22099 CrossRef CAS PubMed.
- Y. Zhang and B. I. Kanner, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1710–1715 CrossRef CAS.
- H. Gutiérrez-de-Terán, A. Massink, D. Rodríguez, W. Liu, G. W. Han, J. S. Joseph, I. Katritch, L. H. Heitman, L. Xia, A. P. Ijzerman, V. Cherezov, V. Katritch and R. C. Stevens, Structure, 2013, 21, 2175–2185 CrossRef PubMed.
- D. Sakharov and C. Lim, J. Am. Chem. Soc., 2005, 127, 4921–4929 CrossRef CAS PubMed.
- N. Manin, M. C. da Silva, I. Zdravkovic, O. Eliseeva, A. Dyshin, O. Yas-ar, D. R. Salahub, A. M. Kolker, M. G. Kiselev and S. Y. Noskov, Phys. Chem. Chem. Phys., 2016, 18, 4191–4200 RSC.
- A. Strasser, H. J. Wittmann, E. H. Schneider and R. Seifert, N. Schmied. Arch. Pharmacol., 2015, 388, 363–380 CrossRef CAS PubMed.
- T. Paramo, P. M. G. E. Brown, M. Musgaard, D. Bowie and P. C. Biggin, J. Biophys., 2017, 113, 2173–2177 CrossRef CAS PubMed.
- Y. Zhao, D. S. Terry, L. Shi, M. Quick, H. Weinstein, S. C. Blanchard and J. A. Javitch, Nature, 2011, 474, 109–113 CrossRef CAS PubMed.
- S. Stolzenberg, M. Quick, C. Zhao, K. Gotfryd, G. Khelashvili, U. Gether, C. J. Loland, J. A. Javitch, S. Y. Noskov, H. Weinstein and L. Shi, J. Biol. Chem., 2015, 290, 13992–14003 CrossRef CAS PubMed.
- D. Menaker, A. Bendahan and B. I. Kanner, J. Neurochem., 2006, 99, 20–28 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 listing all PDB entries of transporter and receptor structures with Na+ bound in the allosteric sites. Table S2 listing the relative % SASA and hydrogen bonds for the Na+-coordinating residues in the representative PDB structures. See DOI: 10.1039/c7sc05284g |
| This journal is © The Royal Society of Chemistry 2018 |