
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinking defects, hierarchical porosity generation and desalination performance in metal–organic frameworks†
Weibin
Liang‡
a,
Lin
Li‡
b,
Jingwei
Hou
 *bc,
Nicholas D.
Shepherd
a,
Thomas D.
Bennett
c,
Deanna M.
D'Alessandro
*a and
Vicki
Chen
b
*bc,
Nicholas D.
Shepherd
a,
Thomas D.
Bennett
c,
Deanna M.
D'Alessandro
*a and
Vicki
Chen
b
aSchool of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: deanna.dalessandro@sydney.edu.au
bUNESCO Center for Membrane Science and Technology, School of Chemical Engineering, The University of New South Wales, NSW 2052, Australia. E-mail: Jingwei.hou@unsw.edu.au
cDepartment of Materials Science and Metallurgy, University of Cambridge, Cambridge CB3 0FS, UK
First published on 5th March 2018
Abstract
Composite membranes with defective metal–organic frameworks (MOFs) connect the emerging fields of MOF topological modification, MOF-polymer interfacial engineering and composite material functionalization. Although defective MOFs can be fabricated via thermal or chemical treatment, the relationship between hierarchical MOF structure and their performance in a polymeric membrane matrix has so far not been investigated. Here we show how a modulator fumarate-based MIL-53(Al) microwave synthesis process results in defective MOFs. This ligand replacement process leads to materials with hierarchical porosity, which creates a higher mesopore volume and Brønsted acidity without compromising the crystalline structure and pH stability. Compared with stoichiometric ratios, increasing the reaction time leads to more effective defect generation. The subsequent incorporation of defective MOFs into polyvinyl alcohol pervaporation membranes can effectively promote the fresh water productivity in concentrated brine treatment, with salt rejection of >99.999%. The membranes also have good long-term operational stability with effective antifouling behavior. We provide evidence that topological engineering of the MOF surface is related to their physical and chemical behaviors in a polymeric matrix, opening up the possibility of MOF defect engineering to realize selective separations, catalysis and sensing within a polymeric matrix.
1. Introduction
Metal–organic frameworks (MOFs) belong to a family of chemically and functionally diverse materials, which have attracted widespread interest across the areas of chemistry, biology, engineering and energy storage, to list but a few examples.1–4 In addition to new materials synthesis, recent research has gradually shifted towards exploring the physical, structural and chemical properties of materials which are already known.5,6 In particular, the engineering control of defects in MOF structures has been considered important in fine-tuning their properties for specific applications.7–9 Fundamentally, the crystalline structures of MOFs are based on a series of relatively weak interactions, e.g. coordination effects, hydrogen bonds, van der Waals' force, π–π interactions and so on, which govern the exhibition of large-scale flexibility, structural disorder and both micro and macroscale defects. Modifying these interactions to facilitate missing ligands and metal ions, or metal reduction and displacement, rather than necessarily adverse effects, can endow MOF materials with specific functions, such as selective adsorption and improved catalytic activity.6,10–13‘On top of’ the microscopic defects within MOF structures, the generation of mesoporous defects with hierarchical porosity has attracted increasing attention.14 Essentially, the introduction of functionalized mesoporous structure on MOFs can be realized by controllable modification of coordinatively unsaturated metal sites.15 For example, the inclusion of monodentate synthetic modulators, e.g. acetic acid within synthetic routines, have been observed to generate ‘spongy’ networks of Fe3+ and Cr3+ based MOFs, or notably, frameworks with UiO-66 type structures.16 Another novel approach to construct the hierarchical structure on MOF is by using a nanosphere monolith template together a double-solvent-induced heterogeneous nucleation approach.17 There is, however, a fine balance between defect generation and loss of mechanical stability,18–20 and the advantageous functions of including defective MOFs within polymeric matrices such as membranes are poorly understood.
In terms of membrane processes, the treatment of highly concentrated brine is technically challenging and energy demanding, as conventional pressure-driven filtration membrane is not a viable option due to the high osmotic pressure that must be overcome.21 The concentrated brine can also lead to rapid scaling and pore blocking for the membrane distillation process. Alternatively, membrane pervaporation is a promising technique, which has been extensively investigated for organic solvent separation and dehydration.22 The dense pervaporation membranes can effectively mitigate the scaling problem; but they still suffer from low desalination productivity and poor operational stability with brine feeds.21
An aluminum fumarate based MOF, MIL-53(Al) (also known as ‘Basolite-100A’) was selected as a possible candidate to link defect generation, hierarchical porosity and membrane performance due to (i) cost efficient synthesis in water with naturally earth abundant sources (fumaric acid and aluminum salts), (ii) high water adsorption capability with good thermal and chemical stability7 and (iii) the retention of initial pore structure upon pore evacuation, unlike the isostructural H2BDC-based MIL-53 (BDC = 1,4-benzenedicarboxylate).23 The framework has previously been studied for applications including natural gas storage, heat transformation and lithium-ion battery technology,7,23,24 but its potential to generate hierarchical structures has not been explored.
Herein, we investigated a controllable defect-generation technique for fumarate-based MIL-53(Al) MOFs (referred as MIL-53(Al) in the following text), drawing relationships between defect content and crystallinity, hierarchical structure and Brønsted acidity. The interaction between the hierarchically porous defective MOFs and polyvinyl (alcohol) (PVA) matrices was studied in the fabrication of nanocomposite pervaporation membranes. Finally, desalination membrane performance was examined with various complex feed solutions including model inland desalination brine, which is otherwise extremely difficult to process with other water treatment techniques.
2. Experimental section
2.1 Synthesis of MIL-53(Al)
Powdered MIL-53(Al)a,t min ([Al(OH)(C4H2O4)]) samples were synthesized using a microwave-assisted solvothermal method in an Anton Paar Monowave 300 microwave oven.25,26 For a general procedure, a 30 mL glass microwave vial was charged with Al2(SO4)3·18H2O (99.99%, Sigma-Aldrich), fumaric acid (99%, Sigma-Aldrich) and urea (≥99.5%, Ajax). Thereafter, Milli-Q water (18 MΩ) was introduced. The mixture was heated with magnetic stirring (600 rpm) to 130 °C within 1 min, and held at this temperature for a desired period of time before cooling to 55 °C within 2 min. The precipitates were vacuum filtered, and repeatedly washed with ethanol to remove the unreacted chemicals. The resulting powder was dried in vacuo. In this work, a series of different stoichiometric ratios (a) and reaction time (t, by default 30 min) were investigated for MIL-53(Al)a,t min synthesis. Detailed information is given in the Section 3.1. A summary of the reaction conditions is provided in Table S1.†2.2 X-ray powder diffraction (XRPD) and Le Bail cell simulation
XRPD measurements were performed on a PANalytical X'pert Pro diffractometer fitted with a solid-state PIXcel detector (45 kV, 40 mA, 1° divergence and anti-scatter slits, and 0.3 mm receiver and detector slits) using Cu-Kα (λ = 1.5406 Å) radiation. Profile fits were performed using the Le Bail extraction method in GSAS.272.3 Potentiometric acid–base titration
Potentiometric titrations were conducted on a benchtop HANNA pH meter (HI2211-02, Basic pH and ORP Benchtop Meter) using procedures similar to those reported for Zr(OH)4 and Zr/Hf-based MOFs.28–30 Prior to analysis, the pH meter was calibrated with commercial buffers (pH 4.00, 7.00, and 10.00, ProSource Scientific pH Buffer Calibration Standards). MOF samples were grounded with a mortar and pestle, and then approximate 50 mg of sample was dispersed in 50 mL of 0.01 M aq. NaNO3 (≥99.0%, Sigma-Aldrich) solution, covered with Parafilm (Parafilm® M), and allowed to equilibrate for 5 h. The MOF suspension was then adjusted to pH 3 using 0.1 M aq. HCl (Merck) under magnetic stirring (1000 rpm) before being titrated with 0.1 M aq. NaOH (Merck) to pH 9.0–9.5. The injection rate of titrant was 20 μL every 30 s. Equivalence points were obtained from the first derivative of the resulting titration curves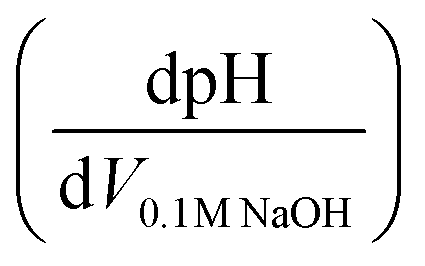 , where the inflection points in the derivative curve represent equivalence points. pKa values were determined as the pH at one-half of the volume of titrant at the equivalence point. After titration, the MIL-53(Al) were filtered, washed with ethanol and dried in vacuo for XRPD analysis.
, where the inflection points in the derivative curve represent equivalence points. pKa values were determined as the pH at one-half of the volume of titrant at the equivalence point. After titration, the MIL-53(Al) were filtered, washed with ethanol and dried in vacuo for XRPD analysis.
2.4 Characterization of MOFs
Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectra were recorded for MIL-53(Al) on a PerkinElmer Spectrum Two IR spectrometer (with UATR Two attachment). TGA measurements were carried out on a DISCOVERY TGA Thermogravimetric Analyzer. Approximately 5 mg of each sample was placed on a platinum pan which was heated under a flow of air at a rate of 10 °C min−1 up to 700 °C. Field-emission SEM (FE-SEM) images were obtained using a Zeiss ULTRA plus microscope (working distance ∼9 mm; acceleration voltage 20 kV). N2 sorption isotherms were recorded on a 3Flex Surface Characterisation Analyser (Micromeritics Instruments Inc.).2.5 Pervaporation membrane fabrication
The composite pervaporation membrane was fabricated with a thin PVA surface coating on PVDF hollow fiber substrates. The substrate PVDF hollow fibre membrane was soaked in Milli-Q water overnight and left to dry at room temperature for at least 12 hours. 2 wt% of PVA was firstly dissolved in water, and then glutaraldehyde and methanol were added to the solution under constant stirring. The molar ratio of glutaraldehyde to PVA repeat unit is maintained at 0.2. To initiate the crosslinking reaction, HCl was added into the solution. A typical coating solution contained 1 g PVA, 2 mL of 10% methanol and 0.4 mL of 1 M HCl. After constant stirring for 60 min, the solution was then dip coated onto the surface of PVDF hollow fiber (coating parameters: 1 cm s−1 lowering speed, 1 min soaking time and 0.4 cm s−1 withdrawn speed). Both ends of the hollow fiber membrane were sealed to prevent the coating solution penetration into the lumen side. After coating the membrane was left to dry in an oven at 50 °C for at least 24 h before use. In terms of the nanocomposite membrane, a certain amount of MIL-53(Al) was added into the PVA solution prior to the dip coating. Probe sonication was applied to ensure a better dispersion of the nanofillers within the membrane matrix. In this work, for characterization purpose, freestanding films were also prepared by pouring the coating solution in a Petri dish, followed by the same drying procedure as above. The pervaporation desalination testing procedure is presented in the ESI.†2.6 FIB-SEM analysis of the MIL-53(Al)/PVA films
Focused Ion Beam-Scanning Electron Microscopy (FIB-SEM) analysis were carried out with Zeiss Auriga Cross-beam FIB system. The mixed matrix films were first coated with a thin layer of chromium and then a Pt protective layer. Then the sample was tiled at an angle of 36°, and a serial of combined with SEM imaging was carried with an automated routine programmed in RunScript software with image drift correction. Slides with nominal thickness of 50 nm (MIL-53(Al)1,15 min/PVA) and 25 nm (MIL-53(Al)1,60 min/PVA) were milled away by FIB. For each sample, 450 SEM images were taken with a magnification of 10 K. Then the collected images were processed with Avizo and Photoshop, and eventually surface rendered with Cinema 4D from Maxon. The FIB-SEM samples contained 30 wt% of MOFs in PVA.3. Results and discussion
3.1 Synthesis of defective MIL-53(Al)
MIL-53(Al)a,t min samples were prepared via a microwave-assisted solvothermal synthesis process, including an Al(III) ion source and fumarate organic linkers, alongside H2O and urea. The in situ hydrolysis of the latter generates ammonium cations which trigger the deprotonation of fumarate ligands and consequently the onset of MIL-53(Al) formation. The generated carbonate ions however act as coordination modulators, i.e., they compete with fumarate ligands and disrupt metal–ligand bond formation (Fig. 1a). The reaction is noted to occur slowly in solution with the reaction rate drastically increasing in response to stimuli such as higher temperatures and concentrations.32–34 Therefore, the defect generation can be readily controlled through variable stoichiometry, concentration, reaction temperature and time.32,33 In this work, a fixed amount of urea (4.6 mmol) was used throughout all synthesis reactions. In a typical synthesis process of MIL-53(Al)1, the reaction was conducted for 30 min with 0.38 mmol Al(III) ions and 0.78 mmol fumaric acid. The effects of reaction stoichiometry, concentration, reaction temperature and time upon defect formation were studied, and the synthetic conditions summarized in Table S1.† Briefly, for MIL-53(Al)a, a indicates the stoichiometric ratios of metal ion and ligand to MIL-53(Al)1, and t describes the reaction time for MIL-53(Al)1,t min (30 min by default).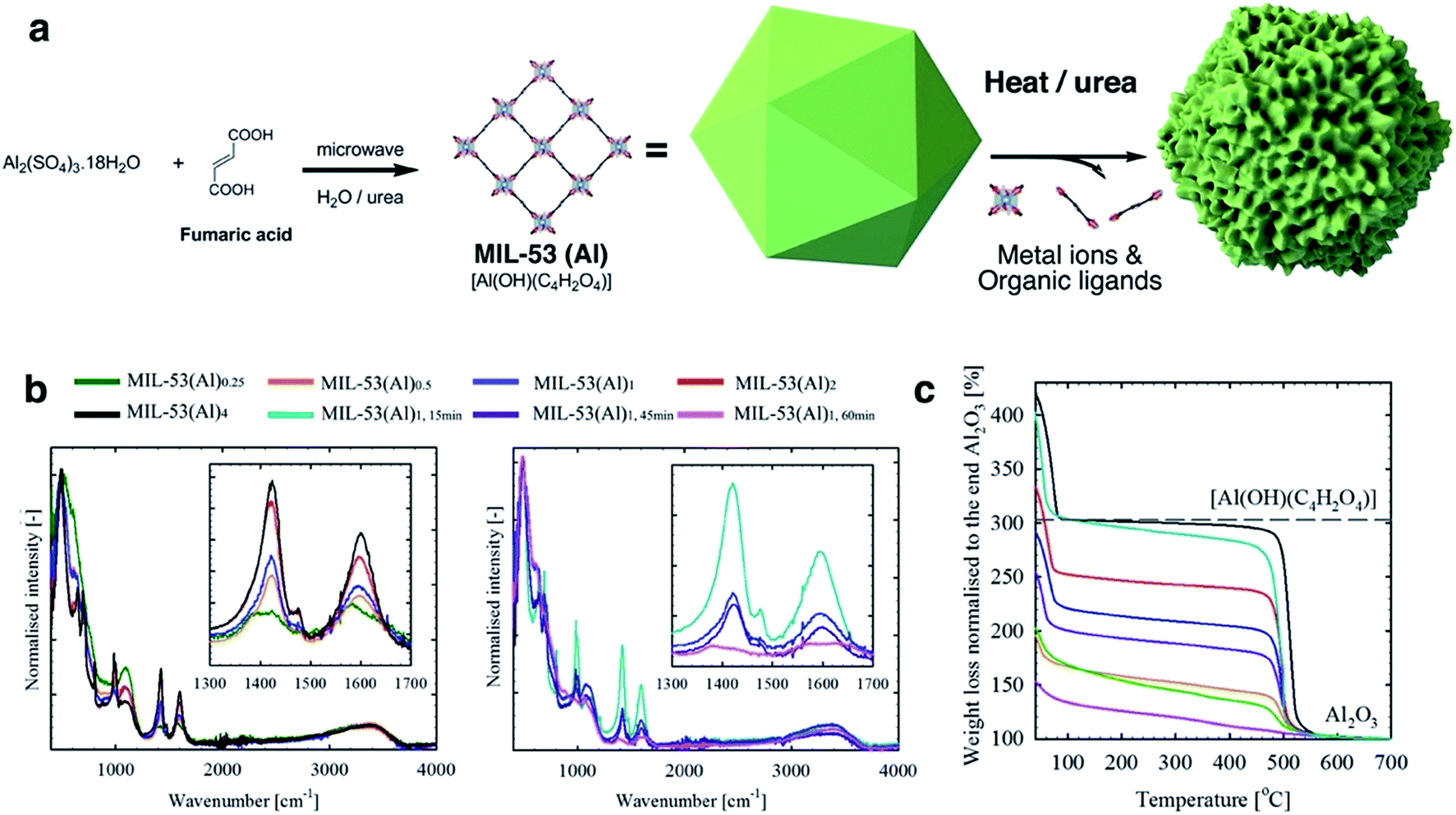 | ||
Fig. 1 (a) Schematic diagram of the MIL-53 synthesis and defect generation; (b) normalised ATR-FTIR spectrum for MIL-53(Al) samples (left: different stoichiometric ratios, and right: different reaction time). Insets show the characteristic signals (anti-symmetric vasym(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and symmetric vsym(CO) vibrations for carboxylate at ca. 1603 and 1426 cm−1,31 respectively) for the coordinating fumarate ligands; (c) normalized TGA curves for MIL-53(Al) samples. Black dashed-line indicates the theoretical weight for perfect MIL-53(Al) ([Al(OH)(C2H2O4)]) at 250 °C. The IR and TGA data are measured for the dried samples. O) and symmetric vsym(CO) vibrations for carboxylate at ca. 1603 and 1426 cm−1,31 respectively) for the coordinating fumarate ligands; (c) normalized TGA curves for MIL-53(Al) samples. Black dashed-line indicates the theoretical weight for perfect MIL-53(Al) ([Al(OH)(C2H2O4)]) at 250 °C. The IR and TGA data are measured for the dried samples. | ||
Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) spectra (Fig. 1b) are consistent with a correlation between the generation of missing linker defects in MIL-53(Al) MOFs and urea exposure. This is based on the observed variable intensity for signals attributable to the fumarate vasym(CO) and vsym(CO) vibrations situated at approximately 1603 and 1426 cm−1.35,36 Spectra for products formed using increasing stoichiometric ratios of urea modulator, and those formed from increasing reaction times, contained signals which decreased successively in intensity (Fig. 1b). Powder X-ray Diffraction (PXRD) measurements confirmed the preservation of structure across reactions (Fig. S1†), although, as observed in other MOFs, increased microwave irradiation times resulted in negligible degradation.37
Le Bail refinement (Fig. S2 and Table S2†) confirmed the structural assignment of the materials as MIL-53(Al),7 with a good agreement found between literature and experimental cell parameters. Thermogravimetric analyses (TGA) data for MIL-53(Al) materials are consistent with the loss of ethanol up to approximately 75 °C, followed by framework decomposition to Al2O3 (Fig. 1c) at ca. 500 °C. Following the methodology of Valenzano et al. in the study of UiO-66,34 defects are observed here as decreased mass loss from solvent and ligand decomposition in comparison to Al2O3 produced. The calculated content of fumarate (Table 1) confirmed that the effect from variation of reaction time is more significant than stoichiometry in generating defects, whilst the overall thermal decomposition temperature of the samples was largely unaffected by defect content. It is important to note that samples with a high concentration of defects (i.e. MIL-53(Al)1,60 min) are likely to contain the crystalline powder MIL-53(Al) and some amorphous Al(OH)x, which cannot be detected by XRD.
| Entry | TGA plateaua (@250 °C) | Approx. molecular formula | pKa1 μ-OH2b | pKa2 μ3-OHc | pKa3 μ-OHd | S BET (m2 g−1) | V total (cm3 g−1) | V micro (cm3 g−1) | V meso (cm3 g−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Data was taken from the normalized aerobic TGA data. b μ-OH2 = water molecules in missing linker defect sites. c μ3-OH = structural hydroxyl group. d μ-OH = hydroxyl groups present for charge stabilization. e Calculated from the N2 adsorption isotherms measured at 77 K. Values in parentheses indicate the uncertainties. f V total represents total pore volume determined using the adsorption brach of the 77 K N2 isotherms at p/p0 = 0.99. g V micro represents the specific micropore volume calculated using the t-plot method. h V meso represents the specific mesopore volume calculated from the N2 adsorption isotherm using the BJH method. | |||||||||
| MIL-53(Al)0.25 | 145.3 | [Al(OH)2.60(C4H2O4)0.20] | 4.55 | 7.00 | 7.72 | 441.4(2) | 0.56 | 0.02 | 0.55 |
| MIL-53(Al)0.5 | 152.5 | [Al(OH)2.54(C4H2O4)0.23] | 4.92 | 6.88 | 7.94 | 500.1(2) | 0.43 | 0.15 | 0.31 |
| MIL-53(Al)1 | 209.3 | [Al(OH)2.02(C4H2O4)0.49] | 4.88 | 7.11 | 7.92 | 907.6(6) | 0.47 | 0.31 | 0.18 |
| MIL-53(Al)2 | 243.2 | [Al(OH)1.72(C4H2O4)0.64] | 5.20 | 6.54 | 8.15 | 989.1(2) | 0.54 | 0.30 | 0.22 |
| MIL-53(Al)4 | 300.0 | [Al(OH)1.22(C4H2O4)0.89] | 4.71 | 7.15 | — | 1224.3(2) | 0.51 | 0.46 | 0.06 |
| MIL-53(Al)1,15 min | 289.7 | [Al(OH)1.30(C4H2O4)0.85] | 5.09 | 7.46 | — | 1213.5(2) | 0.51 | 0.46 | 0.07 |
| MIL-53(Al)1,45 min | 188.4 | [Al(OH)2.20(C4H2O4)0.40] | 4.75 | 6.72 | 7.88 | 801.5(2) | 0.63 | 0.20 | 0.45 |
| MIL-53(Al)1,60 min | 119.6 | [Al(OH)2.82(C4H2O4)0.09] | 4.54 | 6.10 | 7.89 | 426.9(2) | 0.77 | 0.01 | 0.78 |
The Brønsted acidities of the MOF materials were studied via potentiometric titration.38 Three acidic proton sources were generally observed, belonging to (1) water molecules situated in the missing linker defect sites (pKa ≈ 4.83), (2) structural hydroxyl groups (pKa ≈ 6.87) and (3) hydroxyl groups stabilizing the MOF structures (pKa ≈ 7.92) (Fig. 2, Table 1, Fig. S3†). The lower pKa values of the defective MOFs were indicative of a greater degree of structural hydroxyl groups and absorbed water molecules within their porous structures, consistent with the calculated molecular formula (Table 1). The emergence of a proton source within the defective MOFs aligns with our previous research with UiO-66.39 For MIL-53(Al)4 and MIL-53(Al)1,15 min, protons from charge stabilization were not detected, possibly due to the detection limit of the instrument (Fig. S4†). PXRD recorded for MIL-53(Al) materials post-potentiometric analysis indicated that the crystallinity was unaffected under both acidic and basic conditions, as expected based on structural robustness associated with frameworks in general, even after the defects generation (Fig. S5†).
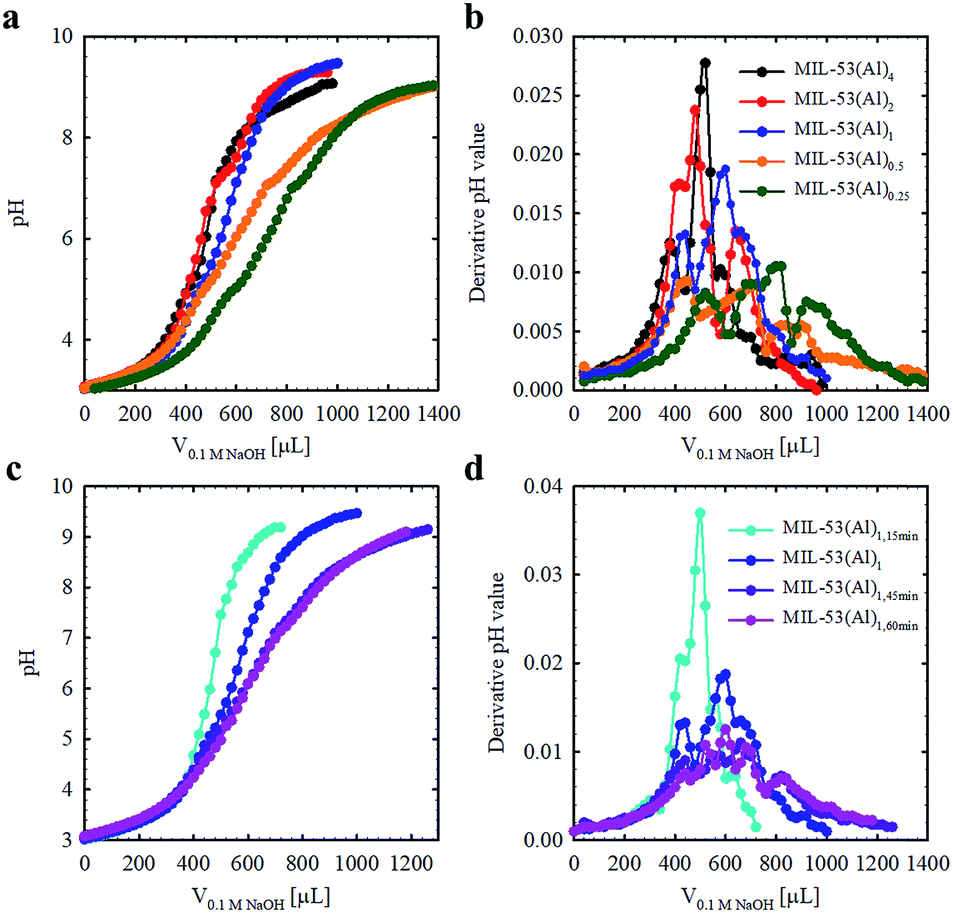 | ||
| Fig. 2 Acid–base titration curves for MIL-53(Al) materials fabricated with variable modulator stoichiometry/concentration (a and b) and exposure time (c and d). | ||
The mesoporous structures of the series of MIL-53(Al)a,t min materials were visualized by scanning electron microscopy (SEM, Fig. 3 and S6†), with pore apertures found to lie within the 20–500 Å literature range,40 the latter being particularly pronounced in MIL-53(Al)0.25, MIL-53(Al)0.5, MIL-53(Al)1,45 min and MIL-53(Al)1,60 min. As these materials are subject to substantial modulator exposure and/or dilution during assembly, the presence of mesopores in the structures is consistent with the aforementioned effects from urea and concentration, as well as previous characterization results (ATR-FTIR, TGA and acid–base titration). This demonstrates the capacity of this technique in tuning the architectural structure of MOFs.41 The introduction of mesoporosity was further substantiated by 77 K N2 isotherms (Fig. 4a–d, S7–S22†). Type I physisorption curves were observed for all samples, with H4 hysteresis observed for MIL-53(Al) materials, in accordance with the presence of mesoporosity.42 The calculated micro- and mesopore profiles (Vmeso and Vmicro) for the MIL-53(Al) series (Fig. 4e–f, Table 1) demonstrate increased mesoporosity with increasing reaction time and urea concentration, although an inverse relationship is noted in for SBET and Vmicro with respect to missing linker defects generated, as reported previously in the MOF literature.43–48
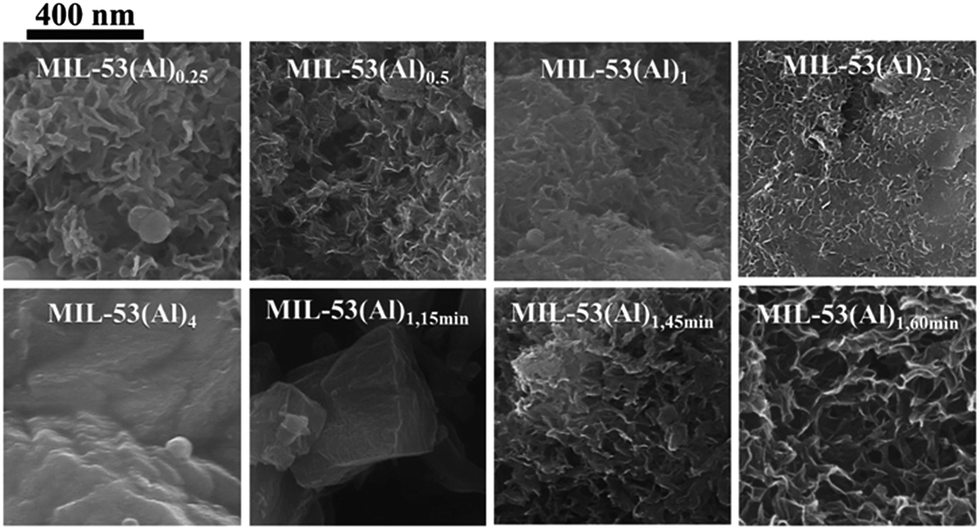 | ||
| Fig. 3 SEM images for MIL-53(Al) materials at high magnification (40 K magnification). | ||
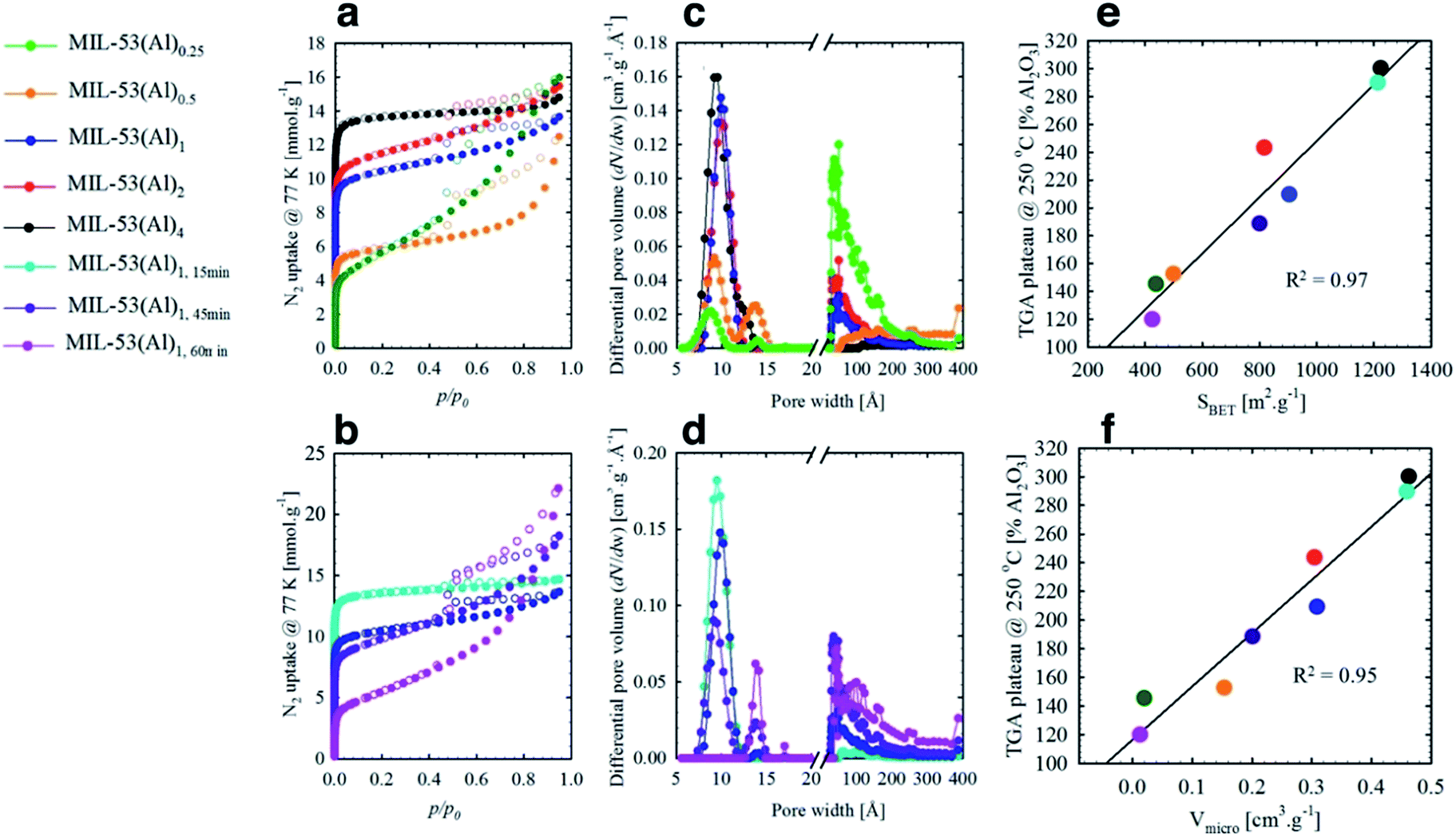 | ||
| Fig. 4 N2 isotherms (77 K) and pore size distributions for MIL-53(Al) materials with adjusted modulator stoichiometry/concentration (a and c) and varied reaction time (b and d); linear fits obtained by plotting (e) the BET surface areas (SBET) and (f) micro pore volume (Vmicro) of all 8 samples against their respective TGA plateau at 250 °C. | ||
At low to medium partial pressures, the water molecules physically adsorb to the surface and withwithin micropores of the material, while at high partial pressure conditions the adsorption mainly occurs within the mesoporous structures, forming a large amount of water clusters via hydrogen bonds.49,50
3.2 Fabrication of MIL-53/PVA composite membranes
The MIL-53(Al) samples fabricated with different reaction times were applied for composite pervaporation membrane fabrication. MOF materials were blended into PVA and then coated onto a PVDF hollow fiber surface (Fig. 5a). Compared with pure PVA coating, which has a smooth morphology with a consistent thickness of 0.8 μm, the incorporation of MOFs generated hierarchical roughness on the membrane surface, without a significant increase of the selective layer thickness (Fig. S23†). Energy dispersive X-ray spectroscopy (EDX) mapping results confirmed the presence of MIL-53(Al) within the selective layers (Fig. S24†), although agglomeration occurred with particularly high loadings. Compared with the PVA/PVDF composite hollow fiber, the membrane with MIL-53(Al) exhibited improved mechanical properties, indicating a good composition between PVA and fillers (Fig. S25†).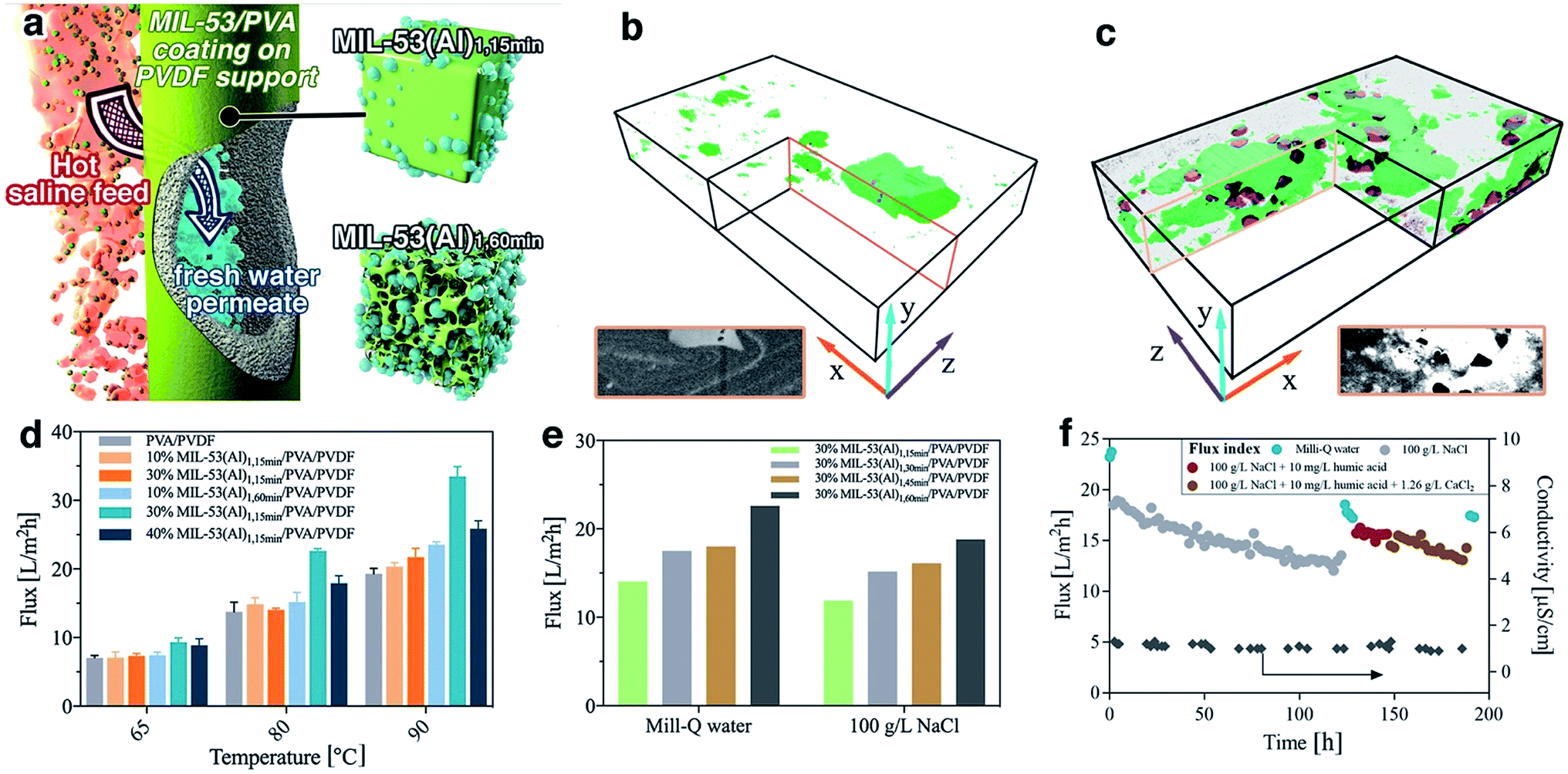 | ||
Fig. 5 (a) Schematic diagram of the nanocomposite pervaporation membrane for brine treatment; (b, c) surface-rendered views of the segmented FIB-SEM tomograms for mixed matrix membranes containing (b) MIL-53(Al)1,15 min and (c) defective type MIL-53(Al)1,60 min in PVA matrix. MOF particles are shown in green, while voids are shown in purple. The dimensions of the boxes shown in (b) and (c) along the x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y:z directions are 9.25, 1.5 and 5.0 μm, respectively. The insert are reference SEM images for the highlighted cross-sectional area; (d, e) pervaporation flux for different membranes. All reported values were the averaged flux of 4 hours' operation after initial stabilisation. (d) Milli-Q water was used as feed. (e) Feed solution temperature was 80 °C. For clarity, MIL-53(Al)1 was denoted as MIL-53(Al)1,30 min in this graph; (f) long term desalination performance with the complex brine feeds at 80 °C using 30% MIL-53(Al)1,60 min/PVA/PVDF membrane. :y:z directions are 9.25, 1.5 and 5.0 μm, respectively. The insert are reference SEM images for the highlighted cross-sectional area; (d, e) pervaporation flux for different membranes. All reported values were the averaged flux of 4 hours' operation after initial stabilisation. (d) Milli-Q water was used as feed. (e) Feed solution temperature was 80 °C. For clarity, MIL-53(Al)1 was denoted as MIL-53(Al)1,30 min in this graph; (f) long term desalination performance with the complex brine feeds at 80 °C using 30% MIL-53(Al)1,60 min/PVA/PVDF membrane. | ||
Given the long-standing issues of mixed matrix membrane compatibility between polymeric matrix and nanofillers, a series of free-standing MIL-53(Al)/PVA films were synthesized in order to probe the interface between MIL-53(Al) nanofiller and the polymeric PVA matrix. FTIR results demonstrated a shift to lower wavenumber of the OH stretching band in the PVA membrane, indicating the potential formation of hydrogen bonds as well as higher water adsorption within the film (Fig. S26†).51 We further examined the water uptake capability for the film (Table S3†). The addition of MIL-53(Al)1,15 min had a negligible improvement on the water adsorption, indicating that the surface OH groups of nanofillers are mostly occupied by the polymeric matrix forming hydrogen bonds. The creation of large scale mesoporous voids in the nanofillers however, e.g. MIL-53(Al)1,60 min, resulted in promotion of water uptake in the films. Such observations are aligned with the glass transition (Tg) shift for PVA films (Fig. S27†), where the Tg is raised by hydrogen bond formation and lowered by water-induced polymer swelling.52,53 More specifically, the pure PVA has a Tg of 63 °C which is comparable to the reference values.53 However, Tg for some film samples were too weak/or too broad to be detected, or they are suppressed due to polymer chain confinement. The incorporation of MIL-53(Al)1,15 min can form hydrogen bond with PVA chains, leading to an improved Tg. In comparison, the addition of defective MOFs can improve the water uptake for the film, leading to PVA chain swelling and subsequently reduced Tg. The presence of crystalline MIL-53(Al) within PVA film was further confirmed by the TGA and XRD results (Fig. S28–S29†).
The internal structure and filler-matrix interfacial morphology of the mixed matrix films were further studied with tomographic focused ion beam scanning electron microscopy (FIB-SEM). A trench was carved on the top surface and a series of SEM images were recorded of cross-sectional directions, and then the surface rendered views after segmentation of different phases (Fig. 5b–c and S30†). The full tomograms are provided as ESI Movies 1 and 2.† Significant differences in the microscale structures were observed: the defective MOF leaves a significant fraction of the composite materials unoccupied, due to their mesoporous structures.
3.3 Nanocomposite membrane pervaporation performance
To further study their technical relevance, the MOF-PVA nanocomposite hollow fiber membranes were tested for their pervaporation performance (Scheme S1†). As shown in Fig. 5d, when using Milli-Q water as feed, the incorporation of MIL-531,15 min (containing fewer defects) into PVA has a negligible effect on the permeation flux as compared with a PVA benchmark membrane. This observation aligns with the above water uptake results. By comparison, the beneficial role of defective MOF (MIL-531,60 min) is immediately apparent with improved water flux. This result is illustrative of the encouraging water transport efficiency for the composite membranes: the presence of micro- or mesovoids at the filler-matrix boundary can effectively promote water dissolution. The improvement is more significant at higher feed temperatures (driving force) where the rate limiting factor is the mass transport across the membrane. However, at high MOF loading (40% MIL-53(Al)1,60 min), the reduced water flux can be attributed to MOF aggregation as discussed above. In addition, the membrane performance with MIL-53 having different level of defects further confirms the functions of the defective structures (Fig. 5e). With the increase of mesoporous volume, the pervaporation flux increases with both pure water and 100 g L−1 NaCl as a feed.The distinct advantage of pervaporation desalination is its capability to process highly concentrated brine solutions. All nanocomposite membranes can effectively desalinate 100 g L−1 NaCl solution with the permeate conductivity at 1–1.5 μS cm−1 for over 120 hours of continuous operation, equivalent to a salt rejection rate of over 99.999% (Fig. S31†). The gradual reduction of permeate flux is attributed to the increased feed concentration, and the flux can be effectively resumed (up to 98.5%) by simple membrane rinsing with Milli-Q water. In addition, the membrane also exhibits satisfactory anti-fouling performance against humic acid and calcium ions (Fig. 5f and S32†) and complex inland desalination brine treatment efficiency (Fig. S33 and Table S4†). Similarly, the water permeation flux after the fouling test or inland desalination brine treatment can be effectively resumed by membrane cleaning using 2 wt% critic acid, indicating that the water flux reduction during the pervaporation test is mainly due to the reversible fouling or inorganic precipitation on the membrane surface. It is worthy to note that during these tests the permeate conductivity was maintained at a level of <2 μm, and the high salt rejection was confirmed by inductively coupled plasma mass spectrometry (ICP-MS) analysis of the inland brine treatment permeate (Table S5†). Although the pore size of MIL-53(Al) is larger than the hydrated diameter of salt ions, it has very minor effect on the salt transport through the membrane due to its relatively low loading. In the composite membrane, the presence of MOF in PVA layer mainly promotes the water uptake, without compromising the salt rejection efficiency, which is dominated by the polymeric PVA section.21 It should be noted that it is difficult to draw a direction comparison with the literature on pervaporation desalination membranes due to different operational parameters applied (Table S6†), the membrane in this work still one of the best performing polymer-based pervaporation membranes, with the highest salt rejection and good scalability. In addition, it should be noted that further process optimization can improve the membrane productivity, e.g. using a liquid nitrogen cold trap.
4. Conclusion
Defect generation in MIL-53(Al) was successfully controlled through the application of a urea modulator, with longer synthetic exposure times being the major factor. Crystalline structures were retained in all cases, and correlated to an increase in pore volume on both the atomic and microscopic scales, improved Brønsted acidity, and the subsequent improvement in performance for pervaporation membranes. Note, however, that the possibility of formation of amorphous Al(OH)x in MIL-53(Al) samples with a high concentration of defects could not be excluded. Our findings underline the intrinsic relationship of nanoscale MOF structure to their functions in a composite material, and highlight the opportunities that defect engineering can provide in MOFs. In this case, it opens up a new route to fine-tuning MOF interactions with a polymer in a composite material, via interfacial topology properties on top of the chemical functionality. Through this work, we expect that the mesoporous-polymer hybrid materials can be expanded for various applications. Alongside the formation of mesoporous MOF materials, this novel system leads to questions that merit further investigation, such as the detailed interaction between mesoporous MOFs and a polymeric matrix, and their subsequent effects on separation, catalysis, drug delivery and sensing performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Scientific Industry and Endowment Fund (SIEF Grant ID RP02-035, CO2MOF) project, the Australian Research Council's Discovery Projects funding scheme (DP130104048), UNSW Faculty of Engineering Silverstar Award, the Australian Institute of Nanoscale Science and Technology (AINST) and the Vibrational Spectroscopy Core Facility at The University of Sydney. We thank Dr Ravichandar Babarao (Royal Melbourne Institute of Technology, Australia) for helpful discussions. NDS gratefully acknowledges the financial support provided by the “Postgraduate Scholarship in Photoactive Metal-Organic Frameworks”. TDB would like to thank the Royal Society for a University Research Fellowship and their support.Notes and references
- O. K. Farha, A. Özgür Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- J. Hou, P. D. Sutrisna, Y. Zhang and V. Chen, Angew. Chem., Int. Ed., 2016, 55, 3947–3951 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, W. C. Isley Iii, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed.
- T. D. Bennett, A. K. Cheetham, A. H. Fuchs and F.-X. Coudert, Nat. Chem., 2017, 9, 11–16 CrossRef CAS PubMed.
- O. Kozachuk, I. Luz, F. X. Llabrés i Xamena, H. Noei, M. Kauer, H. B. Albada, E. D. Bloch, B. Marler, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058–7062 CrossRef CAS PubMed.
- E. Alvarez, N. Guillou, C. Martineau, B. Bueken, B. Van de Voorde, C. Le Guillouzer, P. Fabry, F. Nouar, F. Taulelle, D. de Vos, J.-S. Chang, K. H. Cho, N. Ramsahye, T. Devic, M. Daturi, G. Maurin and C. Serre, Angew. Chem., Int. Ed., 2015, 54, 3664–3668 CrossRef CAS PubMed.
- D. S. Sholl and R. P. Lively, J. Phys. Chem. Lett., 2015, 6, 3437–3444 CrossRef CAS PubMed.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- T.-H. Park, A. J. Hickman, K. Koh, S. Martin, A. G. Wong-Foy, M. S. Sanford and A. J. Matzger, J. Am. Chem. Soc., 2011, 133, 20138–20141 CrossRef CAS PubMed.
- J. Canivet, M. Vandichel and D. Farrusseng, Dalton Trans., 2016, 45, 4090–4099 RSC.
- Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Chem. Commun., 2016, 52, 7806–7809 RSC.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- G. Cai and H.-L. Jiang, Angew. Chem., 2017, 129, 578–582 CrossRef.
- Z. Fang, J. P. Dürholt, M. Kauer, W. Zhang, C. Lochenie, B. Jee, B. Albada, N. Metzler-Nolte, A. Pöppl, B. Weber, M. Muhier, Y. Wang, R. Schmid and R. A. Fischer, J. Am. Chem. Soc., 2014, 136, 9627–9636 CrossRef CAS PubMed.
- B. Bueken, N. Van Velthoven, T. Willhammar, T. Stassin, I. Stassen, D. A. Keen, G. V. Baron, J. F. M. Denayer, R. Ameloot, S. Bals, D. De Vos and T. D. Bennett, Chem. Sci., 2017, 8, 3939–3948 RSC.
- K. Shen, L. Zhang, X. Chen, L. Liu, D. Zhang, Y. Han, J. Chen, J. Long, R. Luque, Y. Li and B. Chen, Science, 2018, 359, 206–210 CrossRef CAS PubMed.
- A. W. Thornton, R. Babarao, A. Jain, F. Trousselet and F. X. Coudert, Dalton Trans., 2016, 45, 4352–4359 RSC.
- M. J. Cliffe, E. Castillo-Martínez, Y. Wu, J. Lee, A. C. Forse, F. C. N. Firth, P. Z. Moghadam, D. Fairen-Jimenez, M. W. Gaultois, J. A. Hill, O. V. Magdysyuk, B. Slater, A. L. Goodwin and C. P. Grey, J. Am. Chem. Soc., 2017, 139, 5397–5404 CrossRef CAS PubMed.
- M. J. Cliffe, J. A. Hill, C. A. Murray, F.-X. Coudert and A. L. Goodwin, Phys. Chem. Chem. Phys., 2015, 17, 11586–11592 RSC.
- L. Li, J. Hou, Y. Ye, J. Mansouri and V. Chen, Desalination, 2017, 422, 49–58 CrossRef CAS.
- Y.-H. Deng, J.-T. Chen, C.-H. Chang, K.-S. Liao, K.-L. Tung, W. E. Price, Y. Yamauchi and K. C. W. Wu, Angew. Chem., Int. Ed., 2016, 55, 12793–12796 CrossRef CAS PubMed.
- Y. Wang, Q. Qu, G. Liu, V. S. Battaglia and H. Zheng, Nano Energy, 2017, 39, 200–210 CrossRef CAS.
- P. G. Yot, L. Vanduyfhuys, E. Alvarez, J. Rodriguez, J.-P. Itie, P. Fabry, N. Guillou, T. Devic, I. Beurroies, P. L. Llewellyn, V. Van Speybroeck, C. Serre and G. Maurin, Chem. Sci., 2016, 7, 446–450 RSC.
- E. Alvarez, N. Guillou, C. Martineau, B. Bueken, B. Van de Voorde, C. Le Guillouzer, P. Fabry, F. Nouar, F. Taulelle, D. de Vos, J.-S. Chang, K. H. Cho, N. Ramsahye, T. Devic, M. Daturi, G. Maurin and C. Serre, Angew. Chem., Int. Ed., 2015, 54, 3664–3668 CrossRef CAS PubMed.
- W. Liang, R. Babarao, T. L. Church and D. M. D'Alessandro, Chem. Commun., 2015, 51, 11286–11289 RSC.
- A. Altomare, M. C. Burla, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Rizzi, J. Appl. Crystallogr., 1999, 32, 339–340 CrossRef CAS.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 CAS.
- T. J. Bandosz, M. Laskoski, J. Mahle, G. Mogilevsky, G. W. Peterson, J. A. Rossin and G. W. Wagner, J. Phys. Chem. C, 2012, 116, 11606–11614 CAS.
- T. G. Glover, G. W. Peterson, J. B. DeCoste and M. A. Browe, Langmuir, 2012, 28, 10478–10487 CrossRef CAS PubMed.
- J. Oomens and J. D. Steill, J. Phys. Chem. A, 2008, 112, 3281–3283 CrossRef CAS PubMed.
- S. Pradhan, V. Sahu and B. G. Mishra, J. Mol. Catal. A: Chem., 2016, 425, 297–309 CrossRef CAS.
- K. A. Fisher, J. J. Meisinger and B. R. James, J. Environ. Qual., 2016, 45, 349–359 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- J. Oomens and J. D. Steill, J. Phys. Chem. A, 2008, 112, 3281–3283 CrossRef CAS PubMed.
- P. J. Larkin, Infrared and Raman Spectroscopy; Principles and Interpretation, Elsevier, 2011 Search PubMed.
- J.-S. Choi, W.-J. Son, J. Kim and W.-S. Ahn, Microporous Mesoporous Mater., 2008, 116, 727–731 CrossRef CAS.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 CAS.
- W. Liang, C. J. Coghlan, F. Ragon, M. Rubio-Martinez, D. M. D'Alessandro and R. Babarao, Dalton Trans., 2016, 45, 4496–4500 RSC.
- Z. Liu, N. Fujita, K. Miyasaka, L. Han, S. M. Stevens, M. Suga, S. Asahina, B. Slater, C. Xiao, Y. Sakamoto, M. W. Anderson, R. Ryoo and O. Terasaki, Microscopy, 2013, 62, 109–146 CrossRef CAS PubMed.
- R. Semino, N. A. Ramsahye, A. Ghoufi and G. Maurin, Microporous Mesoporous Mater., 2017, 254, 184–191 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 1051 CAS.
- O. V. Gutov, M. G. Hevia, E. C. Escudero-Adán and A. Shafir, Inorg. Chem., 2015, 54, 8396–8400 CrossRef CAS PubMed.
- D. Wu, W. Yan, H. Xu, E. Zhang and Q. Li, Inorg. Chim. Acta, 2016, 460, 93–98 CrossRef.
- K. Wang, C. Li, Y. Liang, T. Han, H. Huang, Q. Yang, D. Liu and C. Zhong, Chem. Eng. J., 2016, 289, 486–493 CrossRef CAS.
- C. Atzori, G. C. Shearer, L. Maschio, B. Civalleri, F. Bonino, C. Lamberti, S. Svelle, K. P. Lillerud and S. Bordiga, J. Phys. Chem. C, 2017, 121, 9312–9324 CAS.
- G. C. Shearer, J. G. Vitillo, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 7190–7193 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- B. Bozbiyik, J. Lannoeye, D. E. De Vos, G. V. Baron and J. F. M. Denayer, Phys. Chem. Chem. Phys., 2016, 18, 3294–3301 RSC.
- J. A. Coelho, A. M. Ribeiro, A. F. P. Ferreira, S. M. P. Lucena, A. E. Rodrigues and D. C. S. d. Azevedo, Ind. Eng. Chem. Res., 2016, 55, 2134–2143 CrossRef CAS.
- Z. H. Ping, Q. T. Nguyen, S. M. Chen, J. Q. Zhou and Y. D. Ding, Polymer, 2001, 42, 8461–8467 CrossRef CAS.
- J. Rault, R. Gref, Z. H. Ping, Q. T. Nguyen and J. Néel, Polymer, 1995, 36, 1655–1661 CrossRef CAS.
- K. E. Strawhecker and E. Manias, Chem. Mater., 2000, 12, 2943–2949 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05175a |
| ‡ W. L. and L. L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |