
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic reductive coupling of aldehydes with 1,1-diarylethylenes using an in situ generated pyridine-boryl radical†
Jia
Cao‡
ac,
Guoqiang
Wang‡
a,
Liuzhou
Gao
a,
Xu
Cheng
b and
Shuhua
Li
 *a
*a
aKey Laboratory of Mesoscopic Chemistry of Ministry of Education, Institute of Theoretical and Computational Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, P. R. China. E-mail: shuhua@nju.edu.cn
bInstitute of Chemistry and Biomedical Sciences, Jiangsu Key Laboratory of Advanced Organic Material, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, P. R. China
cSchool of Chemistry and Chemical Engineering, Yan'an University, Yan'an 716000, P. R. China
First published on 28th February 2018
Abstract
A pyridine-boryl radical promoted reductive coupling reaction of aldehydes with 1,1-diarylethylenes has been established via a combination of computational and experimental studies. Density functional theory calculations and control experiments suggest that the ketyl radical from the addition of the pyridine-boryl radical to aldehydes is the key intermediate for this C–C bond formation reaction. This metal-free reductive coupling reaction features a broad substrate scope and good functional compatibility.
Introduction
Carbon–carbon bond formation is the most important transformation in organic synthesis.1 The catalytic reductive coupling of olefins with carbonyl compounds is one of the most economical C–C bond constructing methods, due to the abundant source of olefins and carbonyl compounds.2 Traditionally, transition metal catalysts have played privileged roles in these transformations, including metal-catalyzed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O reductive coupling (Scheme 1, top)3–5 and redox-triggered CO coupling via H2 transfer (Scheme 1, middle).4 However, sensitive organometallic reagents or transition-metal catalysts are usually required in these reactions. In contrast, organocatalytic reductive coupling of olefins with carbonyl derivatives for C–C bond formation in the presence of sensitive functional groups or congested structural environments is still rare.5d
O reductive coupling (Scheme 1, top)3–5 and redox-triggered CO coupling via H2 transfer (Scheme 1, middle).4 However, sensitive organometallic reagents or transition-metal catalysts are usually required in these reactions. In contrast, organocatalytic reductive coupling of olefins with carbonyl derivatives for C–C bond formation in the presence of sensitive functional groups or congested structural environments is still rare.5d
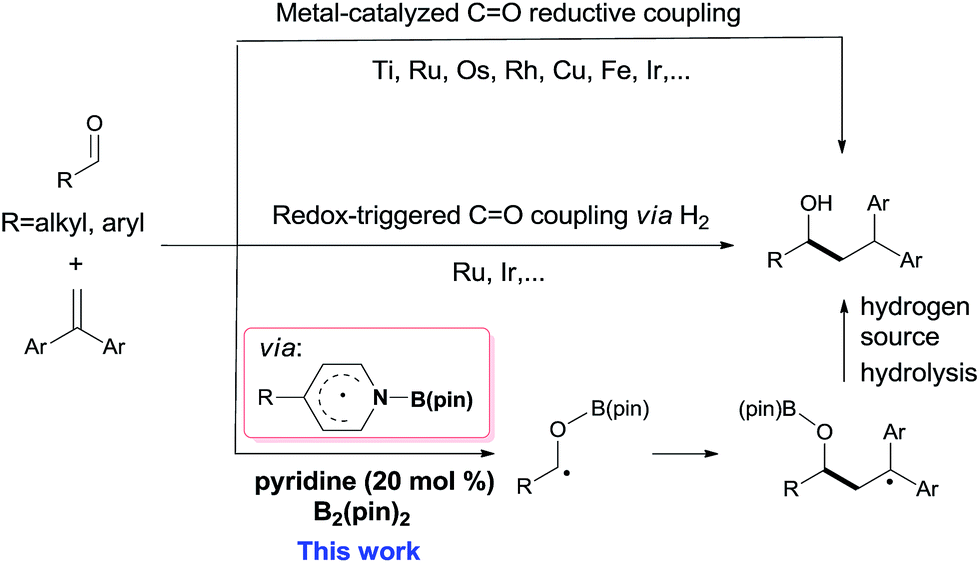 | ||
| Scheme 1 Reductive coupling of carbonyl compounds with olefins. | ||
Boron containing radicals are important reactive intermediates in organic synthesis.6–13 In this context, our group recently revealed that the pyridine-ligated boryl radical (Py-Bpin˙) could be readily generated from (pinacolato)diboron (B2pin2) through a cooperative catalysis involving two 4-cyanopyridine molecules.11 This kind of pyridine-boryl radical was used for the catalytic reduction of azo-compounds11 or as a carbon-centered radical for the synthesis of 4-substituted pyridines.12 Moreover, the pyridine-boryl radical can act as a persistent radical13 for the synthesis of organoboronate derivatives.14 Because the precursors (pyridines and B2pin2) of these pyridine-boryl radicals are inexpensive and stable,15 the development of new chemical transformations with these pyridine-boryl radicals is attractive. In this work, we further explored pyridine-boryl radical chemistry in the organocatalytic reductive coupling of aldehydes with 1,1-diarylalkenes (Scheme 1, bottom), which, to the best of our knowledge, has not been reported previously.
Results and discussion
It will be shown that the reductive coupling of aldehydes and olefins can be promoted by an in situ generated pyridine-boryl radical, following the proposed pathway as shown in Scheme 2. The proposed catalytic cycle consists of the following four steps: (1) activation of the B–B bond of B2pin2 by pyridines to form a pyridine-boryl radical (Int1); (2) the addition of the pyridine-boryl radical to aldehyde 1a to generate a new ketyl radical (Int3), with the regeneration of the pyridine catalyst; (3) the addition of the new ketyl radical to 1,1-diphenylethylene to yield a diaryl-stabilized radical species (Int4); and (4) the hydrogen abstraction of Int4 from an appropriate H-source to yield the final reductive coupling product. In addition, one molecule of Int4 may also abstract a hydrogen atom from another molecule of Int4 to give the reductive coupling product and another disproportionation product. To make this catalytic cycle happen, it is necessary to inhibit the possible radical–radical C–C coupling reaction between the pyridine-boryl radical and the ketyl radical, as observed between α,β-unsaturated ketones and 4-cyanopyridine in the presence of B2pin2.12 Thus, other pyridines with different substituents may be better catalysts than 4-cyanopyridine for the proposed reaction. With a pyridine-boryl radical bearing a suitable substituent, its reactivity might be tuned so that the newly generated ketyl radical could react with 1,1-diphenylethylene to yield a diaryl-stabilized radical species, which then undergoes a hydrogen atom abstraction from an appropriate hydrogen source to produce the reductive coupling product.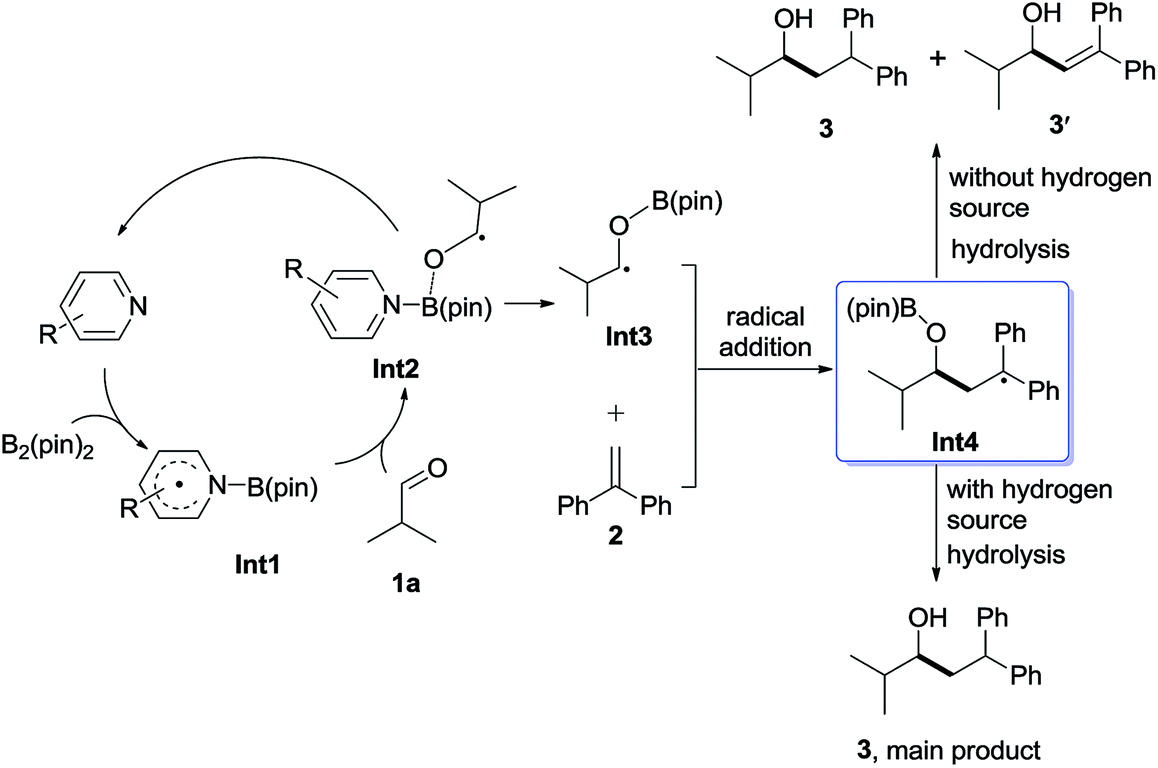 | ||
| Scheme 2 Proposed mechanism for the reductive coupling of isobutyraldehyde and 1,1-diphenylethylene. | ||
To find suitable pyridines which can react with B2pin2 to form the corresponding pyridine-boryl radical under mild conditions, we first performed density functional theory (DFT) calculations with the M06-2X16 functional to screen a series of pyridines. A careful analysis of stationary points revealed that the formation of the pyridine-boryl radical proceed through a [3,3]-sigmatropic rearrangement/homolytic C–C bond cleavage pathway17 rather than via the direct homolytic cleavage of the B–B bond11,18 (see Fig. S1 and S2 in the ESI† for details). As shown in Fig. S1–S12,† the [3,3]-sigmatropic rearrangement is the rate-determining step for the formation of the corresponding pyridine-boryl radical. The activation barrier of this step with different pyridines is highly dependent on the substituent in the pyridine ring. Pyridines with an electron-withdrawing group at the C4 position, such as CN (A), 4-cyano phenyl (B), Ph (C), and CF3 (D), have lower barriers than unsubstituted pyridines and pyridines with an electron-donating group (CH3, CH3O, and N(CH3)2). The pyridine with a CF3 group at the C3 position has a slightly higher activation barrier than that with the same group at the C4 position (D > E). These results indicate that the activation barrier of pyridines correlates closely with the resonance effect and the inductive effect of the substituents. According to the calculated activation barriers, five pyridines (A, B, C, D, and E) with barriers of no more than 28.5 kcal mol−1 are possible candidates for cleaving the B–B bond of B2pin2.
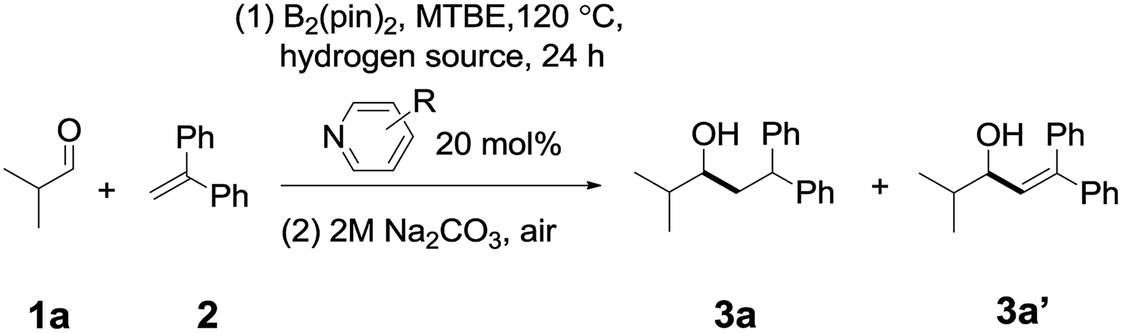
In order to determine a suitable combination of a pyridine catalyst and a hydrogen source, we conducted an initial investigation on the reaction between isobutyraldehyde 1a and 1,1-diphenylethylene 2 (see Tables S1 to S3† for details). As shown in Table 1, by heating a mixture of isobutyraldehyde 1a (1.0 equiv.), 1,1-diphenylethylene (2.0 equiv.), and B2pin2 (1.0 equiv.) in the presence of 1,3,5-trimethyl-1,4-cyclohexadiene (a hydrogen source, 1.0 equiv.) and 4-cyanopyridine A (0.2 equiv.) in tert-butyl methyl ether (MTBE) at 120 °C, the desired reductive coupling product 3a was observed in 28% yield (entry 1), together with a small amount of pyridine-aldehyde adducts (12% yield, see the ESI† for details). When 4-(4-pyridinyl)benzonitrile B was used as the catalyst (entry 2), the NMR yield of 3a improved to 78%, and the yield of a byproduct 3a′ from the disproportionation of the diaryl radical intermediate (Int4) was 6%. However, when other pyridines (for example C, D, or E, entries 3–5) were adopted, the yield of 3a decreased significantly. If Et3SiH was chosen as the hydrogen source, the yield of 3a is somewhat lower than that with 1,3,5-trimethyl-1,4-cyclohexadiene as a hydrogen source (entry 6). In the absence of a hydrogen source (entry 7), the ratio of 3a/3a′ was 52%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16%, suggesting that the addition of a hydrogen source is important for improving the yield of 3a (see Table S2†).
:16%, suggesting that the addition of a hydrogen source is important for improving the yield of 3a (see Table S2†).
|
|
|||
|---|---|---|---|
| Entry | Hydrogen source | Pyridines with different R substituents | Ratio [3a/3a′]b |
| a Reaction conditions: isobutyraldehyde (0.2 mmol), B2(pin)2 (0.2 mmol), catalyst (0.04 mmol), 1,1-diphenylethylene (0.4 mmol), H-donor (0.2 mmol), 24 hours, 120 °C, and MTBE (1 mL). b Yields were determined by 1H-NMR analysis of the crude mixture using CH2Br2 as the internal standard. c Isolated yield of 3a. TMe-1,4-CHD = 1,3,5-trimethyl-1,4-cyclohexadiene. | |||
| 1 | TMe-1,4-CHD | 4-CN (A) | 28%:0% |
| 2 | TMe-1,4-CHD | 4-(4-Cyanophenyl) (B) |
78%:6% (70%)c |
| 3 | TMe-1,4-CHD | 4-Ph (C) | 16%:2% |
| 4 | TMe-1,4-CHD | 4-CF3 (D) | 21%:1% |
| 5 | TMe-1,4-CHD | 3-CF3 (E) | 0% |
| 6 | Et3SiH | 4-(4-Cyanophenyl) (B) | 62%:16% |
| 7 | — | 4-(4-Cyanophenyl) (B) | 52%:16% |
Under the optimum conditions (Table 1, entry 2), we explored the generality of this transformation with a series of alkyl and aryl aldehydes. As shown in Table 2, the reductive coupling reactions of several fully aliphatic aldehydes proceeded with good efficiency (1a–1d). It was noteworthy that aldehydes with CC double bond (1e), methylthio (1f), or furyl (1h) functionalities on the alkyl chain were tolerated, giving the reductive coupling products in moderate to good yields. The α-branched aldehydes (1i–1r), in particular, pivaldehyde (1q) and 1-adamantylcarboxaldehyde (1r), also reacted well to afford the desired products in good yields. It should be mentioned that the substrates with a congested structure environment show less reactivity in transition-metal catalyzed reductive coupling of olefins and aldehydes, possibly because the coordination between the metal centre and the corresponding substrates is difficult.4c However, our method is also suitable for butyl aldehydes (1q and 1r). Beside alkyl aldehydes, aryl aldehydes (1s and 1t) bearing electron-donating groups (CH3 and CH3O) could also serve as the coupling partners, furnishing corresponding products in moderate yields. In addition to aldehydes, alkyl ketones (1u–1w) also reacted smoothly to provide the desired alcohols in 27–42% yield.
|
|
|---|
| a Reaction conditions: aldehyde (0.2 mmol), B2(pin)2 (0.2 mmol), 4-(4-pyridinyl)benzonitrile (0.04 mmol), 1,1-diphenylethylene (0.4 mmol), 1,3,5-trimethyl-1,4-cyclohexadiene (0.2 mmol), MTBE (1.0 mL), 24 h, and 120 °C. Isolated yield. The diastereoselectivities (d. r.) were determined by 1H-NMR analysis of the crude mixture. Boc = tert-butoxycarbonyl. |

|
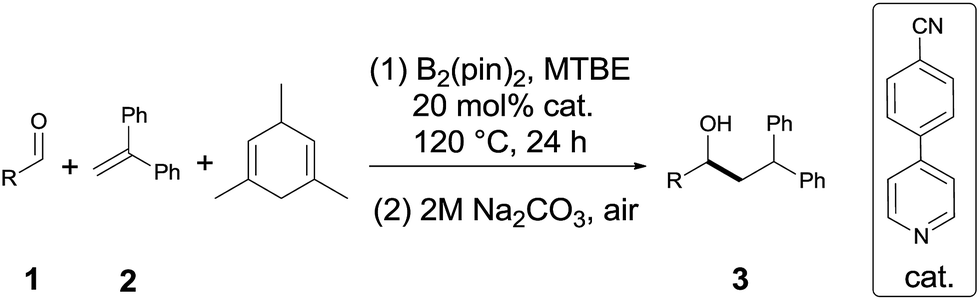
Diarylalkanes are important pharmacophores in drugs.19 It would be attractive to apply this metal-free method in the late stage functionalization of medicinally related molecules. As shown in Table 2(C), an abietic acid derivative (1x) and gemfibrozil derivative (1y) reacted smoothly with 1,1-diphenylethylene to form 3x and 3y in acceptable yields, respectively.
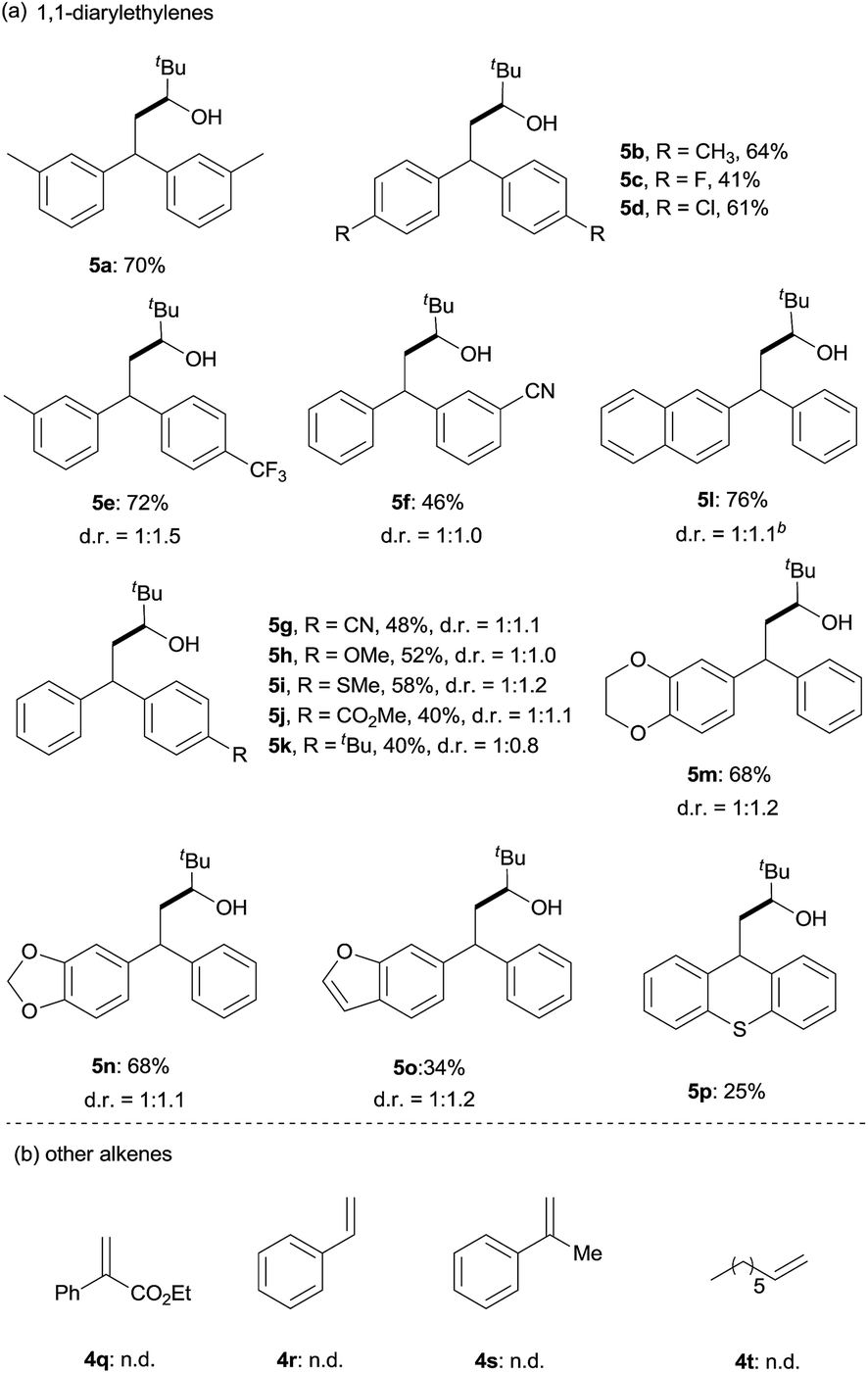
Next, the scope of 1,1-diarylalkenes (4) was examined (Table 3(a)). Both symmetrical (4a–d) and unsymmetrical (4e–o) 1,1-diarylalkenes were converted into the corresponding products 5 in moderate to good yields with modest diastereoselectivities. The reaction tolerated substrates bearing various functional groups on the benzene ring, such as halogen functionalities (4c and 4d), CF3 (4e), CN (4f and 4g), MeO (4h), CH3S (4i), CO2Me (4j), and tBu (4k). More importantly, 1,1-diarylalkenes containing heterocyclic structures (4m–o), such as benzofuran (4o) and thioxanthene (4p), also reacted smoothly to give the expected products in reasonable yields. Additionally, we also tested the reactivity of other alkenes with pivaldehyde 1q (Table 3(b)). However, our results show that other alkenes, including ethyl 2-phenylacrylate (4q), styrenes (4r and 4s) or aliphatic olefin (4t), generally gave little or no desired product. The reason why 1,1-diarylalkenes are suitable coupling partners of ketyl radicals may be due to (1) the radical stabilization effect of two aryl groups, and (2) the less nucleophilicity of present boron-ketyl radicals (compared with typical ketyl radicals).5b Thus, this protocol provides a metal-free reductive coupling method of 1,1-diarylalkenes with aldehydes (via the radical addition mechanism), which traditionally requires transition metal catalysts or organometallic reagents.12–16
|
|
|---|
| a Reaction conditions: pivaldehyde (0.2 mmol), B2(pin)2 (0.2 mmol), 4-(4-pyridinyl)benzonitrile (0.04 mmol), 1,1-diarylethylene (0.4 mmol), 1,3,5-trimethyl-1,4-cyclohexadiene (0.2 mmol), MTBE (1.0 mL), 24 h, and 120 °C. Isolated yield. The diastereoselectivities (d. r.) were determined by 1H-NMR analysis of the crude mixture. b The diastereoselectivities (d. r.) were determined by GC-MS analysis of the crude mixture. tBu = tert-butyl. |
|

To understand the mechanism of the reductive coupling of 1,1-diarylalkenes with aldehydes, we have performed DFT calculations with the M06-2X functional to explore the free energy profile of the proposed mechanism for the reaction between isobutyraldehyde (1a) and 1,1-diphenylethylene (2) in the presence of Int1 as a reactive intermediate. Our theoretical studies have shown that the generation of Int1 from B2pin2 and 4-(4-pyridinyl)benzonitrile is exergonic by 13.4 kcal mol−1 (see Fig. S4†). The calculated free energy profile and transition state structures are displayed in Fig. 1 (the optimized structures of all minimum species are shown in Fig. S12†). First, the coordination of the oxygen atom of isobutyraldehyde to the boron atom of the pyridine-boryl radical Int1 generates a boron-containing intermediate (Int2) viaTS1, with a barrier of 13.3 kcal mol−1. Then, the breaking of the B–N bond in Int2 yields a ketyl radical (Int3) and regenerates the 4-(4-pyridinyl)benzonitrile catalyst. This process is exothermic by 4.5 kcal mol−1, with a barrier of 3.2 kcal mol−1 (relative to Int2), suggesting that the formation of the ketyl radical (Int3) from Int2 is possible. Next, the addition of Int3 to the β-position of 1,1-diphenylethylene to form a diaryl-stabilized radical (Int4) viaTS3 is exothermic by 15.6 kcal mol−1, with a barrier of 15.5 kcal mol−1 (with respect to the radical Int3). Finally, the final product is obtained with a hydrogen atom abstraction from 1,3,5-trimethyl-1,4-cyclohexadiene viaTS4 with a barrier of 26.3 kcal mol−1 (relative to the radical Int4). The whole reductive coupling reaction is exergonic by 11.7 kcal mol−1 (with respect to the reactants 1a and Int1). These results suggest that the studied reaction is thermodynamically and kinetically feasible under the experimental conditions. In addition, our calculations suggest that the direct single electron transfer (SET) process between the pyridine-boryl radical and isobutyraldehyde is highly endergonic (see details in Fig. S13 and S14†). Thus, the SET mechanism for the present reaction can be excluded.
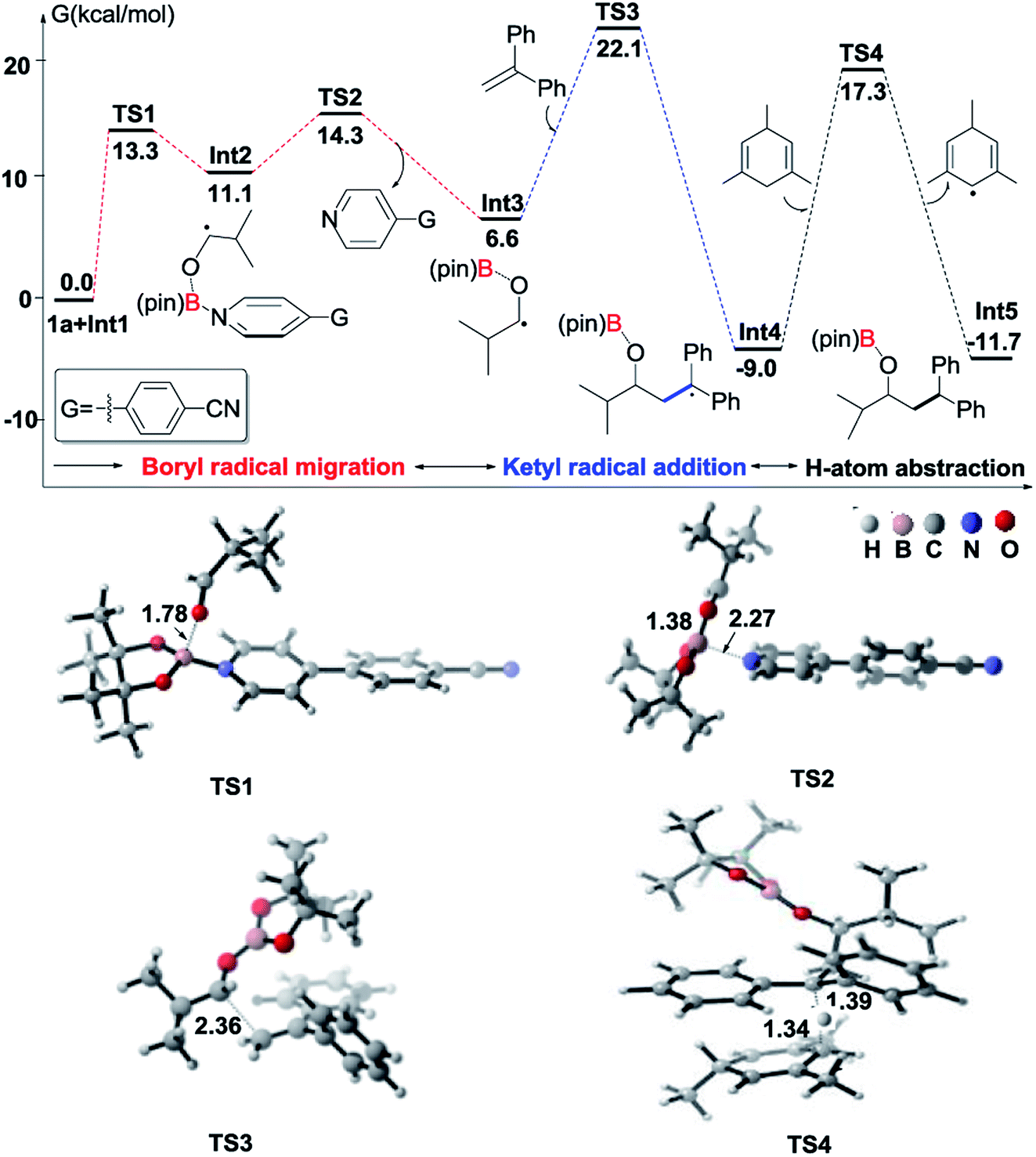 | ||
| Fig. 1 Computed Gibbs free energy profile of the reductive coupling reaction between isobutyraldehyde (1a) and 1,1-diphenylethylene (2) promoted by the 4-(4-pyridinyl)benzonitrile-boryl radical (Int1). The optimized structures of transition states in the reaction path are also displayed. Interatomic distances are in Å. | ||
In addition to DFT calculations described above, we also conducted several experiments to verify the proposed pathway. First, the EPR signal was observed for the reaction of 4-(4-pyridinyl)benzonitrile and B2(pin)2, which supports the formation of the proposed pyridine-boryl radical, as shown in Scheme 3a. Second, the involvement of the ketyl radical was confirmed by a competition experiment (Scheme 3b). It has been reported that thiols are quick hydrogen atom donors that can interfere with the radical reaction.20 When the hydrogen source 1,3,5-trimethyl-1,4-cyclohexadiene was replaced by 3-methylbenzenethiol, the ketyl radical quickly abstracted a hydrogen atom from 3-methylbenzenethiol to yield the reductive product, 3-phenyl-1-propanol, so that its addition to 1,1-diphenylethylene (to form the reductive coupling product) was inhibited (see Page S20†). This result clearly indicated the involvement of the ketyl radical. Third, the generation of the radical species Int4 (or its analogues) via the addition of the ketyl radical to the β-position of arylethene was confirmed by an intermolecular trapping experiment (Scheme 3c). When 2-vinylpyridine and trimethylacetaldehyde were subjected to the standard reaction conditions, species 6 could be detected by HRMS analysis for the crude reaction mixture (see Page S21†). This result suggests that in this reaction, the radical species Int4-like was further trapped by another 2-vinylpyridine molecule. However, in the presence of 2-vinylpyridine as a substrate, the yield of 6 is quite low and its isolation from the reaction mixture was not successful. Besides, we further conducted analysis of the 11B-NMR spectrum and HRMS to detect the formation of the proposed O–boron intermediate (Int6, Fig. S17†). The 11B-NMR of the crude reaction mixture displays resonances at ∼21 ppm, which is consistent with the signal of a boron atom bound to three oxygen atoms.21 In addition, our HRMS analysis (with 4-vinylpyridine as the substrate) also indicates the formation of the O–boron intermediate (Int7), as shown in Fig. S18.† Moreover, we have performed a radical-clock study using cyclopropanecarboxaldehyde as the substrate. The experimental results indicate that some ketyl radicals first convert into the corresponding carbon radicals (via a ring-opening process) and then add to the alkene to form the ring-opening product (Scheme 3d). The experiments described above provide strong evidence on the involvement of a radical addition step between the ketyl radical and 1,1-diarylethylene in this reaction.
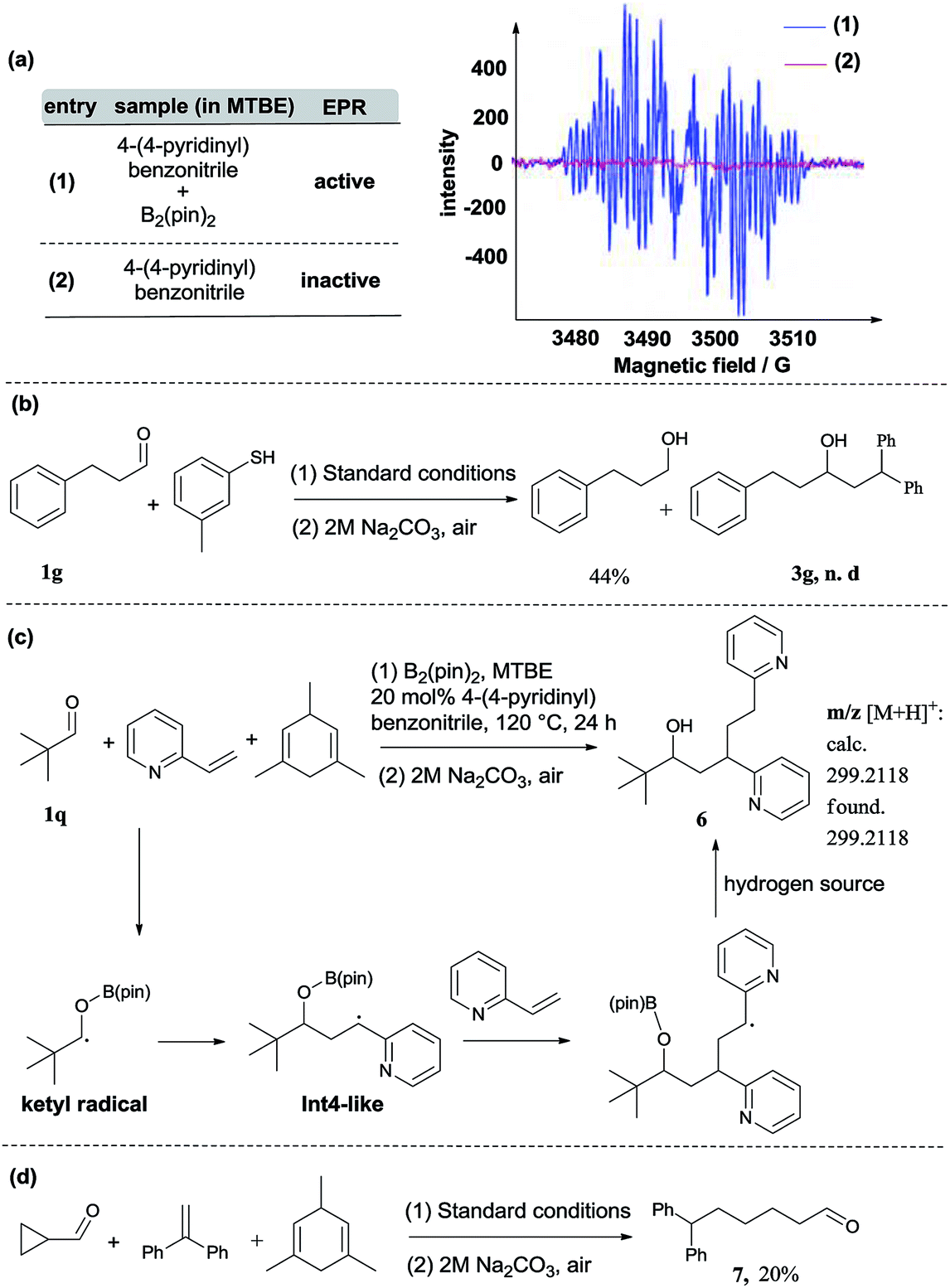 | ||
| Scheme 3 Controlled experiments. | ||
Conclusions
In summary, we have established the organocatalytic reductive coupling of aldehydes with 1,1-diarylalkenes via a combination of computational and experimental studies. This study showed that 4-(4-pyridinyl)benzonitrile is a suitable catalyst for cleaving the B–B bond of B2pin2, and the ketyl radical from the addition of an in situ generated pyridine-boryl radical to aldehydes is a key intermediate for the C–C bond formation. The reaction is practical and applicable to a broad range of aldehydes and 1,1-diarylalkenes with good functional group tolerance. DFT calculations and control experiments were conducted to verify the proposed mechanism. This pyridine-boryl radical promoted radical addition mechanism represents a metal-free reductive coupling reaction of aldehydes with 1,1-diarylalkenes. Further studies will be directed toward the development of new transformations involving readily formed pyridine-boryl radicals with the aid of combined theoretical and experimental studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 21333004 and No. 21361140376, No. 21572099) and China Postdoctoral Science Foundation (Grant No. 2017M620198). All calculations in this work have been done on the IBM Blade cluster system at the High Performance Computing Center of Nanjing University.Notes and references
- (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS PubMed; (b) C. L. Sun and Z. J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed.
- (a) B. Sam, B. Breit and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 3267 CrossRef CAS PubMed; (b) K. D. Nguyen, B. Y. park, T. Luong, H. Sato, V. J. Garza and M. J. Krische, Science, 2016, 354, 6310 CrossRef PubMed; (c) S. W. Kim, W. Zhang and M. J. Kriche, Acc. Chem. Res., 2017, 50, 2371 CrossRef CAS PubMed.
- (a) M. Kimura, A. Ezoe, K. Shibata and Y. Tamaru, J. Am. Chem. Soc., 1998, 120, 4033 CrossRef CAS; (b) J. A. Marshall and N. D. Adamas, J. Org. Chem., 1998, 63, 3812 CrossRef CAS; (c) J. A. Marshall and N. D. Adamas, J. Org. Chem., 1999, 64, 5201 CrossRef CAS; (d) J. A. Marshall and H. R. Chobanian, J. Org. Chem., 2000, 65, 8357 CrossRef CAS PubMed; (e) H. Y. Jang, R. R. Huddleston and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 15156 CrossRef CAS PubMed; (f) J. F. Bower, R. L. Patman and M. J. Krische, Org. Lett., 2008, 10, 1033 CrossRef CAS PubMed; (g) I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 12156 Search PubMed; (h) C. C. Wang, P. S. Lin and C. H. Cheng, J. Am. Chem. Soc., 2002, 124, 9696 CrossRef CAS PubMed; (i) L. M. Geary, S. K. Woo, J. C. Leung and M. J. Krische, Angew. Chem., Int. Ed., 2012, 51, 2972 CrossRef CAS PubMed; (j) L. M. Geary, J. C. Leung and M. J. Krische, Chem.–Eur. J., 2012, 18, 16823 CrossRef CAS PubMed.
- (a) S. Y. Zhang, Y. Q. Tu, C. A. Fan, F. M. Zhang and L. Shi, Angew. Chem., Int. Ed., 2009, 48, 8761 CrossRef CAS PubMed; (b) J. M. Ketcham, I. Shin, T. P. Montgomery and M. J. Krische, Angew. Chem., Int. Ed., 2014, 53, 9142 CrossRef CAS PubMed; (c) Y. L. Zheng, Y. Y. Liu, Y. M. Wu, Y. X. Wang, Y. T. Lin and M. Ye, Angew. Chem., Int. Ed., 2016, 55, 6315 CrossRef CAS PubMed.
- (a) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828 CrossRef CAS PubMed; (b) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312 CrossRef CAS PubMed; (c) K. N. Lee, Z. Lei and M.-Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003 CrossRef CAS PubMed; (d) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652 CrossRef CAS PubMed.
- (a) B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25 RSC; (b) B. Carboni and L. Monnier, Tetrahedron, 1999, 55, 1197 CrossRef CAS; (c) C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415 CrossRef CAS PubMed; (d) Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, 1st edn, 2001, pp. 11–27 Search PubMed; (e) P. P. Power, Chem. Rev., 2003, 103, 789 CrossRef CAS PubMed; (f) P. Renaud, A. Beauseigneur, A. Brecht-Forster, B. Becattini, V. Darmency, S. Kandhasamy, F. Montermini, C. Ollivier, P. Panchaud, D. Pozzi, E. M. Sccanlan, A.-P. Schaffner and W. Weber, Pure Appl. Chem., 2007, 79, 223 CrossRef CAS; (g) D. Crich, D. Grant, V. Krishnamurthy and M. Patel, Acc. Chem. Res., 2007, 40, 453 CrossRef CAS PubMed; (h) A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023 CrossRef CAS PubMed; (i) D. Lu, C. Wu and P. Li, Chem.–Eur. J., 2014, 20, 1630 CrossRef CAS PubMed; (j) A. Martí-Sómer, O. Mó and M. Yáῆez, ChemPhysChem, 2014, 15, 2288 CrossRef PubMed; (k) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed; (l) D. Zhao and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 3166 CrossRef CAS PubMed.
- For selected examples on N-heterocyclic carbine-boryl radical, see: (a) S. H. Ueng, M. M. Brahmi, É. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, J. Am. Chem. Soc., 2008, 130, 10082 CrossRef CAS PubMed; (b) J. C. Walton, M. M. Brahmi, L. Fensterbank, E. Lacôte, M. Malacria, Q. Chu, S. H. Ueng, A. Solovyev and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 2350 CrossRef CAS PubMed; (c) J. C. Walton, M. M. Brahmi, J. Monot, L. Fensterbank, M. Malacria, D. P. Curran and E. Lacôte, J. Am. Chem. Soc., 2011, 133, 10312 CrossRef CAS PubMed; (d) X. Pan, A. L. Vallet, S. Schweizer, K. Dahbi, B. Delpech, N. Blanchard, B. Graff, S. J. Geib, D. P. Curran, J. Lalevée and E. Lacôte, J. Am. Chem. Soc., 2013, 135, 10484 CrossRef CAS PubMed; (e) T. Kawamoto, S. J. Geib and D. P. Curran, J. Am. Chem. Soc., 2015, 137, 8617 CrossRef CAS PubMed; (f) G. Tambutet and Y. Guindon, J. Org. Chem., 2016, 81, 11427 CrossRef CAS PubMed; (g) S. C. Ren, F. L. Zhang, J. Qi, Y. S. Huang, A. Q. Xu, H. Y. Yan and Y. F. Wang, J. Am. Chem. Soc., 2017, 139, 6050 CrossRef CAS PubMed; (h) M. F. Silva, P. Schweyen, D. Gisinger, T. Bannenbergm, M. Freytag, C. Kleeberg and M. Tamm, Angew. Chem., Int. Ed., 2017, 56, 1135 CrossRef PubMed; (i) J. W. Taylor, A. M. Skimming, C. F. Guzman and W. H. Will, J. Am. Chem. Soc., 2017, 139, 11032 CrossRef PubMed.
- For selected examples on the amine-boryl radical, see: (a) J. A. Baban, V. P. J. Marti and B. P. Robert, J. Chem. Soc., Perkin Trans. 2, 1985, 1723 RSC; (b) K. M. Johnson, J. Nicholas Kirwan and B. P. Robert, J. Chem. Soc., Perkin Trans. 2, 1990, 1125 RSC; (c) J. Lalevée, N. Blanchard, A. C. Chany, M. A. Tehfe, X. Allonas and J.-P. Fouassier, J. Phys. Org. Chem., 2009, 22, 986 CrossRef; (d) J. Lalevée, N. Blanchard, M. A. Tehfe, A. C. Chany and J. P. Fouassier, Chem.–Eur. J., 2010, 16, 12920 CrossRef PubMed; (e) Y. Aramaki, H. Omiya, M. Yamashita, K. Nakabayashi, S. i. Ohkoshi and K. NozaKI, J. Am. Chem. Soc., 2012, 134, 19989 CrossRef CAS PubMed; (f) Y. J. Yu, F. L. Zhang, J. Cheng, J. H. Hao, W. T. Deng and Y. F. Wang, Org. Lett., 2018, 20, 24 CrossRef CAS PubMed; (g) L. Zhang and L. Jiao, Chem. Sci., 2018 10.1039/c8sc00008e.
- For selected examples on the phosphine-boryl radical, see: (a) D. H. R. Barton and M. Jacob, Tetrahedron Lett., 1998, 39, 1331 CrossRef CAS; (b) A. J. Rosenthal, M. Devillard, K. Miqueu, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 9198 CrossRef CAS PubMed.
- Recent examples on radical reactions involving B2pin2, see: (a) F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. B. Wang, Angew. Chem., Int. Ed., 2010, 49, 1846 CrossRef CAS PubMed; (b) A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 2985 CrossRef CAS PubMed; (c) W. B. Liu, X. B. Yang, Y. Gao and C. J. Li, J. Am. Chem. Soc., 2017, 139, 8621 CrossRef CAS PubMed; (d) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283 CrossRef CAS PubMed; (e) L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440 CrossRef CAS PubMed.
- G. Wang, H. Zhang, J. Zhao, W. Li, J. Cao, C. Zhu and S. Li, Angew. Chem., Int. Ed., 2016, 55, 5985 CrossRef CAS PubMed.
- G. Wang, J. Cao, L. Gao, W. Chen, W. Huang, X. Cheng and S. Li, J. Am. Chem. Soc., 2017, 139, 3904 CrossRef CAS PubMed.
- For reviews on the persistent radical, see: (a) A. Studer, Chem.–Eur. J., 2001, 7, 1159 CrossRef CAS PubMed; (b) A. Studer, Chem. Soc. Rev., 2004, 33, 267 RSC; (c) H. Fischer, Chem. Rev., 2001, 101, 3581 CrossRef CAS PubMed; (d) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- (a) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607 CrossRef CAS PubMed; (b) W.-M. Cheng, R. Shang, B. Zhao, W.-L. Xing and Y. Fu, Org. Lett., 2017, 19, 4291 CrossRef CAS PubMed.
- (a) I. Beletskaya and C. Moberg, Chem. Rev., 2006, 106, 2320 CrossRef CAS PubMed; (b) J. Takaya and N. Iwasawa, ACS Catal., 2012, 2, 1993 CrossRef CAS; (c) R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594 RSC; (d) T. Ohmura, Y. Morimasa and M. Suginome, J. Am. Chem. Soc., 2015, 137, 2852 CrossRef CAS PubMed; (e) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS PubMed; (f) A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415–430 RSC; (g) M. B. Ansell, O. Navarro and J. Spencer, Coord. Chem. Rev., 2017, 1, 54 CrossRef.
- (a) Y. Zhao, N. E Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef PubMed; (b) Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed; (c) Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 13126 CrossRef CAS PubMed.
- D. P. Chen, G. Q. Xu, Q. H. Zhou, L. W. Chung and W. J. Tang, J. Am. Chem. Soc., 2017, 139, 9767 CrossRef CAS PubMed.
- This new mechanism on the formation of the pyridine-boryl radical in the 4-cyanopyridine/B2pin2 system is different from that reported in ref. 11.
- (a) A. Vasilopoulos, S. L. Zultanski and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 7705 CrossRef CAS PubMed; (b) E. Yamamoto, M. J. Hilton, M. Orlandi, V. Saini, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 15877 CrossRef CAS PubMed.
- X. C. Pan, E. Lacôte, J. Lalevée and D. P. Curran, J. Am. Chem. Soc., 2012, 134, 5669 CrossRef CAS PubMed.
- R. Shintani and K. Nozaki, Organometallics, 2013, 32, 2459–2462 CrossRef CAS; D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05225a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |