
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
cis-Oxoruthenium complexes supported by chiral tetradentate amine (N4) ligands for hydrocarbon oxidations†
Chun-Wai
Tse
 ab,
Yungen
Liu
ac,
Toby
Wai-Shan Chow
a,
Chaoqun
Ma
c,
Wing-Ping
Yip
a,
Xiao-Yong
Chang
a,
Kam-Hung
Low
a,
Jie-Sheng
Huang
a and
Chi-Ming
Che
*abc
ab,
Yungen
Liu
ac,
Toby
Wai-Shan Chow
a,
Chaoqun
Ma
c,
Wing-Ping
Yip
a,
Xiao-Yong
Chang
a,
Kam-Hung
Low
a,
Jie-Sheng
Huang
a and
Chi-Ming
Che
*abc
aState Key Laboratory of Synthetic Chemistry, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: cmche@hku.hk
bHKU Shenzhen Institute of Research and Innovation, Shenzhen, Guangdong 518053, China
cDepartment of Chemistry, Southern University of Science of Technology, Shenzhen, Guangdong 518055, China
First published on 15th February 2018
Abstract
We report the first examples of ruthenium complexes cis-[(N4)RuIIICl2]+ and cis-[(N4)RuII(OH2)2]2+ supported by chiral tetradentate amine ligands (N4), together with a high-valent cis-dioxo complex cis-[(N4)RuVI(O)2]2+ supported by the chiral N4 ligand mcp (mcp = N,N′-dimethyl-N,N′-bis(pyridin-2-ylmethyl)cyclohexane-1,2-diamine). The X-ray crystal structures of cis-[(mcp)RuIIICl2](ClO4) (1a), cis-[(Me2mcp)RuIIICl2]ClO4 (2a) and cis-[(pdp)RuIIICl2](ClO4) (3a) (Me2mcp = N,N′-dimethyl-N,N′-bis((6-methylpyridin-2-yl)methyl)cyclohexane-1,2-diamine, pdp = 1,1′-bis(pyridin-2-ylmethyl)-2,2′-bipyrrolidine)) show that the ligands coordinate to the ruthenium centre in a cis-α configuration. In aqueous solutions, proton-coupled electron-transfer redox couples were observed for cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) and cis-[(pdp)RuIII(O3SCF3)2]CF3SO3 (3c′). Electrochemical analyses showed that the chemically/electrochemically generated cis-[(mcp)RuVI(O)2]2+ and cis-[(pdp)RuVI(O)2]2+ complexes are strong oxidants with E° = 1.11–1.13 V vs. SCE (at pH 1) and strong H-atom abstractors with DO–H = 90.1–90.8 kcal mol−1. The reaction of 1b or its (R,R)-mcp counterpart with excess (NH4)2[CeIV(NO3)6] (CAN) in aqueous medium afforded cis-[(mcp)RuVI(O)2](ClO4)2 (1e) or cis-[((R,R)-mcp)RuVI(O)2](ClO4)2 (1e*), respectively, a strong oxidant with E(RuVI/V) = 0.78 V (vs. Ag/AgNO3) in acetonitrile solution. Complex 1e oxidized various hydrocarbons, including cyclohexane, in acetonitrile at room temperature, affording alcohols and/or ketones in up to 66% yield. Stoichiometric oxidations of alkenes by 1e or 1e* in tBuOH/H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) afforded diols and aldehydes in combined yields of up to 98%, with moderate enantioselectivity obtained for the reaction using 1e*. The cis-[(pdp)RuII(OH2)2]2+ (3c)-catalysed oxidation of saturated C–H bonds, including those of ethane and propane, with CAN as terminal oxidant was also demonstrated.
:1 v/v) afforded diols and aldehydes in combined yields of up to 98%, with moderate enantioselectivity obtained for the reaction using 1e*. The cis-[(pdp)RuII(OH2)2]2+ (3c)-catalysed oxidation of saturated C–H bonds, including those of ethane and propane, with CAN as terminal oxidant was also demonstrated.
Introduction
The selective oxidations of hydrocarbons1 including alkanes and alkenes, and oxidation of alcohols,2 catalysed by metal complexes under mild conditions are important reactions in chemical synthesis. Iron and manganese complexes bearing tetradentate pyridylmethyl amine or quinolylamine N4 ligands1b,h,i,k–o,r constitute one of the platforms for performing efficient and selective C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C functionalizations, wherein the widely employed N4 ligands include mcp, pdp and bqcn ligands and their derivatives (examples depicted in Fig. 1).3–6 These acyclic chiral tetradentate amine (N4) ligands, in most scenarios, coordinate to metal ions in a cis-α configuration to form octahedral metal complexes (Fig. 2), leaving a pair of cis sites for oxidant activation or substrate binding. Stereoretentive C–H hydroxylation,3a,b,f,4a,b enantioselective epoxidation5c–f,6a,c–e and asymmetric cis-dihydroxylation (AD) of alkenes5g,6b have been achieved under limiting substrate conditions. One type of proposed active metal–oxo intermediates in these oxidation reactions catalysed by metal chiral N4 complexes is the corresponding high-valent cis-dioxo complexes, i.e., cis-M(O)2 species supported by chiral N4 ligands.6b In the iron-catalysed systems, cis-[(N4)FeV(O)(OR)]2+ (R = H or acyl) active intermediates,5h,7,8,9 and a cis-[(N4)FeV(O)2]+ active intermediate in alkene cis-dihydroxylation,10 have been proposed; isolation of these active species and elucidation of the reaction mechanisms in these iron systems have often been difficult because of the extraordinary reactivity of high-valent iron–oxo complexes. A proposed cis-[(N4)MnV(O)2]+ intermediate, cis-[((S,S)-bqcn)MnV(O)2]+,6b in manganese-catalysed enantioselective cis-dihydroxylation of alkenes was also found to be insufficiently stable for isolation. While several cis-[(N4)ReV(O)2]+ complexes and a cis-[(N4)ReVI(O)2]2+ complex have been isolated and structurally characterized in our recent work,11 the former were not reactive towards organic substrates, and concerning hydrocarbon oxidation reactivity, the latter only reacted with weak C–H bonds (bond dissociation energy: ∼76 kcal mol−1) of 1,4-cyclohexadiene, 9-10-dihydroanthracene and xanthene at 80 °C to give dehydrogenation or ketone products.
C functionalizations, wherein the widely employed N4 ligands include mcp, pdp and bqcn ligands and their derivatives (examples depicted in Fig. 1).3–6 These acyclic chiral tetradentate amine (N4) ligands, in most scenarios, coordinate to metal ions in a cis-α configuration to form octahedral metal complexes (Fig. 2), leaving a pair of cis sites for oxidant activation or substrate binding. Stereoretentive C–H hydroxylation,3a,b,f,4a,b enantioselective epoxidation5c–f,6a,c–e and asymmetric cis-dihydroxylation (AD) of alkenes5g,6b have been achieved under limiting substrate conditions. One type of proposed active metal–oxo intermediates in these oxidation reactions catalysed by metal chiral N4 complexes is the corresponding high-valent cis-dioxo complexes, i.e., cis-M(O)2 species supported by chiral N4 ligands.6b In the iron-catalysed systems, cis-[(N4)FeV(O)(OR)]2+ (R = H or acyl) active intermediates,5h,7,8,9 and a cis-[(N4)FeV(O)2]+ active intermediate in alkene cis-dihydroxylation,10 have been proposed; isolation of these active species and elucidation of the reaction mechanisms in these iron systems have often been difficult because of the extraordinary reactivity of high-valent iron–oxo complexes. A proposed cis-[(N4)MnV(O)2]+ intermediate, cis-[((S,S)-bqcn)MnV(O)2]+,6b in manganese-catalysed enantioselective cis-dihydroxylation of alkenes was also found to be insufficiently stable for isolation. While several cis-[(N4)ReV(O)2]+ complexes and a cis-[(N4)ReVI(O)2]2+ complex have been isolated and structurally characterized in our recent work,11 the former were not reactive towards organic substrates, and concerning hydrocarbon oxidation reactivity, the latter only reacted with weak C–H bonds (bond dissociation energy: ∼76 kcal mol−1) of 1,4-cyclohexadiene, 9-10-dihydroanthracene and xanthene at 80 °C to give dehydrogenation or ketone products.
 | ||
| Fig. 1 Structures and abbreviations of chiral N4 ligands used in this study. Ligands were prepared as either the (R,R)-enantiomer or a racemic mixture. | ||
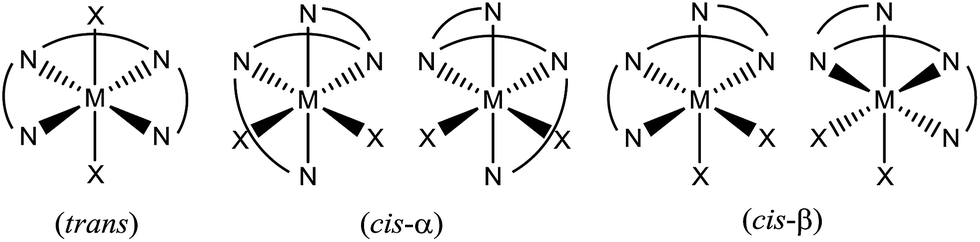 | ||
| Fig. 2 Different wrapping modes of acyclic tetradentate N4 ligands in an octahedral environment. | ||
To search for isolable cis-dioxo metal complexes that are supported by the abovementioned chiral N4 ligands and are reactive towards hydrocarbon oxidation, including the cis-dihydroxylation of alkenes and the oxidation of strong C–H bonds at room temperature, we directed our efforts to ruthenium systems. High-valent Ru–oxo complexes are generally more stable than their iron counterparts due to their lower redox potentials as well as substitutional inertness of auxiliary ligands.12cis-Dioxoruthenium(VI) complexes can have a delicate balance between stability and reactivity that allows them to be isolated/characterized12,13 or even studied in reactions with organic substrates in a stoichiometric manner.14–16 Several cationic cis-dioxoruthenium(VI) complexes, including cis-[(Tet-Me6)RuVI(O)2]2+ (Tet-Me6 = N,N,N′,N′-3,6-hexamethyl-3,6-diazooctane-1,8-diamine),14acis-[(Me3tacn)RuVI(O)2(O2CCF3)]+ (Me3tacn = 1,4,7-trimethyl-triazacyclononane),15acis-[(6,6′-Cl2bpy)2RuVI(O)2]2+ (6,6′-Cl2bpy = 6,6′-dichloro-2,2′-bipyridine),16acis-[(bpy)2RuVI(O)2]2+ (bpy = 2,2′-bipyridine)17 and cis-[(dmp)2RuVI(O)2]2+ (dmp = 2,9-dimethyl-1,10-phenanthroline),18 have been isolated and/or spectroscopically characterized. Among them, cis-[(Me3tacn)RuVI(O)2(O2CCF3)]+ and cis-[(6,6′-Cl2bpy)2RuVI(O)2]2+ are known to react with simple saturated alkanes (e.g., cyclohexane) stoichiometrically.15,16 The related catalytic oxygenation of cyclohexane with tBuOOH could be performed with [(Me3tacn)RuIIICl3]19 and cis-[(Cl2bpy)2RuII(OH2)2]2+ as catalysts.16b Du Bois and co-workers recently demonstrated selective C–H functionalization catalysed by [(Me3tacn)RuIIICl3] with ceric ammonium nitrate (CAN) as a terminal oxidant to give tertiary C–H hydroxylation products.20 Using bis(bipyridine)Ru catalysts, the selective functionalization of amine derivatives was attainable with various oxidants and acid additives.21 These studies highlight the amendable oxidation capabilities of the cis-dioxoruthenium(VI) moiety and the underdeveloped potential of ruthenium catalysts in C–H oxidation.
Thus far, studies on highly oxidizing cis-dioxoruthenium(VI) complexes have focused on tridentate Me3tacn and simple bidentate aromatic diimine ligands.15–18 The Me3tacn ligand is not flexible for structure modification.21 Ruthenium complexes with aromatic diamine ligands may undergo cis–trans isomerization22 and ligand loss in a high oxidation state.17 These difficulties can potentially be resolved by utilizing the abovementioned chiral N4 ligands: the first coordination sphere is highly tuneable by ligand modification, as revealed by recent works from White,3d Costas,3b,4c,d and their co-workers; the higher rigidity and denticity can provide better conformational stability under catalytic conditions.
In this work, we aim to (i) isolate/generate cis-dioxoruthenium(VI) complexes bearing chiral tetradentate amine (N4) ligands, (ii) study the redox potentials and hydrocarbon oxidation reactions of these cis-[(N4)RuVI(O)2]2+ complexes, and (iii) gain insight into the activity of chiral Ru(N4) complex in asymmetric oxidation reactions. Until now, studies on ruthenium complexes supported by chiral N4 ligands (mcp, pdp, bqcn and their derivatives) have been limited,23–25 including a report involving some data of [RuII(mcp)Cl2]-catalysed oxidation of thioanisole with H2O2,25a another report involving the synthesis and crystallographic characterization of [RuII((R,R)-pdp)(NCMe)2]2+,25b and density functional calculations on a hypothetical monooxo ruthenium(IV) species cis-[(bqcn)RuIV(O)(NCMe)]2+.26 No examples of the corresponding cis-dioxo ruthenium chiral N4 complexes have been reported.23–26 Herein, we describe the syntheses, characterization, and electrochemical and reactivity studies of a series of chiral Ru(N4) complexes, including a highly reactive chiral cis-[(N4)RuVI(O)2]2+ complex that can perform dihydroxylation of alkenes and oxidation of strong C–H bonds of alkanes (including cyclohexane) and oxidation of alcohols at room temperature. The studies on cis-[(N4)RuVI(O)2]2+ complexes provide insight into the reactivity and electrochemical properties of the analogous highly oxidizing cis-[(N4)M(O)2]n+ (n = 1 or 2; M = Fe or Mn) species.6b,10
Results
Synthesis and characterization
In this work, a series of ruthenium complexes bearing six chiral tetradentate amine N4 ligands (Fig. 1) and different auxiliary ligands were prepared (Schemes 1 and 2). The reaction of K2[RuIIICl5(OH2)] with the mcp, Me2mcp, pdp or Me2pdp ligand in ethanol under refluxing conditions (Scheme 1) gave the corresponding cis-[(N4)RuIIICl2]+ complex (1a, 2a, 3a or 4a) in 32–97% yield.27 The reaction of 1a with Zn/Hg in distilled water at 80 °C for 30 min, followed by subsequent treatment of the solution with AgOTf and 0.2 M CF3CO2H, afforded cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) in 20% yield (Scheme 1).28 To prepare ruthenium complexes containing the bqcn and Me2bqcn ligands, an alternative synthetic method was developed. Treatment of bqcn or Me2bqcn with a slight excess (1.2 equiv.) of [RuII(OH2)6](OTs)2 under Ar in THF furnished the OTs− salt of cis-[(N4)RuII(OH2)2]2+ (5c or 6c) in good yield (up to 71%). A similar treatment using pdp or Me2pdp gave the OTs− salt of 3c or 4c. Recrystallization of 5c·OTs or 6c·OTs in acetonitrile in the presence of LiClO4 produced cis-[(bqcn)RuII(NCMe)2](ClO4)2 (5d) and cis-[(Me2bqcn)RuII(NCMe)2](ClO4)2 (6d), respectively (Scheme 2).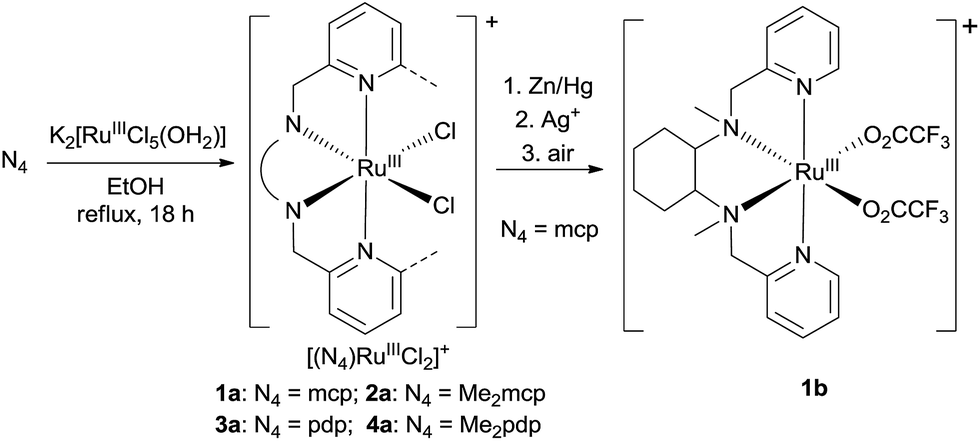 | ||
| Scheme 1 Preparation of 1a–4a and 1b. | ||
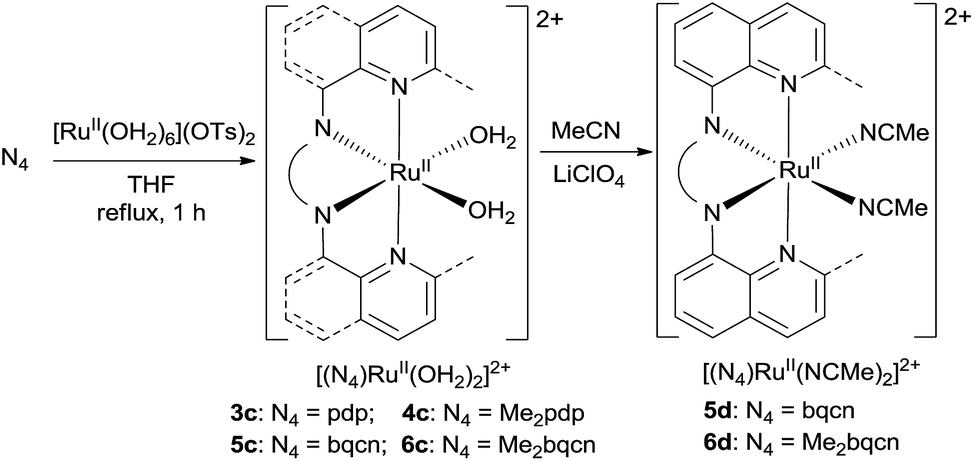 | ||
| Scheme 2 Preparation of 3c–6c, 5d and 6d. | ||
The structures of 1a, 2a, 3a, 5d and 6d (as ClO4− salts) were established by X-ray crystallography. All these complexes, except 5d, adopt a cis-α configuration (Fig. 3 and S1–S5, ESI†), where the two terminal pyridyl/quinolyl groups are positioned trans to each other. For 5d, its crystal structure showed that the cis-α and cis-β isomers are present in a 1:1 ratio in the unit cell.29 The two isomers could not be separated by repeated recrystallizations. Fig. 3a depicts the structure of cis-α-5d; its two methyl groups on the cyclohexane-1,2-diamine nitrogen atoms are oriented anti to each other (C40 and C47). In the cis-β isomer (Fig. 3b), the corresponding two methyl groups (C10 and C17) show the opposite (syn) orientation.
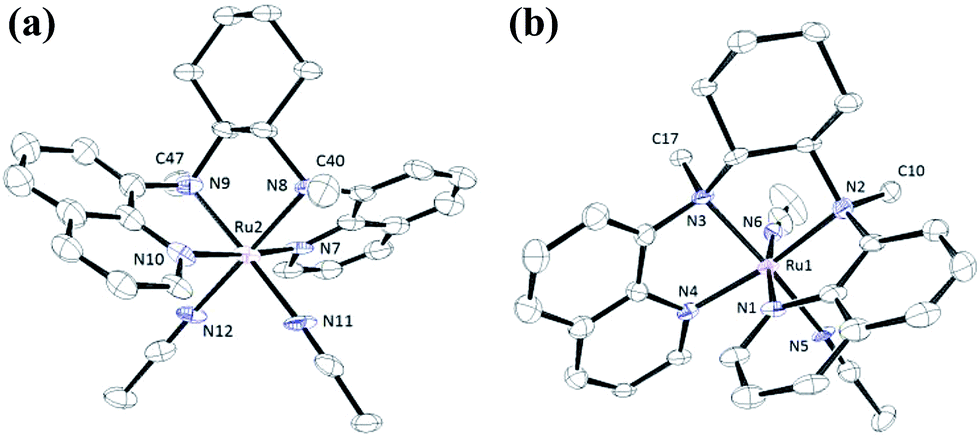 | ||
| Fig. 3 ORTEP drawings of the complex cations of cis-[(bqcn)RuII(NCMe)2](ClO4)2 (5d). (a, left) cis-α isomer (α-5d). (b, right) cis-β isomer (β-5d). Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 30% probability level. | ||
The 1H NMR spectra of 3c, 4c (in CD3CN) and 6d show signals indicative of a cis-α configuration (Fig. 2) with C2 symmetry (see Experimental section); no interconversion to the cis-β conformer was observed by standing the solution at room temperature for days. In contrast, the 1H NMR spectrum of 5d comprises a mixture of signals from the cis-α and cis-β isomers.
The UV-Vis absorption spectra of the cis-[(N4)RuIIICl2]+ complexes (1a, 2a, 3a and 4a) in acetonitrile solution are characterized by pπ(Cl) → Ru(III) LMCT transition band at λ 400–450 nm (ε = 900–2000 dm3 mol−1 cm−1, Fig. S6, ESI†).27 In aqueous solutions, the cis-[(N4)RuII(OH2)2]2+ complexes (3c, 4c, 5c and 6c) show intense absorption bands at 361–477 nm (ε = 4700–6900 dm3 mol−1 cm−1, Fig. S7, ESI†) assignable to dπ (Ru) → pπ* (pyridyl or quinolyl) MLCT transitions.
The cis-dichlororuthenium(III) complexes display different cyclic voltammetric behaviours in acetonitrile solutions (Fig. S9, ESI†). Complexes 1a and 3a display a reversible couple at E1/2 = ca. 0 V vs. SCE. This is assigned to the RuIII/II couple: cis-[(N4)RuIIICl2]+ + e− → cis-[(N4)RuIICl2]0. The RuIV/III couple was not observed at potentials up to 1.6 V vs. SCE. For 2a, where the N4 ligand possesses a methyl substituent on the pyridyl moiety, the RuIII/II couple is irreversible. The irreversible reduction of cis-[(N4)RuIIICl2]+ occurs at Epc = −0.01 V; upon the reverse scan, an oxidation wave appears at Epa = 0.69 V, which is attributed to the oxidation of cis-[(Me2mcp)RuIICl(NCMe)]+ to cis-[(Me2mcp)RuIIICl(NCMe)]2+ after a ligand exchange reaction of [(Me2mcp)RuIICl2]0 with the solvent.30,31 The cyclic voltammograms of 5d and 6d in MeCN display reversible oxidation couples at E1/2 = 1.35 V and 1.36 V vs. SCE, respectively (Fig. S11, ESI†). The electrochemical reaction is assigned to: cis-[(N4)RuIII(NCMe)2]3+ + e− → cis-[(N4)RuII(NCMe)2]2+.
Aqueous electrochemistry of cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) and cis-[(pdp)RuIII(O3SCF3)2]CF3SO3 (3c′)
The cyclic voltammogram of cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) at pH 1 displays three reversible/quasi-reversible couples (i), (ii) and (iii) at E1/2 = 0.37, 0.92 and 1.11 V vs. SCE, respectively (Fig. 4a). Using rotating-disk electrode voltammetry, the coulombic stoichiometries of the redox couples were determined to be 1.0, 1.9 and 1.1 for couples (i), (ii) and (iii), respectively (Fig. 4b). With reference to previous work,27 these couples could be assigned to RuIII/II, RuV/III and RuVI/V redox processes, and the electrochemical reactions (1)–(3) are depicted in Scheme 3. At pH 5, the E1/2 of couples (i) and (iii) shift to 0.25 and 0.98 V, respectively, and couple (ii) splits into two reversible one-electron couples (iv) and v at 0.65 and 0.77 V, respectively (Fig. S12, ESI†). Couples (iv) and (v) are assigned to RuIV/III and RuV/IV couples (eqn (4) and (5) in Scheme 3). The cathodic shift in the E1/2 of couple (iii) with an increasing pH is in accordance with other dioxoruthenium(VI) complexes.14a,32,33,34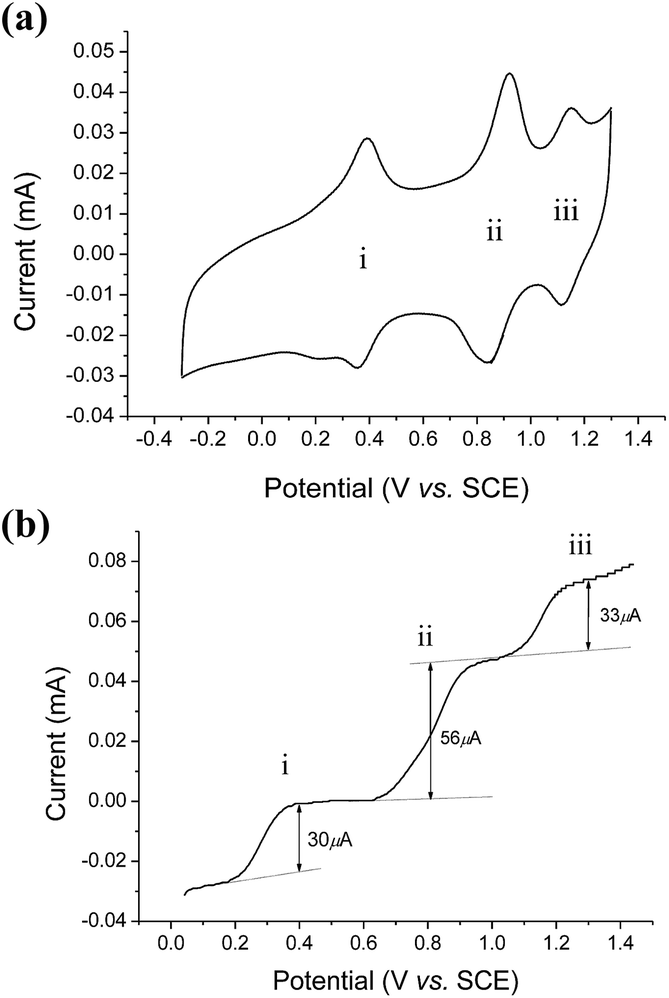 | ||
| Fig. 4 (a, upper) Cyclic voltammogram of cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) at pH 1. (b, lower) Rotating-disk electrode voltammogram of cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) at pH 1. Working electrode: edge-plane pyrolytic graphite for CV; glassy carbon for RDEV. Rest potentials: ca. 0.55 V. | ||
 | ||
| Scheme 3 Proposed redox couples for cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) in different pH buffer solutions. The cis-sign is omitted for clarity. | ||
The electrochemical properties of cis-[(pdp)RuIII(O3SCF3)2]-CF3SO3 (3c′, Scheme S1, ESI†) in 0.1 M CF3SO3H at pH 1 are reminiscent of that of 1b. As depicted in Fig. 5a, 3c′ shows a reversible couple I at E1/2 = 0.36 V and a quasi-reversible couple III at E1/2 = 1.13 V (Epa = 1.19 V) vs. SCE. Notably, at the foot of couple III, there is a less defined couple II at E1/2 = 0.95 V. Couple I (ΔEp ∼ 60 mV; ipa/ipc ∼ 1) is attributed to a RuIII/II couple (eqn (6) in Scheme 4). Couple II is assigned as a RuIV/III couple (eqn (7)). Its much smaller current measured relative to the RuIII/II couple is attributed to the rate-determining deprotonation of [RuIII(OH)] or [RuIII(OH2)] prior to the oxidation of RuIII to RuIV.35 Couple III is assigned as a RuVI/IV couple (eqn (8)).36 The natures of couples I, II and III were examined by rotating-disk electrode voltammetry (Fig. 5b), showing that the limiting current/number of electrons involved in couples I and (II and III) has a ratio of 1 to 2.7.37
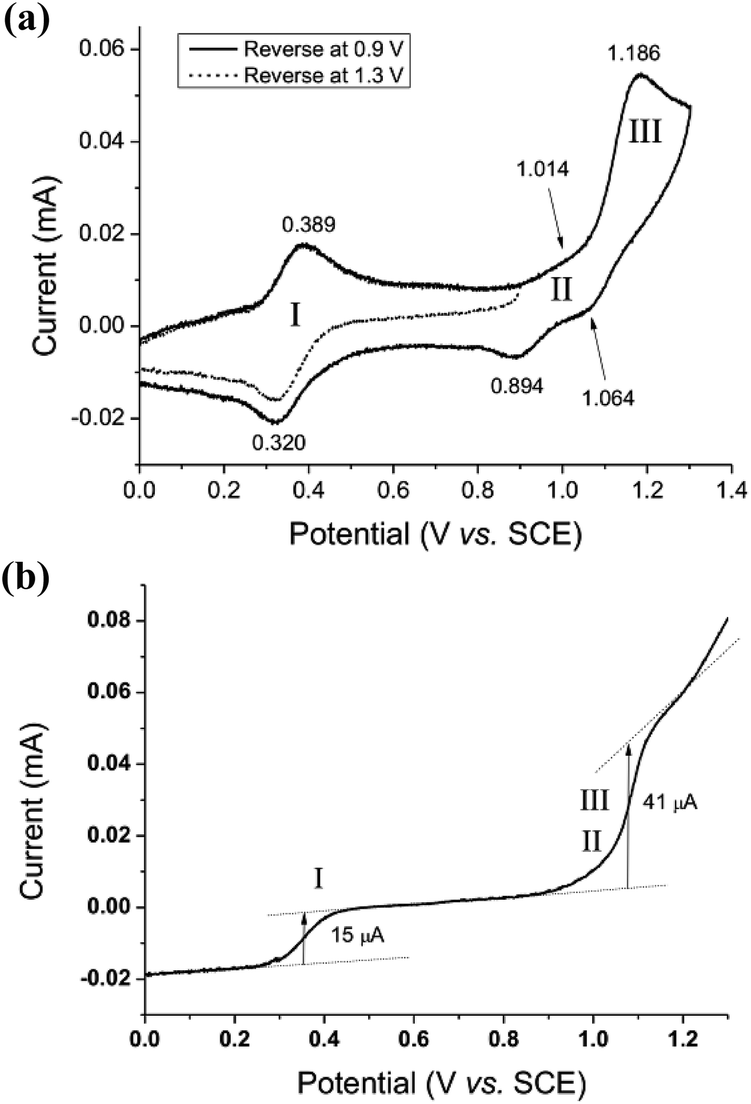 | ||
| Fig. 5 Cyclic voltammogram (a, upper) at 0.1 V s−1 and rotating-disk-electrode voltammogram (b, lower) at 100 rpm of 3c′ in 0.1 M CF3SO3H (pH 1). Working electrode: edge-plane pyrolytic graphite for CV; glassy carbon for RDEV. | ||
 | ||
| Scheme 4 Proposed redox couples for cis-[(pdp)RuIII(O3SCF3)2]CF3SO3 (3c′) in different pH buffer solutions. The cis-sign is omitted for clarity. | ||
The complex cis-[(pdp)RuII(OH2)2](OTs)2 (3c·OTs) similarly shows a reversible couple at E1/2 = 0.36 V and an irreversible oxidation wave at Epa = 1.14 V at pH 1 (Fig. S14a, ESI†).38 At pH 1, cis-[(bqcn)RuII(OH2)2](OTs)2 (5c·OTs) shows a reversible RuIII/II couple at E1/2 = 0.45 V and a shoulder oxidation wave at Epa = 1.15 V (Fig. S14b, ESI†), while cis-[(Me2bqcn)RuII(OH2)2](OTs)2 (6c·OTs) shows a reversible RuIII/II couple at E1/2 = 0.49 V and a shoulder oxidation wave at Epa = 1.08 V (Fig. S14c, ESI†). The σ-donating ability of the N4 ligands follows the order of mcp ≈ pdp > bqcn > Me2bqcn, as revealed by the E1/2 values of the RuIII/II couples (Table 1).39 However, varying the structure of the N4 ligand has a minor effect on the redox potentials of the electrochemically generated cis-dioxoruthenium(VI) complexes (ΔEpa ∼ 70 mV).
| Complex | E 1/2 of RuIII/II couple (V vs. SCE) | RuVI/V or RuVI/IV oxidation (V vs. SCE) |
|---|---|---|
| 1b (N4 = mcp) | 0.37 | E 1/2 = 1.11 |
| 3c′ (N4 = pdp) | 0.36 | E 1/2 = 1.13 |
| 5c·OTs (N4 = bqcn) | 0.45 | E pa = 1.15 |
| 6c·OTs (N4 = Me2bqcn) | 0.49 | E pa = 1.08 |
Variable-pH cyclic voltammetry of 3c′ was conducted in Britton–Robinson buffer.40–42 Selected voltammograms at pH = 2.56, 5.02 and 6.37 are displayed in Fig. S15 (ESI†). Above pH 1.98, couple III splits into two one-electron couples (RuV/IV and RuVI/V); the former, which merges with couple II to form a new couple IV, can be assigned as a RuV/III couple (eqn (9)). The latter one is designated as couple V (eqn (10)). The Pourbaix diagram from pH 1 to 7.96 is shown in Fig. 6. For couple I (RuIII/II), there are two straight-line fragments with slopes of −56 and −122 mV per pH unit at 1 < pH < 6.37 and 6.37 < pH < 7.24, respectively, corresponding to the electrochemical reactions described in eqn (6) and (11). The breakpoint (pH = 6.4) of the plot for couple I is logically the pKa value of cis-[(pdp)RuIII(OH)(OH2)]2+, which is comparable to that of cis-[(Tet-Me6)RuIII(OH)(OH2)]2+ (pKa = 6.5).14a For couple IV (RuV/III), three linear segments with slopes of −57, −85 and −52 mV per pH unit are found at 1.98 < pH < 5.72, 5.72 < pH < 6.37 and 6.37 < pH < 7.96, respectively. The corresponding electrochemical reactions are described in eqn (9), (12) and (13). For couple V (RuVI/V), its potential shifts cathodically with a slope of −51 mV pH−1 at 1.98 < pH < 5.02. This is in line with its one-proton one-electron nature (equation (10)). At 5.02 < pH < 7.96, it becomes insensitive to pH, suggesting a one-electron process that does not involve proton loss (equation (14)). From this observation, together with the breakpoint of the plot of couple IV, the pKa value of cis-[(pdp)RuV(O)(OH)]2+ is estimated to be 5.6.
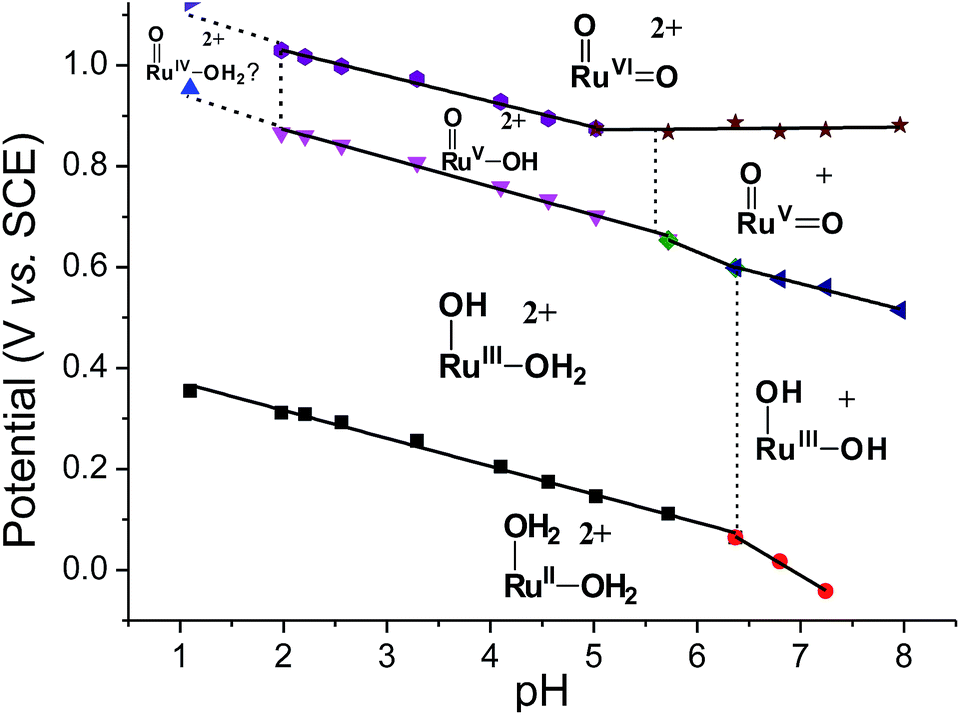 | ||
| Fig. 6 Pourbaix diagram of 3c′. Data points at pH 1 were extracted from the cyclic voltammogram in 0.1 M CF3SO3H (i.e., Fig. 5a), while data points at pH ≥ 2 were extracted from variable-pH measurements in Britton–Robinson buffer. | ||
With the above electrochemical information in hand, the bond dissociation energy (DO–H) for cis-[(pdp)RuV(O)(O–H)]2+ to form cis-[(pdp)RuVI(O)2]2+ can be obtained from eqn (15), based on the thermochemical method developed by Mayer and Bordwell.43,44 The DO–H value is calculated to be 90.8 kcal mol−1 for cis-[(pdp)RuVI(O)2]2+ (Scheme 5) and 90.1 kcal mol−1 for cis-[(mcp)RuVI(O)2]2+.
| DO–H = 23.06E° + 1.37pKa + C45 | (15) |
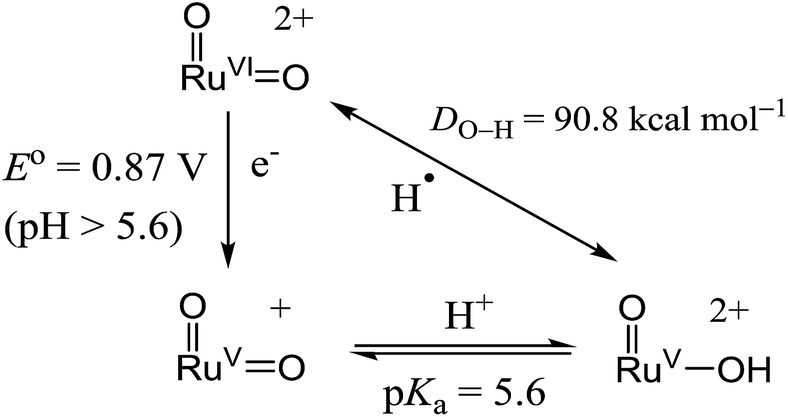 | ||
| Scheme 5 Thermochemical cycle of cis-[(pdp)RuVI(O)2]2+. | ||
Isolation or generation of cis-dioxoruthenium(VI) complexes via chemical oxidation
Treatment of cis-[(mcp)RuIII(O2CCF3)2]+ (1b) with excess CAN in aqueous solution gave cis-[(mcp)RuVI(O)2]2+ (1e), which was isolated as a pale green perchlorate salt in 66% yield (Scheme 6, see Experimental section for details). The UV-visible absorption spectrum of a freshly prepared solution of cis-[(mcp)RuVI(O)2](ClO4)2 (Fig. 7) in acetonitrile shows a prominent absorption peak at λmax = 260 nm (ε = 8700 dm3 mol−1 cm−1), a broad shoulder band at 340 nm (ε = 2210 dm3 mol−1 cm−1) and a weak absorption band at 700 nm (ε = 80 dm3 mol−1 cm−1). The high-resolution ESI mass spectrum of 1e shows a prominent ion species centred at m/z = 229.0631 that matches the formulation and the isotope distribution pattern of [(mcp)Ru(O)2]2+ (Fig. S16, ESI†). The IR spectrum of 1e shows two peaks at 845 and 868 cm−1, which are assigned to the symmetric and asymmetric stretches of the cis-dioxoruthenium(VI) moiety.14a,15a,16a Complex 1e is diamagnetic, as revealed by its 1H NMR signals. Notably, 1e is stable at −15 °C under argon for a few hours but decomposes in aqueous tert-butanol or acetonitrile within 30 min to give a dark brown solution, the ESI-MS analysis of this solution showed peaks centred at m/z = 460.1, which corresponds to [(mcp)Ru(OH)2]+ in aqueous tert-butanol, and m/z = 254.1, which corresponds to [(mcp)Ru(NCMe)2]2+ in acetonitrile. In aqueous solution at pH 1, cis-[(mcp)RuVI(O)2](ClO4)2 (1e) shows an identical cyclic voltammogram as that as 1b. The cyclic voltammogram of 1e in acetonitrile shows a reversible one-electron couple at E1/2 = 0.78 V vs. Ag/AgNO3 (0.1 M in MeCN), attributed to a RuVI/V couple: cis-[(mcp)RuVI(O)2]2+ + e− → [(mcp)RuV(O)2]+. Based on this redox potential, 1e is a stronger oxidant than cis-[(Tet-Me6)RuVI(O)2](ClO4)2 (E1/2 = 0.53 V vs. Ag/AgNO3).14a | ||
| Fig. 7 UV-Vis absorption spectrum of cis-[(mcp)RuVI(O)2](ClO4)2 (1e) in acetonitrile. | ||
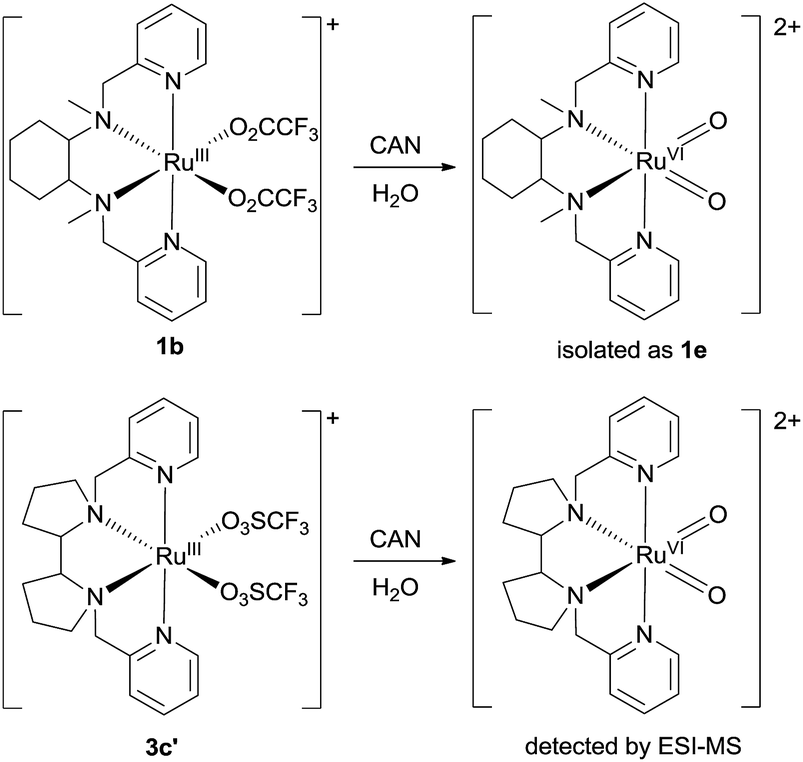 | ||
| Scheme 6 Preparation of 1e and generation of cis-[(pdp)RuVI(O)2]2+. | ||
Similar oxidation of cis-[(pdp)RuIII(O3SCF3)2]+ (3c′) or in situ generated cis-[(pdp)RuII(OH2)2]2+ by CAN did not furnish isolable cis-[(pdp)RuVI(O)2](ClO4)2 (Scheme 6). Upon addition of the CAN solution into an ice-cooled solution of 3c′, a dark brown solution resulted, and subsequent addition of ClO4− or PF6− did not induce solid formation. Small-scale reactions of 3c·CF3SO3 with a CeIV oxidant were performed in water and monitored by high-resolution ESI-MS. Under dilute conditions ([Ru] = 1 × 10−4 M), treatment of 3c·CF3SO3 with 4 equiv. of CeIV(ClO4)4 generated predominant ruthenium signals at m/z = 220.05 and 228.56. These signals are attributed to [(pdp)RuIV(O)]2+ and [(pdp)RuV(O)(OH)]2+ species (Fig. S17, ESI†). When a slight excess of CeIV(ClO4)4 (6 equiv.; 150% for a 4e− oxidation process) was employed, a new signal that corresponds to {[(pdp)RuVI(O)2]ClO4}+ was observed at m/z = 555.05 (Fig. 8). Notably, its signal intensity dropped significantly after ca. 1 min (Fig. S18, ESI†). A cis-dioxo-Ru(V) species was also detected at m/z = 456.10 (Fig. S19a, ESI†), with its signal intensity remaining constant for at least 3 min (Fig. S19b†). At a higher concentration of 3c·CF3SO3 ([Ru] = 1 × 10−3 M), a complicated spectrum dominated by noise signals was obtained with just 4 equiv. of CeIV(ClO4)4. Most likely, the decomposition of Ru(pdp) complexes under oxidizing condition is significantly fast with [Ru] ≥ 1 mM. This may account for the difficult isolation of cis-[(pdp)RuVI(O)2](ClO4)2 in the large-scale preparative experiment.
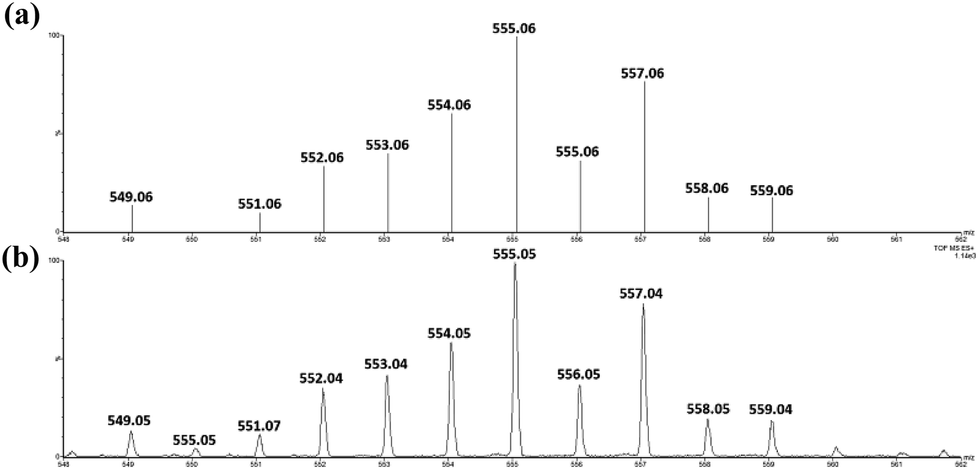 | ||
| Fig. 8 (Upper) Simulated ESI-MS pattern of {[(pdp)RuVI(O)2]ClO4}+. (Lower) Experimental ESI-MS signals for a reaction mixture of 3c·CF3SO3 and 6 equiv. of CeIV(ClO4)4, [Ru] = 1 × 10−4 M. | ||
Stoichiometric oxidation of hydrocarbons by cis-[(mcp)RuVI(O)2](ClO4)2 (1e)
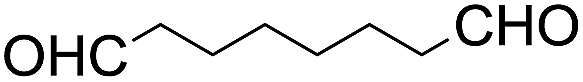
The results of the electrochemical studies suggest that cis-[(mcp)RuVI(O)2](ClO4)2 (1e) is a strong oxidant (E° = 1.11 V vs. SCE at pH 1). In aqueous tert-butanol, freshly prepared 1e could stoichiometrically oxidize cyclooctene to give a mixture of cis-cyclooctane-1,2-diol (27%) and 1,8-octanedialdehyde (70%) (Table 2, entry 1).46 Compared with our previous works, [(Me3tacn)RuVI(O)2(O2CCF3)]ClO4 oxidized cyclooctene stoichiometrically to give cis-cyclooctane-1,2-diol and 1,8-octanedialdehyde in 85% and 5% yields, respectively, whereas use of cis-[(Tet-Me6)RuVI(O)2](ClO4)2 gave 22% cis-cyclooctane-1,2-diol and 60% 1,8-octanedialdehyde.47 Apart from the organic products, a green ruthenium compound was isolated at the end of the reaction of 1e with cyclooctene. ESI-MS analysis revealed a prominent ion peak at m/z = 460.1; its m/z ratio and isotopic distribution pattern are consistent with a [(mcp)RuIII(OH)2]+ formulation.| Entry | Alkene subtrate | Product(s) | % Yieldb (% ee) |
|---|---|---|---|
|
a Reaction conditions: 1e* (0.3 mmol), substrate (30 mmol), tBuOH/H2O (5:1 v/v, 12 mL), under argon, room temperature, and 30 min.
b Isolated yield, calculated as mmol of product per mmol of 1e.
c Determined by GC.
|
|||
| 1 |
|
|
70c |
| cis-Diol | 27 | ||
| 2 |
|
syn-Diol | 20 (24) |
| anti-Diol | 28 (35) | ||
| PhCHO | 45c | ||
| 3 |
|
syn-Diol | 21 (30) |
| anti-Diol | 25 (36) | ||
| PhCHO | 52c | ||
| 4 |
|
syn-Diol | 19 (28) |
| anti-Diol | 26 (33) | ||
| PhCHO | 53c | ||
| 5 |
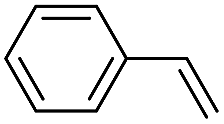
|
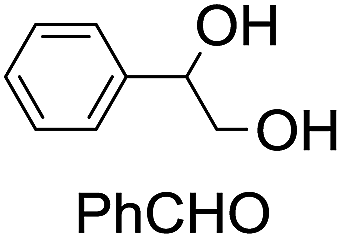
|
42 (27) |
| 50c | |||
| 6 |
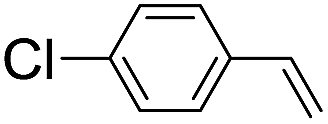
|
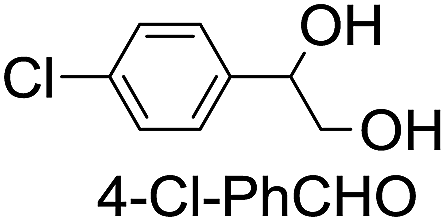
|
39 (33) |
| 51c | |||
| 7 |
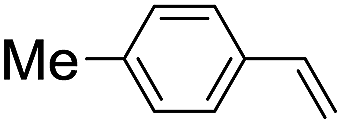
|
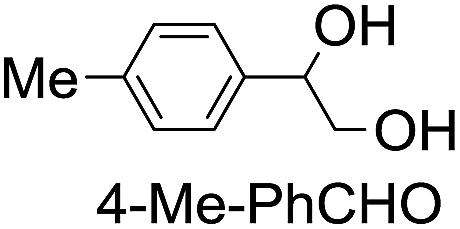
|
43 (28) |
| 48c | |||
| 8 |
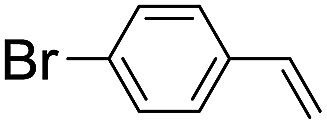
|
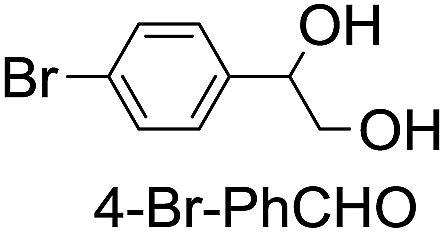
|
45 (34) |
| 47c | |||
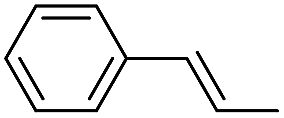
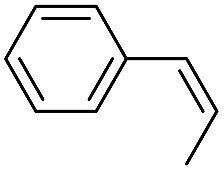
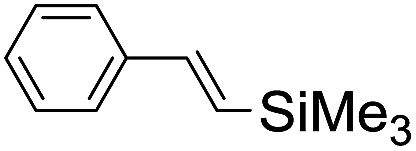
Using chiral (R,R)-mcp as a ligand, the chiral cis-dioxoruthenium(VI) complex, cis-[((R,R)-mcp)RuVI(O)2](ClO4)2 (1e*), was prepared. Several stoichiometric alkene oxidation reactions were performed by reacting 1e* (0.3 mmol) with excess alkene substrate (30 mmol, 100 equiv.) in a degassed (5:1 v/v) tert-butanol/H2O mixture (12 mL) under argon at room temperature for 30 min (Table 2). Aryl alkenes were oxidized to their corresponding diols (39–48% yields) with ee values ranging from 24 to 36%, accompanied by the formation of CC bond cleavage products in considerable amounts (45–53%). In the reaction of 1e* with styrene, for instance, a 42% yield of styrene glycol (27% ee) and 50% yield of benzaldehyde were obtained (entry 5, Table 2). Similarly, trans-β-(trimethylsilyl)styrene reacted with 1e* to afford a 19% yield of syn-diol (28% ee) and 26% yield of anti-diol (33% ee) along with a 53% yield of benzaldehyde (entry 4, Table 2). There is no major difference in the reactions of 1e* with trans-β-methylstyrene and with cis-β-methylstyrene, which afforded the enantio-enriched syn-diol in 20% yield (24% ee) and 21% yield (30% ee), anti-diol in 28% yield (35% ee) and 25% yield (36% ee), benzaldehyde in 45% and 52% yields, respectively (entries 2 and 3, Table 2). The effects of para-substituents on the enantioselectivity of p-substituted styrenes in the reaction with 1e* were examined (entries 5–8, Table 2); the para-substituents CH3, Cl and Br had no significant effect on either the yields (39–45%) or ee (28–34%) of the diol products.
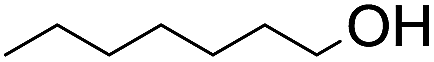
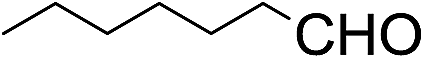
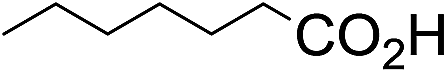
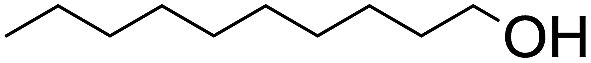
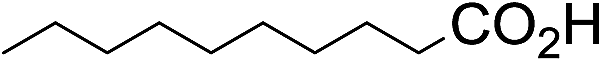
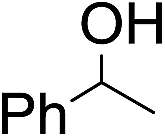
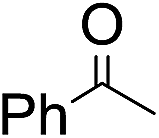
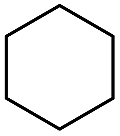
The stoichiometric oxidations of alcohols and alkanes by 1e were studied. When 1e was treated with benzyl alcohol (100 equiv.) in acetonitrile at room temperature for 30 min, benzaldehyde was formed in 90% yield (Table 3, entry 1). Similarly, other primary alcohols such as 1-heptanol and 1-octanol were oxidized by 1e to give a mixture of aldehyde and carboxylic acid (entries 2 and 3, Table 3). Under these conditions, cyclooctene reacted with 1e to afford cyclooctene oxide and 1,8-octanedialdehyde in 30% and 58% yields, respectively (entry 4, Table 3). Complex 1e could also oxidize saturated C–H bonds. For instance, ethylbenzene (BDEC–H = 85.4 kcal mol−1)48 was oxidized by 1e in acetonitrile to give acetophenone (55% yield) and 1-phenylethanol (26% yield) (entry 5, Table 3). Notably, cyclohexane (BDEC–H = 99.5 kcal mol−1)48 was oxidized to give cyclohexanone in 62% yield (entry 6, Table 3). Similar to the reported cis-dioxoruthenium(VI) complexes,14a,16a when adamantane was employed as a substrate, C–H oxidation occurred primarily at the 3° carbon; 1-adamantanol was formed as the sole product in 58% yield (entry 7, Table 3). The oxidation of cis-4-methylcyclohexyl benzoate afforded the tertiary alcohol in moderate yield (66%) with complete retention of the configuration; no epimerized product was observed (entry 8, Table 3). Reaction of 1e* with the two racemic substrates in entries 9 and 10 (Table 3) predominantly gave oxygenated products at the tertiary C–H bonds; however, chiral HPLC analysis of the tertiary alcohol product revealed no kinetic resolution effect (ee <2%). These organic transformations were accompanied by the reduction of cis-dioxoruthenium(VI) to cis-[(mcp)RuII(NCMe)2](ClO4)2 (1d), which was isolated and characterized (ESI†).
| Entry | Substrate | Product(s) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: 1e (0.3 mmol), substrate (30 mmol), MeCN (12 mL), under argon, room temperature, and 30 min. b Isolated yield, calculated as mmol of product per mmol of 1e. c Determined by GC. d 1e* instead of 1e was used. | |||
| 1 |
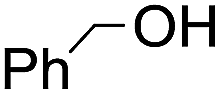
|
PhCHO | 90c |
| 2 |
|
|
60 |
|
22 | ||
| 3 |
|

|
55 |
|
26 | ||
| 4 |
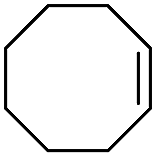
|

|
58c |
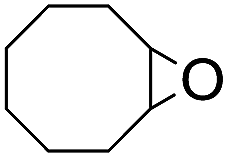
|
30 | ||
| 5 |
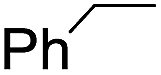
|
|
26c |
|
55c | ||
| 6 |
|

|
62c |
| 7 |
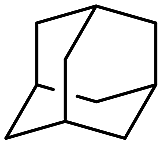
|
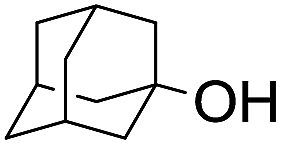
|
58 |
| 8 |
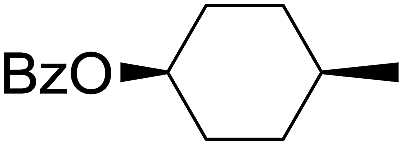
|

|
66 |
| 9d |
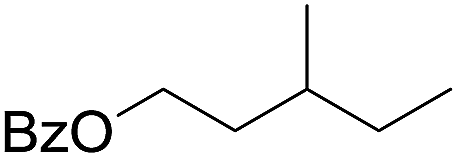
|
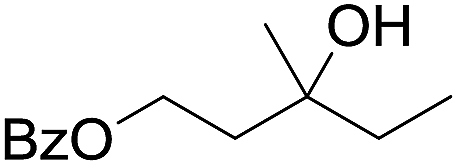
|
58 |
| 10d |
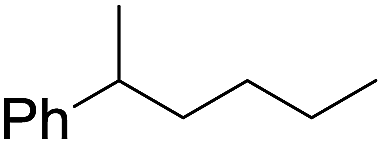
|
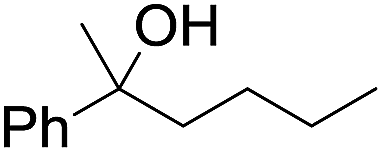
|
60 |
Catalytic oxidation of alkanes with CAN mediated by cis-[(pdp)RuII(OH2)2]2+ (3c)
The catalytic activities of the cis-[(mcp)RuIII(O2CCF3)2]+ (1b) and cis-diaquoruthenium(II) complexes (3c–6c) towards the hydroxylation of C(sp3)–H bonds were examined using CAN as a terminal oxidant. cis-1,2-Dimethylcyclohexane (S1) was chosen as an initial test substrate (Table 4). The reaction of S1 with 2 mol% 3c·OTs and 3 equiv. of CAN for 15 min at room temperature in aqueous tert-butanol afforded a tertiary alcohol product (P1) in 64% yield based on 80% conversion (entry 2, Table 4).49 The stereogenic centres are retained in the alcohol product, indicating that the hydroxylation reaction does not involve long-lived carbon-based radicals that can epimerize. Among the screened ruthenium catalysts, 3c·OTs showed the highest catalytic activity. When 4c·OTs or 6c·OTs was employed as the catalyst, particularly, the substrate conversion was <10% (entries 3 and 5, Table 4).50 Therefore, subsequent studies focused on the use of 3c·OTs as a catalyst. In a control experiment, in which the ruthenium catalyst was replaced by [RuII(OH2)6](OTs)2, S1 remained intact for a 30 min reaction (Table 5, entry 2). Subsequent addition of 3c·OTs to this reaction mixture followed by stirring for 15 min afforded P1 in 64% yield based on 61% conversion.| Entry | Catalyst | Reaction time (min) | Conversion (%) | Product yield (%) based on conversion |
|---|---|---|---|---|
|
a Reaction conditions: substrate (0.25 mmol), catalyst (2 mol%), CAN (0.75 mmol), tBuOH/H2O (1:1 v/v, 4 mL), and room temperature.
|
||||
| 1 | 1b | 15 | 60 | 62 |
| 2 | 3c·OTs | 15 | 80 | 64 |
| 3 | 4c·OTs | 30 | 8 | 55 |
| 4 | 5c·OTs | 15 | 73 | 61 |
| 5 | 6c·OTs | 30 | 6 | 61 |
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Reaction time | Conversion (%) | Products (yield in % based on conversion) |
|
a Reaction conditions: substrate (0.25 mmol), catalyst (2 mol%), CAN (0.75 mmol), tBuOH/H2O (1:1 v/v, 4 mL), and room temperature.
b [RuII(OH2)6](OTs)2 was used as the catalyst.
c
t
BuOH/H2O (3:1 v/v, 4 mL) was used as the solvent.
d CF3CH2OH/H2O (3:1 v/v, 4 mL) was used as the solvent.
e 1.5 mmol CAN was used.
|
||||
| 1 | S1 | 15 min | 80 | P1 (64) |
| 2b | S1 | 30 min | <1 | — |
| 3c | S2 | 45 min | 51 | P2 (96) |
| 4d | S3 | 1 h | 52 | P3 (84) |
| 5c | S4 | 1.5 h | 74 | P4a (47), P4b (3), P4c (32) |
| 6d | S5 | 1 h | 59 | P5 (83) |
| 7 | S6 | 1.5 h | 40 | P6 (80) |
| 8 | S7 | 1 h | 60 | P7 (85) |
| 9 | S8 | 1 h | 65 | P8 (89) |
| 10 | S9 | 15 min | 28 | P9 (91) |
| 11c,e | S10 | 40 min | 84 | P10 (89) |
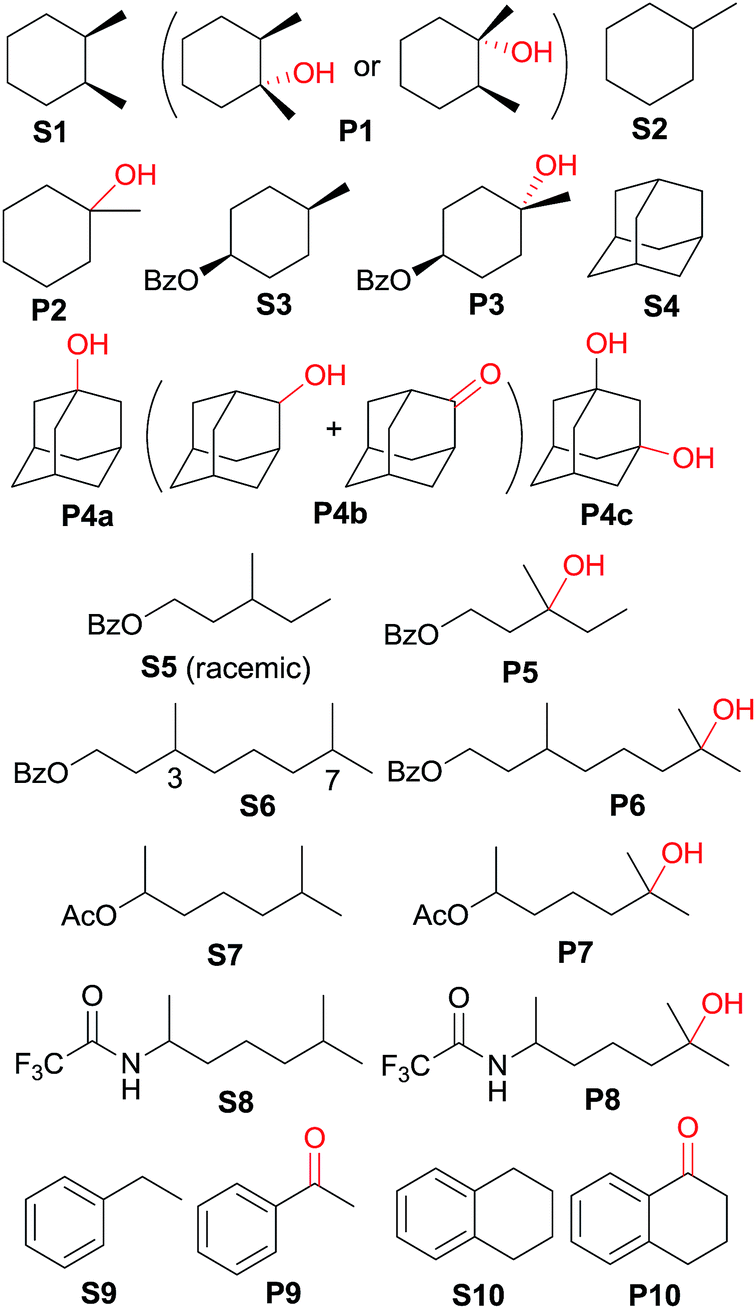
Oxidation of methylcyclohexane (S2) gave a tertiary alcohol product (P2) with high selectivity (96%) based on 52% conversion (Table 5, entry 3). Similarly, S3 was oxidized to P3 with good selectivity (entry 4, Table 5). For the oxidation of adamantane (S4), apart from ordinary oxygenation products, such as Ad-1-ol (P4a, 47% yield) and “Ad-2-ol + Ad-2one” (P4b, 3% yield), adamantan-1,3-diol (P4c) was also formed in 32% yield (entry 5, Table 5). Most likely, the initial hydroxylation of S4 gives P4a; the latter, being more soluble, was efficiently further hydroxylated to yield P4c.51 The normalized 3°/2° selectivity is as high as 79:1, showing the strong preference of the active oxidant to attack 3° over 2° C–H bonds. Following this preference, the oxidation of racemic S5 produced P5 in 48% isolated yield (entry 6, Table 5).52 Compound S6 has two possible sites (C3 and C7) for tertiary C–H hydroxylation. Analysis of the crude reaction mixture by 1H NMR spectroscopy revealed the C7:C3 selectivity to be a ratio of >10:1. After purification, a C7-hydroxylated product (P6) was obtained in 80% yield (entry 7, Table 5). Reactions of S7 and S8 similarly occurred at the 3° C–H bond which were remote from the electron-withdrawing ester/amide groups, in 51% and 58% isolated yields, respectively (entries 8 and 9, Table 5). In entries 10 and 11 (Table 5), the benzylic C–H bonds in ethylbenzene and tetralin were oxidized to give acetopheone (P9) and tetralone (P10), respectively. No aromatic ring degradation products were found. Thus, the possible involvement of RuO4 was unlikely as it is known to degrade aromatic rings.53 A kinetic isotope effect (KIE) of kH/kD = 5.2 was found in the competitive oxidation of an equimolar mixture of ethylbenzene and d10-ethylbenzene, indicative of C–H bond cleavage in the rate-determining step (RDS) or in a product-determining step following the RDS.54
For more complex substrates, the catalyst loading was increased to 5 mol% to furnish oxidation products in isolated yields ranging from 37% to 76% (55–93% based on conversion, Table 6). In general, sterically unhindered tertiary C–H bonds were preferred over unactivated methylene centres (entries 1 and 4, Table 6). For the oxidation of S12 that contains both tertiary and benzylic C–H bonds, only the ketone product P12 was formed (entry 2, Table 6). Notably, the reaction of S13 gave desaturation product P13 (entry 4, Table 6), presumably via an alcohol intermediate.55
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Reaction time | Conversion (%) | Products (yield in % based on conversion) |
|
a Reaction conditions: substrate (0.2 mmol), catalyst (5 mol%), CAN (1.2 mmol), tBuOH/H2O (1:1 v/v, 4 mL), and room temperature.
|
||||
| 1 | S11 | 40 min | 50 | P11 (84) |
| 2 | S12 | 40 min | 82 | P12 (93) |
| 3 | S13 | 20 min | 68 | P13 (55) |
| 4 | S14 | 13 h | 73 | P14 (78) |

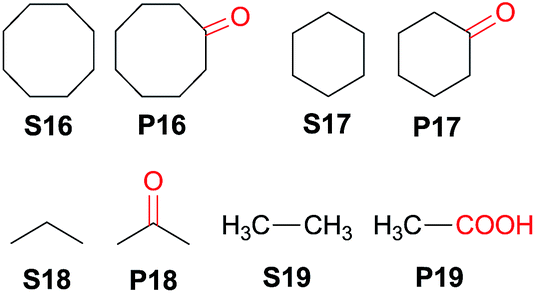
Interestingly, this catalytic protocol was also found capable of oxidizing strong secondary and primary C–H bonds of light alkanes (Table 7). The oxidation of cyclooctane (BDEC–H = 95.7 kcal mol−1)48 for 1.5 h gave cyclooctanone in 95% yield based on 40% conversion (entry 1, Table 7). Similarly, cyclohexane (BDEC–H = 99.5 kcal mol−1)48 was oxidized to cyclohexanone with a turnover number (TON) of 9 (entry 2, Table 7). The oxidation of propane (BDEC–H = 98.1 kcal mol−1)48 afforded acetone with a TON = 8 for a 3 h reaction (entry 3, Table 7). Lastly, oxidation of ethane (BDEC–H = 100.5 kcal mol−1)48 afforded acetic acid with a TON = 3 (entry 4, Table 7).56
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Reaction time (h) | Conversion (%) | Products (yield in % based on conversion) |
|
a Reaction conditions: substrate (0.25 mmol), catalyst (2 mol%), CAN (0.75 mmol), tBuOH/H2O (1:1 v/v, 4 mL), and room temperature.
b
t
BuOH/H2O (3:1 v/v, 4 mL) was used as the solvent.
c 1.5 mmol CAN was used.
d Conversion was not determined because of the high volatility of the substrate.
e Gaseous substrate used in excess (100 psi).
|
||||
| 1b | S16 | 1.5 | 40 | P16 (95) |
| 2b,c | S17 | 1.5 | —d | P17 (TON = 9) |
| 3e | S18 | 3 | — | P18 (TON = 8) |
| 4e | S19 | 3 | — | P19 (TON = 3) |
General remarks/discussion
General properties of the ruthenium N4 complexes
Two series of ruthenium complexes, cis-[(N4)RuIIICl2]+ (1a–4a) and cis-[(N4)RuII(OH2)2]2+ (3c–6c), were prepared. Owing to the lability of aqua ligands, [RuII(OH2)6](OTs)2 is an efficient precursor for the synthesis of cis-[(N4)RuII(OH2)2]2+ complexes (3c–6c). The cis-diaquoruthenium(II) complexes were isolated as ditosylate (OTs−) salts and are air sensitive. In aqueous solutions under aerobic conditions, they are susceptible to oxidation, as determined by the depletion of the characteristic MLCT transition band at 360–480 nm. Accompanying the UV-Vis spectral changes, the predominant species observed in ESI-MS analysis changed from [(N4)RuII(OTs)]+ to [(N4)RuIII(OH)2]+. Complexes without ortho-methyl substituents on the pyridyl/quinolyl moieties (3c and 5c) are less prone to aerobic oxidation; the process requires hours to complete. In contrast, complexes 4c and 6c are readily oxidized to Ru(III) species within 1 h.The structural analyses of the cis-[(N4)RuIIICl2]+ complexes by X-ray crystallography show that the cis-α configuration is the predominantly preferred geometry. 1H NMR spectroscopy of the cis-[(N4)RuII(OH2)2]2+ complexes in CD3CN or the bis(acetonitrile)ruthenium(II) complex revealed that the coordination geometry depends on the ligand structure. In particular, the bqcn ligand coordinates to the ruthenium centre in an unselective manner affording a mixture of cis-α and cis-β isomers, which do not interconvert in acetonitrile solution. In the X-ray crystal structures of cis-α-5d and cis-β-5d, the N-methyl groups have different orientations (anti or syn). Thus, interconversion between the two isomers requires (i) breakage of the Ru–N(quinolyl) bond, (ii) breakage of the Ru–N(amine) bond, and (iii) epimerization of the N-methyl group followed by migration of the acetonitrile ligand. These are expected to have large kinetic barriers, therefore, interconversion between the two isomeric forms is slow, and the ligand topology is likely determined at the synthetic stage of 5c·OTs. Similar arguments have been addressed by Nam, Shin and co-workers; they found that cis-α- or cis-β-[(bqcn)FeII(NCMe)2]2+ could be independently obtained with different synthetic methods and that these isomers do not interconvert in solution at room temperature.57
Electrochemistry/reduction potentials
Aqueous electrochemical measurements (at pH 1) of cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) and cis-[(pdp)RuIII(O3SCF3)2]-CF3SO3 (3c′) revealed the strong oxidizing powers of their corresponding cis-dioxoruthenium(VI) species. The highly anodic redox potentials (E° = 1.11–1.13 V vs. SCE) are comparable to those of cis-[(6,6′-Cl2bpy)2RuVI(O)2]2+ (1.17 V)58 and electrochemically generated cis-[(TPA)RuVI(O)2]2+ (1.1 V, TPA = tris(2-pyridylmethyl)amine).42The aqueous electrochemical data allow the determination of the hydrogen-atom affinity of the cis-dioxoruthenium(VI) complexes. The DO–H values are calculated to be 90.8 kcal mol−1 for cis-[(pdp)RuVI(O)2]2+ and 90.1 kcal mol−1 for cis-[(mcp)RuVI(O)2]2+. Referring to Table 8, these values are comparable to those of cis-[(bpy)2RuVI(O)2]2+ (93.5 kcal mol−1)17 and [(TSMP)FeIV(O)] (90 kcal mol−1, H2TSMP = meso-tetrakis(sulfonatomesityl)porphyrin),59,60 but are considerably larger than those of several (mono)oxoruthenium(IV) complexes (82.7–84.8 kcal mol−1),61,62trans-dioxoruthenium(VI) complexes (76.3–82.8 kcal mol−1),63,64 another cis-dioxoruthenium(VI) complex supported by the Me3tacn ligand (87.5 kcal mol−1)65 and several Mn–oxo complexes (79–84.3 kcal mol−1).66–69
| Complex | D O–H | Reference |
|---|---|---|
| a Calculated from the reported electrochemical data. | ||
| cis-[(bpy)2RuVI(O)2]2+ | 93.5a | 17 |
| cis-[(pdp)RuVI(O)2]2+ | 90.8 | This work |
| cis-[(mcp)RuVI(O)2]2+ | 90.1 | This work |
| [(TSMP)FeIV(O)] | 90 | 59, 60 |
| cis-[(Me3tacn)RuVI(O)2(O2CCF3)]2+ | 87.5a | 65 |
| [(N4Py)RuIV(O)(OH2)]2+ | 84.8 | 62 |
| [(bpy)2(py)RuIV(O)]2+ | 84 | 61 |
| trans-[(N2O2)RuVI(O)2]2+ | 82.8 | 63 |
| [(TPA)RuIV(O)(OH2)]2+ | 82.7 | 62 |
| [(Me2EBC)MnIV(O)(OH)]+ | 84.3 | 66, 67 |
| MnVIIO4− | 80 | 68 |
| [(phen)2MnIV(μ-O)2MnIII(phen)2]3+ | 79 | 69 |
| trans-[(14-TMC)RuVI(O)2]2+ | 76.3 | 64 |
Another piece of interesting information can be extracted from the pH-dependent cyclic voltammogram of 3c′, where the RuV/III couple was observed over the pH range of 1.98 to 7.96. At pH 4.1, for example, the potential of the redox couple cis-[(pdp)RuV(O)(OH)]2+ + 2e− + 2H+ → cis-[(pdp)RuIII(OH)(OH2)]2+ occurs at 0.76 V vs. SCE. This can provide a basis to estimate the redox potential of the putative cis-[(pdp)FeV(O)(OH)]2+ or cis-[(pdp)FeV(O)2]+ species, which are perceived to be strong oxidants but have not been reported in the literature. We previously reported a density functional theory (DFT) study of trans-dioxo complexes of iron, ruthenium and osmium, trans-[(NH3)2(NMeH2)2MVI(O)2]2+ (M = Fe, Ru, Os), where the reduction potentials of the corresponding FeVI/V and RuVI/V couples were estimated to be 1.3 V and 0.56 V vs. NHE, respectively.70 This theoretical study implies that a OFeO complex would be ∼0.7 V more oxidizing than the corresponding ORuO complex with the same ligand system. If the same relationship can be applied to Fe/Ru(N4) complexes in a cis-configuration, the potential of the redox couple cis-[(pdp)FeV(O)(OH)]2+ + 2e− + 2H+ → cis-[(pdp)FeIII(OH)(OH2)]2+ (or cis-[(pdp)FeV(O)2]+ + 2e− + 2H+ → cis-[(pdp)FeIII(OH)2]+, depending on the pKa value) would occur at approximately 1.4–1.5 V vs. SCE at pH 4.1, which is equivalent to 1.6–1.7 V vs. SCE at pH 1. This suggests that a cis-dioxoiron(V) species, if it exists, would be much more reactive than the cis-dioxoruthenium(VI) counterpart. The highly anodic/oxidizing reduction potential of the cis-dioxoiron(V) species may not be favourable for alkene dihydroxylation, as side reactions (e.g., CC cleavage) may become dominant. In comparison, the FeV/III couple of cis-[(L-N4Me2)FeV(O)2]+ (L-N4Me2 = N,N′-dimethyl-2,11-diaza[3.3](2,6)pyridinophane), an intermediate proposed to be involved in alkene dihydroxylation,10 was computed to occur at 1.34 V vs. SCE at pH 1.71
Reactivity of cis-dioxoruthenium(VI)
The results presented in this work show the strong oxidizing power of cis-dioxoruthenium(VI) complexes containing chiral N4 ligands by electrochemical analysis and their reactivity with hydrocarbons. Although several cis-dioxoruthenium(VI) complexes are known,14a,15a,16a,17 chiral ones have not been reported in the literature to the best of our knowledge. In this work, we isolated and spectroscopically characterized the chiral complex cis-[((R,R)-mcp)RuVI(O)2](ClO4)2 (1e*). Complex 1e* could effect the stoichiometric oxidations of alcohols, alkanes and alkenes, as was found for other cis-dioxoruthenium(VI) complexes. In the reaction of 1e* with alkenes, considerable amounts of dihydroxylation products were obtained with moderate enantioselectivities (∼30% ee, Table 2), albeit with the predominant products being CC bond cleavage ones, such as carbonyl compounds. In addition, a mixture of syn- and anti-diols was obtained, which possibly indicates the non-concerted nature of the dihydroxylation reaction.72,73 The reactivity/selectivity in the Ru((R,R)-mcp)-mediated asymmetric cis-dihydroxylation (AD) reaction of alkenes is in great contrast to some of the known, highly efficient chiral Fe(N4) or Mn(N4) catalysts. For instance, cis-[((R,R)-Me2bqcn)FeII(OTf)2] and cis-[((S,S)-bqcn)MnIICl2] gave cis-diols in up to 95% yields and 99.8% ee via proposed cis-[((R,R)-Me2bqcn)FeIII(OOH)]2+ and cis-[((S,S)-bqcn)MnV(O)2]+ intermediates, respectively.5g,6b
Based on the stoichiometric reaction of cis-[(mcp)RuVI(O)2]2+ (1e) with alkenes, a related catalytic reaction was developed using NaIO4 as a terminal oxidant. cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) turned out to be an efficient catalyst for the oxidative scission of aryl alkenes to carbonyl compounds (Table S5, ESI†, 6 examples). At a catalyst loading of 1 mol%, aryl CC bonds are cleaved to aldehydes or ketones in high conversions (83–100%) and high yields (89–100%).74 Over-oxidation of aldehydes to carboxylic acids was not observed by controlling the stoichiometry of NaIO4 (10% excess). The timespan of the reaction (1 h) is comparable to that (30 min) reported by Bera and co-workers using an abnormal-NHC–Ru(II) catalyst.75
Using cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) as a catalyst and H2O2 as a terminal oxidant, we also developed a catalytic protocol for the oxidation of alcohols (Table S6, ESI†, 14 examples). Alcoholic substrates were effectively oxidized to carbonyl compounds or carboxylic acids in yields up to 98% (see the ESI† for a more detailed description). ESI-MS analysis of a mixture of 1b and H2O2 did not reveal formation of 1e or other high-valent ruthenium–oxo complexes. The active intermediate could be hydroperoxo- or peroxo-Ru(III) species, which has yet to be clarified.
Reports on the oxidation of alkanes catalysed by ligand-supported ruthenium complexes are sparse in the literature.76 In 2010, Du Bois and co-workers developed a RuCl3/pyridine/KBrO3 protocol for the hydroxylation of various substituted alkane substrates;77 In 2012, they improved the yield and allowed a lower catalyst loading by employing [(Me3tacn)RuIIICl3] as a catalyst in combination with AgClO4 as an additive and CAN as a terminal oxidant.20 Using desorption electrospray ionization mass spectrometry (DESI-MS), cis-[(Me3tacn)RuVI(O)2(OH)]+ was identified to be a plausible reactive hydroxylating agent, but the possible involvement of Ru(V) and/or Ru(IV) species could not be discounted.78 In this work, stoichiometric reactions between 1e and several alkane substrates (Table 3) provided direct evidence that cis-dioxoruthenium(VI) preferentially oxidizes the tertiary C–H bonds in hydrocarbons. The same selectivity was observed in catalytic experiments. Aqueous electrochemical and ESI-MS experiments showed that cis-[(pdp)RuVI(O)2]2+ is accessible via the successive oxidative deprotonation of a low-valent precursor, such as 3c or 3c′. The DO–H values of cis-[(Me3tacn)RuVI(O)2(O2CCF3)]2+ and cis-[(pdp)RuVI(O)2]2+ are calculated to be 87.5 and 90.8 kcal mol−1, respectively (vide supra). We anticipate that the Ru(pdp) complex, with an additional driving force of 3.3 kcal mol−1, would be as reactive as the Ru(Me3tacn) complex in alkane oxidation reactions. Additionally, the use of a chiral N4 supporting ligand might incorporate chirality into the oxygenated products.3c,4d A catalytic system for the oxidation of alkanes by cis-[(pdp)RuII(OH2)2]2+ (3c) with CAN is herein reported. Compared to the [(Me3tacn)RuIIICl3]/AgClO4/CAN system, our system avoids the use of a Ag+ salt as a chloride scavenger, and no pretreatment of the catalyst is required.79 In general, 2–5 mol% catalyst (3c) and 3–6 equiv. of CAN afforded 3° C–H hydroxylated products in isolated yields of ca. 50% (Tables 4 and 5), which are comparable to those in other Ru-catalysed C–H hydroxylation protocols (e.g., [(Me3tacn)RuIIICl3]/AgClO4/CAN and cis-[(tBu2bpy)2RuIICl2]/H5IO6/CF3SO3H).20,21 In C–H oxidation with substrates containing a mixture of tertiary and secondary C–H bonds, the reaction occurs preferentially at the tertiary position and is highly stereoretentive (e.g., oxidation of S1 to P1 in Table 5, S11 to P11 in Table 6), which is a fundamentally defining feature in C–H functionalization rendering this method of synthetic value. When the substrate contains multiple tertiary C–H bonds (S8–S10, Table 5), hydroxylation preferentially occurs at the most electron-rich site, as was also observed in other Fe/Mn-catalysed C–H hydroxylation systems (e.g., cis-[(pdp)FeII(NCMe)2]2+/H2O2/AcOH).3a,b,4b This similar reactivity pattern implies the common electrophilic nature of cis-dioxoruthenium(VI) and the active oxidant in Fe(pdp)-catalysed reactions. In literature, the identity of the latter was investigated by multiple research groups which has led to different formulations.5d,7,8,9 Talsi, Bryliakov and co-workers identified an S = 1/2 species by EPR and assigned it to [(pdp)FeV(O)(OAc)]2+.5d Based on computational results, Wang, Que, Shaik and co-workers suggested a cyclic Fe(III) peracetate complex that undergoes O–O bond cleavage to a transient oxoiron(IV)–AcO˙ species which performs efficient C–H hydroxylations.7
We also demonstrated the strong oxidizing power of this catalytic system in the reaction with propane and ethane (Table 7). Although the turnover numbers are not impressive, identification of appreciable amounts of the various oxidation products is significant, as light alkanes often exhibit resistance to oxidation. To the best of our knowledge, this represents a rare example of ruthenium-catalysed/mediated oxidation of light alkanes (<C4), except Drago's reported work on the cis-[(dmp)RuII(S)2]2+ (S = MeCN or H2O)-catalysed hydroxylation of methane with H2O2.80
Some issues remain to be resolved/explored that are worth being addressed. First, the stability/robustness of the highly oxidizing cis-dioxoruthenium(VI) species is of concern. In CAN-driven catalytic oxidation of alkanes, the turnover number based on 3c is typically less than 30. Post-reaction analysis of the mixture revealed that the catalyst had degraded/decomposed almost completely. A likely deactivation pathway of the catalyst is the oxidation of the ligand by the strongly oxidizing Ru–oxo intermediate. Indeed, it was noted that complexes 4c and 6c showed much poorer activities than 3c and 5c (Tables 4 and S4, ESI†), presumably due to the intramolecular oxidation of the ortho-Me group by the Ru–oxo moiety.50 Although our recent work on the Fe(N4)-catalysed AD reaction showed that installation of an ortho-Me group could substantially improve the catalyst activity (particularly the enantioselectivity),5g this strategy cannot be directly transplanted to the ruthenium chemistry. For the Fe((R,R)-Me2bqcn)-catalysed AD reaction, the active intermediate was proposed to be [((R,R)-Me2bqcn)FeIII(OOH)]2+ rather than dioxoiron(V).5g From ESI-MS experiments, it was also demonstrated that the decomposition of Ru(pdp) complexes under oxidizing condition is considerably fast when [Ru] ≥ 1 mM. Thus, a delicate balance between the oxidizing power and stability of the active intermediate is yet to be achieved for efficient ruthenium-catalysed C–H oxidation. Moreover, either stoichiometrically or catalytically, the studied chiral ruthenium complexes (1e*, 3c–6c*) did not show noticeable enantioselectivity in reactions with racemic tertiary alkane substrates (e.g., entries 9, 10, Table 3; entry 6, Table 5). This suggests, without any directing group,3c there is not sufficient chiral differentiation between the two isomeric forms by kinetic resolution at the chiral ruthenium centre.
Conclusions
In this work, we reported the preparation and electrochemistry of several ruthenium complexes bearing tetradentate N4 ligands including cis-[(mcp)RuIII(O2CCF3)2]ClO4 (1b) and cis-[(pdp)RuIII(O3SCF3)2]CF3SO3 (3c′). Complex cis-[(mcp)RuVI(O)2](ClO4)2 (1e) was obtained from CAN oxidation of 1b in aqueous solution. Complex 1e is a powerful oxidant with E(RuVI/V) = 0.78 V (vs. Ag/AgNO3) in acetonitrile or E° = 1.11 V (vs. SCE) at pH 1. In aqueous tert-butanol, [((R,R)-mcp)RuVI(O)2](ClO4)2 (1e*) underwent stoichiometric alkene cis-dihydroxylation to afford cis-diol in 24% ee for trans-β-methylstyrene oxidation. With high hydrogen-atom affinities (DO–H = 90.1–90.8 kcal mol−1), 1e and chemically generated cis-[(pdp)RuVI(O)2]2+ are active oxidants for C–H oxidation. cis-[(pdp)RuII(OH2)2]2+ (3c), in combination with CAN as a terminal oxidant, catalysed the oxidation of unactivated C–H bonds including those of some pharmaceutical ingredients and natural product derivatives. This work demonstrates that efficient oxidation catalysts can be constructed based on the cis-dioxoruthenium(VI) moiety on a N4 ligand platform. The diversity and flexibility of chiral N4 ligand design will direct subsequent efforts to improve the reaction selectivities.4d Further studies are also directed to gain a better understanding of the reaction mechanism in hydrocarbon oxidations and to explore other catalytic activities of chiral Ru(N4) complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Hong Kong Research Grants Council General Research Fund (17303815, 17301817) and Basic Research Program-Shenzhen Fund (JCYJ20170412140251576, JCYJ20150629151046879). We thank the X-Ray Crystallography Laboratory in the Department of Chemistry at The University of Hong Kong for instrumental support (Bruker D8 Venture).Notes and references
- Selected reviews: (a) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed; (b) L. Que Jr and W. B. Tolman, Nature, 2008, 455, 333 CrossRef PubMed; (c) S. I. Murahashi and D. Zhang, Chem. Soc. Rev., 2008, 37, 1490 RSC; (d) M. Zhou and R. H. Crabtree, Chem. Soc. Rev., 2011, 40, 1875 RSC; (e) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950 RSC; (f) M. Costas, Coord. Chem. Rev., 2011, 255, 2912 CrossRef CAS; (g) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (h) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (i) E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418 CrossRef CAS; (j) H. Srour, P. Le Maux, S. Chevance and G. Simonneaux, Coord. Chem. Rev., 2013, 257, 3030 CrossRef CAS; (k) K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2014, 276, 73 CrossRef CAS; (l) W. N. Oloo and L. Que Jr, Acc. Chem. Res., 2015, 48, 2612 CrossRef CAS PubMed; (m) O. Cussó, X. Ribas and M. Costas, Chem. Commun., 2015, 51, 14285 RSC; (n) G. B. Shul'pin, Catalysts, 2016, 6, 50 CrossRef; (o) G. Olivo, O. Cussó and M. Costas, Chem.–Asian J., 2016, 11, 3148 CrossRef CAS PubMed; (p) J. A. Labinger, Chem. Rev., 2017, 117, 8483 CrossRef CAS PubMed; (q) J. He, M. Wasa, K. S. L. Chan, O. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (r) K. P. Bryliakov, Chem. Rev., 2017, 117, 11406 CrossRef CAS PubMed.
- Selected reviews: (a) I. Arends, P. Gamez and R. A. Sheldon, Adv. Inorg. Chem., 2006, 58, 235 CrossRef CAS; (b) C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547 RSC; (c) M. N. Kopylovich, A. P. C. Ribeiro, E. C. B. A. Alegria, N. M. R. Martins, L. M. D. R. S. Martins and A. J. L. Pombeiro, in Adv. Organomet. Chem., 2015, vol. 63, p. 91 Search PubMed; (d) C. Parmeggiani, C. Matassini and F. Cardona, Green Chem., 2017, 19, 2030 RSC; (e) R. H. Crabtree, Chem. Rev., 2017, 117, 9228 CrossRef CAS PubMed.
- Fe examples on C–H hydroxylation: (a) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS PubMed; (b) L. Gomez, I. Garcia-Bosch, A. Company, J. Benet-Buchholz, A. Polo, X. Sala, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2009, 48, 5720 CrossRef CAS PubMed; (c) M. A. Bigi, S. A. Reed and M. C. White, J. Am. Chem. Soc., 2012, 134, 9721 CrossRef CAS PubMed; (d) P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052 CrossRef CAS PubMed; (e) J. M. Howell, K. Feng, J. R. Clark, L. J. Trzepkowski and M. C. White, J. Am. Chem. Soc., 2015, 137, 14590 CrossRef CAS PubMed; (f) D. Font, M. Canta, M. Milan, O. Cussó, X. Ribas, R. J. M. Klein Gebbink and M. Costas, Angew. Chem., Int. Ed., 2016, 55, 5776 CrossRef CAS PubMed.
- Mn examples on C–H functionalization: (a) K. Nehru, S. J. Kim, I. Y. Kim, M. S. Seo, Y. Kim, S.-J. Kim, J. Kim and W. Nam, Chem. Commun., 2007, 4623 RSC; (b) R. V. Ottenbacher, D. G. Samsonenko, E. P. Talsi and K. P. Bryliakov, Org. Lett., 2012, 14, 4310 CrossRef CAS PubMed; (c) M. Milan, G. Carboni, M. Salamone, M. Costas and M. Bietti, ACS Catal., 2017, 7, 5903 CrossRef CAS; (d) M. Milan, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 196 CrossRef CAS PubMed.
- Fe examples on CC functionalization:
(a) M. Costas, A. K. Tipton, K. Chen, D.-H. Jo and L. Que Jr, J. Am. Chem. Soc., 2001, 123, 6722 CrossRef CAS PubMed;
(b) K. Suzuki, P. D. Oldenburg and L. Que Jr, Angew. Chem., Int. Ed., 2008, 47, 1887 CrossRef CAS PubMed;
(c) M. Wu, C.-X. Miao, S. Wang, X. Hu, C. Xia, F. E. Kühn and W. Sun, Adv. Synth. Catal., 2011, 353, 3014 CrossRef CAS;
(d) O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196 CrossRef CAS;
(e) O. Cussó, I. Garcia-Bosch, X. Ribas, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2013, 135, 14871 CrossRef PubMed;
(f) O. Cussó, X. Ribas, J. Lloret-Fillol and M. Costas, Angew. Chem., Int. Ed., 2015, 54, 2729 CrossRef PubMed;
(g) C. Zang, Y. Liu, Z.-J. Xu, C.-W. Tse, X. Guan, J. Wei, J.-S. Huang and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 10253 CrossRef CAS PubMed;
(h) M. Borrell and M. Costas, J. Am. Chem. Soc., 2017, 139, 12821 CrossRef CAS PubMed.
- Mn examples on CC functionalization:
(a) A. Murphy, G. Dubois and T. D. P. Stack, J. Am. Chem. Soc., 2003, 125, 5250 CrossRef CAS PubMed;
(b) T. W.-S. Chow, Y. Liu and C.-M. Che, Chem. Commun., 2011, 47, 11204 RSC;
(c) O. Cussó, I. Garcia-Bosch, D. Font, X. Ribas, J. Lloret-Fillol and M. Costas, Org. Lett., 2013, 15, 6158 CrossRef PubMed;
(d) R. V. Ottenbacher, D. G. Samsonenko, E. P. Talsi and K. P. Bryliakov, ACS Catal., 2014, 4, 1599 CrossRef CAS;
(e) C. Miao, B. Wang, Y. Wang, C. Xia, Y.-M. Lee, W. Nam and W. Sun, J. Am. Chem. Soc., 2016, 138, 936 CrossRef CAS PubMed;
(f) D. Shen, C. Saracini, Y.-M. Lee, W. Sun, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 15857 CrossRef CAS PubMed.
- Y. Wang, D. Janardanan, D. Usharani, K. Han, L. Que Jr and S. Shaik, ACS Catal., 2013, 3, 1334 CrossRef CAS.
- A. M. Zima, O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2017, 7, 60 CrossRef CAS.
- O. Cussó, J. Serrano-Plana and M. Costas, ACS Catal., 2017, 7, 5046 Search PubMed.
- T. W.-S. Chow, E. L.-M. Wong, Z. Gou, Y. Liu, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2010, 132, 13229 CrossRef CAS PubMed.
- V. Y.-M. Ng, C.-W. Tse, X. Guan, X. Chang, C. Yang, K.-H. Low, H. K. Lee, J.-S. Huang and C.-M. Che, Inorg. Chem., 2017, 56, 15066 CrossRef CAS PubMed.
- C.-M. Che and T.-C. Lau, Ruthenium and Osmium: High Oxidation States, in Comprehensive Coordination Chemistry II, Elsevier Ltd, 2004, vol. 5, p. 733 Search PubMed.
- T. Ishizuka, H. Kotani and T. Kojima, Dalton Trans., 2016, 45, 16727 RSC.
- (a) C.-K. Li, C.-M. Che, W.-F. Tong, W.-T. Tang, K.-Y. Wong and T.-F. Lai, J. Chem. Soc., Dalton Trans., 1992, 2109 RSC; (b) W.-C. Cheng, W.-Y. Yu, C.-K. Li and C.-M. Che, J. Org. Chem., 1995, 60, 6840 CrossRef CAS.
- (a) W.-C. Cheng, W.-Y. Yu, K.-K. Cheung and C.-M. Che, Chem. Commun., 1994, 1063 RSC; (b) S. L.-F. Chan, H.-Y. Kan, K.-L. Yip, J.-S. Huang and C.-M. Che, Coord. Chem. Rev., 2011, 899 CrossRef CAS.
- (a) C.-M. Che and W.-H. Leung, J. Chem. Soc., Chem. Commun., 1987, 1376 RSC; (b) C.-M. Che, K.-W. Cheng, M. C. W. Chan, T.-C. Lau and C.-K. Mak, J. Org. Chem., 2000, 65, 7996 CrossRef CAS PubMed.
- J. C. Dobson and T. J. Meyer, Inorg. Chem., 1988, 27, 3283 CrossRef CAS.
- C. L. Bailey and R. S. Drago, J. Chem. Soc., Chem. Commun., 1987, 179 RSC.
- W.-C. Cheng, W.-H. Fung and C.-M. Che, J. Mol. Catal. A: Chem., 1996, 113, 311 CrossRef CAS.
- E. McNeill and J. Du Bois, Chem. Sci., 2012, 3, 1810 RSC.
- J. B. C. Mack, J. D. Gipson, J. Du Bois and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 9503 CrossRef CAS PubMed.
- B. D. Durham, S. R. Wilson, D. J. Hodgson and T. J. Meyer, J. Am. Chem. Soc., 1980, 102, 600 CrossRef CAS.
- W.-P. Yip, PhD thesis, The University of Hong Kong, 2003 Search PubMed.
- T. W.-S. Chow, PhD thesis, The University of Hong Kong, 2010 Search PubMed.
- (a) T. Soundiressane, S. Selvakumar, S. Menage, O. Hamelin, M. Fontecave and A. P. Singh, J. Mol. Catal. A: Chem., 2007, 270, 132 CrossRef CAS; (b) Y. Popowski, I. Goldberg and M. Kol, Chem. Commun., 2016, 52, 7932 RSC.
- (a) Z. Tang, Y. Wang, X. Cui, Y. Yang, J. Tian, X. Fei and S. Lv, Inorg. Chim. Acta, 2016, 443, 235 CrossRef CAS; (b) Z. Tang, Y. Wang and P. Zhang, J. Coord. Chem., 2017, 70, 417 CrossRef CAS.
- C.-K. Li, W.-T. Tang, C.-M. Che, K.-Y. Wong, R.-J. Wang and T. C.-W. Mak, J. Chem. Soc., Dalton Trans., 1991, 1909 RSC.
- Attempts to prepare the analogous complex [(Me2mcp)RuIII(O2CCF3)2]ClO4 (2b) by a similar protocol was unsuccessful. Treatment of 2a with Zn/Hg at 80 °C in water, followed by metathesis with AgOTf afforded a light green solution. However, the corresponding trifluoroacetato complex 2b was not isolated upon addition of NaClO4.
- Remark: due to the small size of crystal sample (0.2 × 0.04 × 0.01 mm), the data were collected at a low resolution of 1 Å.
- A similar scenario is also observed for 4a although the RuIII/II couple (E1/2 = −0.01 V) is quasi-reversible instead of irreversible. The oxidation of cis-[(Me2pdp)RuIICl(NCMe)]+ to cis-[(Me2pdp)RuIIICl(NCMe)]2+ occurs at Epa = 0.64 V..
- The reversibility of the reduction wave of 2a in acetonitrile is partially restored at high scan rates (Fig. S10, ESI†).
- (a) C.-M. Che, W.-T. Tang, W.-O. Lee, W.-T. Wong and T.-F. Lai, J. Chem. Soc., Dalton Trans., 1989, 2011 RSC; (b) C.-M. Che, W.-T. Tang and C.-K. Li, J. Chem. Soc., Dalton Trans., 1990, 3735 RSC.
- K. J. Takeuchi, G. J. Samuels, S. W. Gersten, J. A. Gilbert and T. J. Meyer, Inorg. Chem., 1983, 22, 1407 CrossRef CAS.
- Attempts were made to study in details the effect of pH on the redox couples of 1b over the pH range of 1–10. However, ill-defined/irreversible redox couples were recorded at pH 3 and 4 and in alkaline medium.
- (a) R. C. McHatton and F. C. Anson, Inorg. Chem., 1984, 23, 3935 CrossRef CAS; (b) J. A. Gilbert, D. S. Eggleston, W. R. Murphy, D. A. Geslowitz, S. W. Gersten, D. J. Houston and T. J. Meyer, J. Am. Chem. Soc., 1985, 107, 3855 CrossRef CAS; (c) C. Ho and C.-M. Che, J. Chem. Soc., Dalton Trans., 1990, 967 RSC.
- In the presence of organic substrates such as ethanol or propan-2-ol (0.4–2.0 M), couple III of 3c′ is replaced by a large catalytic oxidative wave at ca. 1.2 V. A mild catalytic current was observed in the case of tosylic acid (0.4 M) (Fig. S13, ESI†).
- Couples II and III, having a potential difference of <200 mV at pH 1, are not well-separated in rotating-disk electrode voltammetric measurement.
- The return wave of couple III of 3c·OTs is less reversible, attributed to the oxidation of the benzylic C–H bonds in the tosylate anion (OTs−) by the electrochemically generated high-valent cis-dioxoruthenium(VI) species. See also ref. 36.
- Well-defined redox couples have not been observed for 4c·OTs.
- H. T. K. Britton and R. A. Robinson, J. Chem. Soc., 1931, 1456 RSC.
- B. Radaram, J. A. Ivie, W. M. Singh, R. M. Grudzien, J. H. Reibenspies, C. E. Webster and X. Zhao, Inorg. Chem., 2011, 50, 10564 CrossRef CAS PubMed.
- Y. Hirai, T. Kojima, Y. Mizutani, Y. Shiota, K. Yoshizawa and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 5772 CrossRef CAS PubMed.
- (a) J. M. Mayer, Acc. Chem. Res., 1998, 31, 441 CrossRef CAS; (b) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961 CrossRef CAS PubMed.
- F. G. Bordwell, J. P. Cheng and J. A. Harrelson, J. Am. Chem. Soc., 1988, 110, 1229 CrossRef CAS.
-
D
O–H is the bond dissociation free energy of the O–H bond of (Mred–OH), E° is the standard 1e− reduction potential of (MoxO)/(Mred–O−) couple, pKa is the acid dissociation constant of (Mred–OH), and C is a constant of 63.1 kcal mol−1 (for aq. solution with E° vs. SCE).
- No change in product yields or product distribution was observed when the reaction was conducted under air.
- W.-P. Yip, W.-Y. Yu, N. Zhu and C.-M. Che, J. Am. Chem. Soc., 2005, 127, 14239 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- The remaining mass balance is ascribed to the formation of secondary C–H oxidation products such as 2,3-dimethylcyclohexanone and 3,4-dimethylcyclohexanone where the yields were estimated to be 20–30% by GC analysis.
- We examined a reaction mixture of 4c·OTs (0.1 mM) and CAN (10 equiv.) in water and observed a new species at m/z 483.2, attributable to an intramolecularly oxidized Ru(Me2pdp) species (see Fig. S20, ESI† for details).
- In a control experiment where the substrate (adamantane) was replaced by adamantan-1-ol (P4a), P4c was formed in 79% yield based on 83% conversion. A yet to be confirmed highly polar side product was also obtained in ca. 15%. This side product has a m/z value of 184 in GC-MS analysis and is likely adamantan-1,3,5-triol..
- As was found in stoichiometric oxidation by 1e*, no kinetic resolution effect was observed by chiral HPLC analysis (ee <1%).
- P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- Attempts to extend the substrate scope to more complex hydrocarbon artemisinin only afforded small amount of oxygenated products based on <5% conversion.
- In these reactions, the corresponding alcohol product could not be detected.
- S. Hong, Y.-M. Lee, K.-B. Cho, K. Sundaravel, J. Cho, M. J. Kim, W. Shin and W. Nam, J. Am. Chem. Soc., 2011, 133, 11876 CrossRef CAS PubMed.
- K.-Y. Wong, W.-O. Lee, C.-M. Che and F. C. Anson, J. Electroanal. Chem., 1991, 319, 207 CrossRef CAS.
- D. Wang, M. Zhang, P. Bühlmann and L. Que Jr, J. Am. Chem. Soc., 2010, 132, 7638 CrossRef CAS PubMed.
- M. Wolak and R. van Eldik, Chem.–Eur. J., 2007, 13, 4873 CrossRef CAS PubMed.
- J. R. Bryant and J. M. Mayar, J. Am. Chem. Soc., 2003, 125, 10351 CrossRef CAS PubMed.
- T. Ishizuka, S. Ohzu and T. Kojima, Synlett, 2014, 25, 1667 CrossRef.
- D. T. Y. Yiu, M. F. W. Lee, W. W. Y. Lam and T.-C. Lau, Inorg. Chem., 2003, 42, 1225 CrossRef CAS PubMed.
- W. W. Y. Lam, W.-L. Man and T.-C. Lau, Coord. Chem. Rev., 2007, 251, 2238 CrossRef CAS.
- W.-C. Cheng, PhD thesis, The University of Hong Kong, 1995.
- G. Yin, A. M. Danby, D. Kitki, J. D. Carter, W. M. Scheper and D. H. Busch, J. Am. Chem. Soc., 2007, 129, 1512 CrossRef CAS PubMed.
- G. Yin, A. M. Danby, D. Kitki, J. D. Carter, W. M. Scheper and D. H. Busch, J. Am. Chem. Soc., 2008, 130, 16245 CrossRef CAS PubMed.
- K. A. Gardner, L. L. Kuehnert and J. M. Mayer, Inorg. Chem., 1997, 36, 2069 CrossRef CAS PubMed.
- K. Wang and J. M. Mayer, J. Am. Chem. Soc., 1997, 119, 1470 CrossRef CAS.
- G. S. M. Tong, E. L.-M. Wong and C.-M. Che, Chem.–Eur. J., 2008, 14, 5495 CrossRef CAS PubMed.
- W.-P. To, T. W.-S. Chow, C.-W. Tse, X. Guan, J.-S. Huang and C.-M. Che, Chem. Sci., 2015, 6, 5891 RSC.
- Both the syn- and anti-diols are enantio-enriched. Additionally, no 18O-incorporation into the anti-diol product was observed when the stoichiometric reaction between 1e and trans-β-methylstyrene was conducted in tBuOH/H218O, indicating that the anti-diol did not arise from ring opening of epoxide. Lloret-Fillol, Costas and co-workers reported trans-2-octene oxidation with CAN catalysed by iron complex bearing a chiral pdp derivative, which gave syn-diol by enantioselective cis-dihydroxylation, together with racemic anti-diol by ring-opening of epoxide (see ref. 73).
- I. Garcia-Bosch, Z. Codolà, I. Prat, X. Ribas, J. Lloret-Fillol and M. Costas, Chem.–Eur. J., 2012, 18, 13269 CrossRef CAS PubMed.
- Alkyl alkenes preferentially undergo epoxidation instead of CC oxidative scission by the “1b + NaIO4” protocol. For example, reaction of cis-cyclooctene gave 95% cis-cyclooctene oxide based on 100% conversion.
- P. Daw, R. Petakamsetty, A. Sarbajna, S. Laha, R. Rampapanicker and J. K. Bera, J. Am. Chem. Soc., 2014, 136, 13987 CrossRef CAS PubMed.
- V. S. Thirunavukkarasu, S. I. Kozhushkov and L. Ackermann, Chem. Commun., 2014, 50, 29 RSC.
- E. McNeill and J. Du Bois, J. Am. Chem. Soc., 2010, 132, 10202 CrossRef CAS PubMed.
- C. Flender, A. M. Adams, J. L. Roizen, E. McNeill, J. Du Bois and R. N. Zare, Chem. Sci., 2014, 5, 3309 RSC.
- During the course of our study, Du Bois, Sigman, and co-workers recently reported a bis(bipyridine)Ru-catalysed process where cis-[RuII(ligand)2Cl2] could be directly used as catalyst without Ag+ pre-treatment (see ref. 21).
- A. S. Goldstein and R. S. Drago, J. Chem. Soc., Chem. Commun., 1991, 21 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization, Scheme S1, Tables S1–S6, Fig. S1–S20. CCDC 1589975 (1a), CCDC 1589976 (2a), CCDC 1589977 (3a), CCDC 1589978 (5d), CCDC 1589979 (6d). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc05224c |
| This journal is © The Royal Society of Chemistry 2018 |