
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAddition of dihydrogen to a borylborenium center†
Junhao
Zheng
,
Zhen Hua
Li
 * and
Huadong
Wang
*
* and
Huadong
Wang
*
Department of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Handan Road 220, Shanghai, 200433, China. E-mail: huadongwang@fudan.edu.cn; lizhenhua@fudan.edu.cn
First published on 28th November 2017
Abstract
The activation of a H–H bond, the simplest covalent bond, is a fundamentally important process. Addition of H2 to an elemental center typically occurs on low valent transition metal or main group complexes through oxidative addition to afford metal dihydride complexes. In contrast, activation of H2 on a high valent center generally results in heterolytic cleavage of the H–H bond to a proton and hydride. Here, we report experimental and computational evidence for the addition of H2 to a borenium center in an N-heterocyclic carbene (NHC) coordinated borylborenium cation, which leads to the formation of a dihydroborenium complex accompanied by the elimination of two σ-bonded substituents, namely mesityl (Mes) and pinacolboryl (Bpin) groups, as mesitylboronic ester.
Introduction
The activation of dihydrogen is a key step for hydrogenation reactions, which are considered as “the largest-volume human-made chemical reactions in the world”.1,2 In the activation process, the H–H σ bonding orbital is depopulated along with population of the H–H σ* anti-bonding orbital. Possessing empty/filled d orbitals suitable for interaction with H2 orbitals, low valent transition metal complexes can activate H–H bonds through oxidative addition, affording metal-dihydride complexes (Scheme 1a).1 Such oxidative addition is not limited to transition metal complexes. Low valent main group complexes, such as carbenes,3 silylenes4 and Al(I)5 complexes, can also activate H2 through oxidative addition due to the presence of an empty p orbital and a lone pair on the central element which can synergistically interact with H2 orbitals. On the other hand, high valent transition metal or main group complexes cannot undergo oxidative addition when treated with H2 due to their lack of filled d orbitals or lone pairs. Instead, they can activate H2 through either σ bond metathesis or base-assisted activation (Scheme 1b and c). The former pathway mainly involves d0 or d0fn metal complexes, resulting in hydrogenolysis of one M–E σ bond and the formation of a hydride species and H–E compound.6 The latter pathway includes synergistic interaction of a Lewis acid and base with H2, exemplified by the well-studied Noyori catalysts7 and the recently emerging frustrated Lewis pairs (FLPs),8 which leads to the formation of a hydride species and a protonated base. In contrast, the addition of H2 to a high valent element center to afford dihydride species still remains unknown (Scheme 1d).9 Such a reaction pathway would involve the cleavage of three σ-bonds with the concomitant formation of three new ones. | ||
| Scheme 1 Mechanisms for the activation of H2. | ||
Recently, we10 and the group of Nikonov11 independently discovered that electrophilic hydroboranes can activate H2 in the absence of Lewis bases through σ bond metathesis at 60 °C. Miqueu, Bouhadir and Bourissou et al. reported that phosphine coordinated mesityborenium complexes can react with H2 at 80 °C to afford corresponding hydroborenium complexes and mesitylene.12,13 Our theoretical investigation suggests that the charge transfer from the bonding orbital of a B–E bond to the σ* anti-bonding orbital of H2 plays an important role in the H2 activation process.14 To increase the reactivity of organoboranes against H2, we synthesized an N-heterocyclic carbene (NHC) coordinated borylborenium cation (1),15,16 anticipating that the boryl substituent would enhance the interaction between the B–B bonding orbital and the σ* anti-bonding orbital of H2. Indeed, borenium 1 was found to be able to activate H2 at a temperature as low as −40 °C. Additionally, to our surprise, the borenium center was inserted into the H–H bond, affording a dihydroborenium complex. Herein we report the synthesis of borenium 1 and its reactivity against H2 along with experimental and theoretical investigation of the reaction mechanism.
Results and discussion
Synthesis of 1 and its reactivity against H2
To prepare the precursor for borenium 1, IMe4 borane derivative IMe4-B(H)2Mes (2, IMe4 = 1,3,4,5-tetramethylimidazol-2-ylidene) was treated with I2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 to afford IMe4-B(H)(I)Mes, which was not isolated but directly reacted with IPrCuBpin (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, Bpin = pinacolboryl),18 producing the unsymmetrical diboron complex 3 in 52% yield (Scheme 2). The 11B NMR spectrum of 3 features one singlet at 40.9 ppm and one doublet at −29.5 ppm with the former assigned to the Bpin unit and the latter to the NHC coordinated BH moiety. Hydride abstraction from 3 with one equivalent of [Ph3C][B(C6F5)4] in toluene at 25 °C resulted in near quantitative formation of the borylborenium complex 1. After purification, complex 1 could be isolated in 96% yield and was characterized using NMR spectroscopy, high resolution mass spectrometry (HRMS) and elemental analysis. In the 11B NMR spectrum of 1, the resonance corresponding to the NHC coordinated boron atom appears at 78.0 ppm as a broad singlet, which is consistent with the formation of a borenium cation.15 An upfield shift of the Bpin moiety (32.8 ppm) compared to 3 was also observed. The identity of 1 was further confirmed using derivatization. Treatment of 1 with one equivalent of 4-dimethylaminopyridine (DMAP) gave DMAP coordinated boronium complex 4, which was characterized using X-ray diffraction. When the amount of DMAP was increased to two equivalents, complex 4 was still the only observable product and no bis-DMAP adduct was produced.
17 to afford IMe4-B(H)(I)Mes, which was not isolated but directly reacted with IPrCuBpin (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, Bpin = pinacolboryl),18 producing the unsymmetrical diboron complex 3 in 52% yield (Scheme 2). The 11B NMR spectrum of 3 features one singlet at 40.9 ppm and one doublet at −29.5 ppm with the former assigned to the Bpin unit and the latter to the NHC coordinated BH moiety. Hydride abstraction from 3 with one equivalent of [Ph3C][B(C6F5)4] in toluene at 25 °C resulted in near quantitative formation of the borylborenium complex 1. After purification, complex 1 could be isolated in 96% yield and was characterized using NMR spectroscopy, high resolution mass spectrometry (HRMS) and elemental analysis. In the 11B NMR spectrum of 1, the resonance corresponding to the NHC coordinated boron atom appears at 78.0 ppm as a broad singlet, which is consistent with the formation of a borenium cation.15 An upfield shift of the Bpin moiety (32.8 ppm) compared to 3 was also observed. The identity of 1 was further confirmed using derivatization. Treatment of 1 with one equivalent of 4-dimethylaminopyridine (DMAP) gave DMAP coordinated boronium complex 4, which was characterized using X-ray diffraction. When the amount of DMAP was increased to two equivalents, complex 4 was still the only observable product and no bis-DMAP adduct was produced.
 | ||
| Scheme 2 Synthesis of 1 and 4, and the molecular structure of 4 (the hydrogen atoms and counter anion B(C6F5)4− are omitted for clarity). | ||
When treated with 1.5 bar H2 in a toluene solution at room temperature, borenium 1 was quantitatively converted to a new species 5 in less than one minute. This reaction can take place at a much lower temperature. Stirring of the solution of 1 under 4.0 bar H2 for 2 hours at −40 °C also resulted in the quantitative formation of 5 (Scheme 3). This represents the mildest conditions for the activation of H2 by an organoboron species.19,20 Although complex 5 decomposes to unidentified complexes in bromobenzene solution after 1 day at room temperature, it was fully characterized using NMR and HRMS analysis. In the 1H NMR spectrum of 5, a broad signal at δ 2.87 ppm was observed, which was assigned to a BH2 moiety after comparison with the corresponding 1H{11B} NMR spectrum. We also noticed that the signal for the pinacol moiety appears as a broad singlet at δ 1.26 ppm, which prompted us to carry out a VT-NMR study (Fig. 1). At 60 °C, the signal for the pinacol moiety appears as a sharp singlet at δ 1.30 ppm. Upon cooling to −20 °C, this signal decoalesces to two broad singlets (δ 1.39 and 1.12 ppm), implying the existence of inequivalent methyl groups in the pinacol moiety. The signals corresponding to the Mes moiety also become broad during the cooling process, and those for IMe4 are largely temperature-invariant. In the 11B NMR spectrum of 5 at 25 °C, besides the signal from the anion [B(C6F5)4]− at δ −15.9 ppm, two broad singlets at δ 33.3 and −7.6 ppm were observed. As the former is in the range of a tricoordinate boron and the latter indicates the formation of a tetracoordinate boron, we assigned compound 5 as the adduct of [IMe4BH2][B(C6F5)4] and MesBpin with the oxygen atom of the pinacol moiety coordinating to the boron center of [IMe4BH2]+. A similar coordinate mode was observed in a Bi(III) complex in which the Bi(III) center is coordinated by the oxygen atom of a borate ester moiety.21 In agreement with this assignment, HRMS analysis of 5 revealed the formation of fragments [IMe4BH2]+ and [MesBpinLi]+ (Li ion was added as an ionization source). Our assignment was further confirmed by the independent synthesis of compound 5 from the borenium salt [IMe4BH2][B(C6F5)4]22 and MesBpin, which shows identical NMR spectra. The MesBpin moiety in 5 is readily replaced by a stronger base such as DMAP, affording the literature known boronium salt [IMe4B(H)2(DMAP)][B(C6F5)4]22 and free MesBpin. Treatment of 1 with D2 resulted in the formation of the corresponding deuterated analog 5-D2. The 2H NMR spectrum of 5-D2 displays a broad resonance at δ 2.86 ppm originating from the BD2 moiety.23 After quenching with DMAP, [IMe4B(D)2(DMAP)][B(C6F5)4] and MesBpin were obtained, confirming that the two hydride substituents in 5 originate from H2. To the best of our knowledge, this is the first time that the insertion of a boron center to a H–H bond has been observed.24 In a related study, Stephan and Bertrand reported that an acyclic(alkyl)(amino)carbene supported borylene can undergo a 1,2-addition reaction when treated with H2 and a dihydroboron species was proposed as a transient intermediate from the oxidative addition of H2 on the boron center.25 Recently, the groups of Wagner26 and Yamashita27 discovered that diboron species, such as reduced 9,10-dihydro-9,10-diboraanthracene and (o-Tol)2BB(o-Tol)2, can function as FLPs to react with H2, affording hydroborane complexes.
 | ||
| Scheme 3 Reaction of borylborenium 1 with H2 or D2. | ||
 | ||
| Fig. 1 VT-NMR spectra of 5 in C6D5Br (■: Bpin, ▲: IMe4, *: Mes). | ||
Experimental mechanism studies
To probe the reaction mechanism, we carried out a kinetic study of the reaction between 1 and H2 which was monitored using 1H NMR spectroscopy. Besides the starting material 1, H2 and the final product 5, no intermediate was observed in the reaction mixture. The rate of disappearance of 1 under a 10-fold excess of H2 followed pseudo-first-order kinetics. A linear dependence of the observed rate constant kobs on the H2 pressure at −15 °C suggested a first-order dependence on H2 pressure. These data were consistent with an overall second-order rate law for the H2 activation reaction. Kinetic measurements at temperatures ranging from −45 to −5 °C allow the determination of activation parameters (ΔH≠ = 6.2(2) kcal mol−1 and ΔS≠ = −33.8(6) cal mol−1 K−1) (Fig. 2a). The low value of ΔS≠ implies an associative mechanism. Furthermore, an inverse kinetic isotope effect (KIE = 0.80) was obtained by measuring the rate constant of the reaction between 1 and D2 (Fig. 2b), which indicates the existence of a dihydrogen complex in a rapid equilibrium before the H–H bond is cleaved.1 Observations of inverse KIEs for H2 activation reactions are rare. We only noticed two examples in the literature, which involve Ir28 and Fe29 complexes. | ||
| Fig. 2 (a) Eyring plot of the reaction between 1 and H2; (b) plots of the reactions between 1 and H2 or D2 at 258 K. | ||
Based on the kinetic study, three possible mechanisms could be considered for the formation of complex 5 from borenium 1 and H2. One mechanism (Scheme 4, path I) is that borenium 1 undergoes reductive elimination to afford borinylium intermediate 6, followed by the oxidative addition of H2 and coordination of MesBpin to form complex 5. The second proposed mechanism (Scheme 4, path II) involves the initial formation of [IMe4B(H)Mes][B(C6F5)4] (7) and HBpin,26 which subsequently go through ligand exchange to form complex 5. A third mechanism (Scheme 4, path III) would be that the addition of H2 and elimination of MesBpin occur in a concomitant manner. Both of the latter two pathways could proceed via initial coordination of H2 to the borenium center of 1.
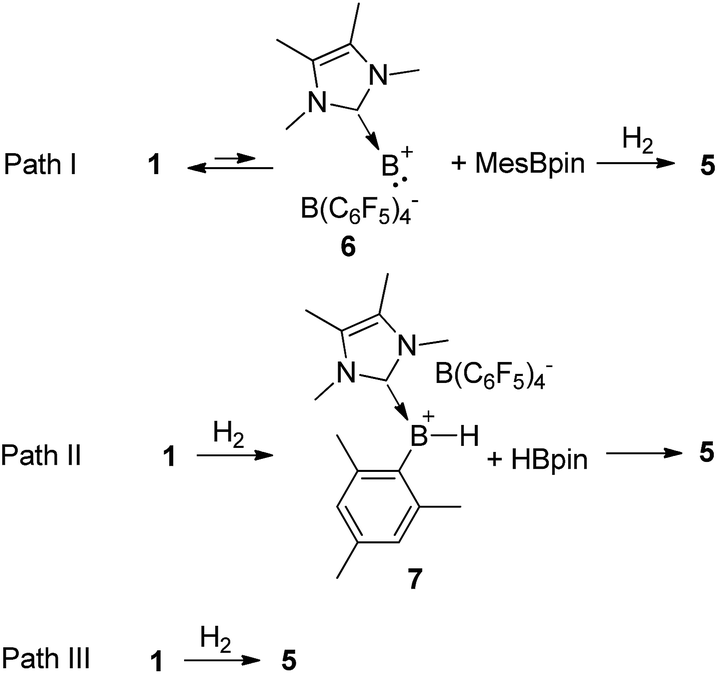 | ||
| Scheme 4 Possible reaction pathways for the formation of complex 5. | ||
If path I is operative, the reductive elimination and the reverse reaction, oxidative addition, must be very facile. To test this hypothesis, we mixed equimolar amounts of borenium 1 and (Mes-d9)(Bpin-d12) in toluene and no H/D scrambling was observed even at 80 °C. Therefore, path I can be ruled out. To test the feasibility of path II, we synthesized borenium 7 and studied its reactivity against HBpin. Mixing equimolar amounts of 7 and HBpin at room temperature led to the quantitative formation of 5 in one minute. When this reaction took place at −78 °C, complete conversion to 5 was observed after 10 min. No adduct of 7 and HBpin was observed while monitoring this reaction at −78 °C using 11B NMR spectroscopy (Fig. S45†). Subsequent kinetic study revealed that the activation parameters ΔH≠ and ΔS≠ for this reaction are 2.5(1) kcal mol−1 and −44.9(3) cal mol−1 K−1, respectively.
To distinguish paths II and III, we examined the reaction of an equimolar mixture of 1 and the deuterated analog [IMe4B(Bpin-d12)(Mes-d9)][B(C6F5)4] (1-D) with H2 in toluene (no H/D scrambling was observed for this mixture before H2 was introduced). If path II is effective, one would anticipate the observation of the non-crossover products (MesBpin and (Mes-d9)(Bpin-d12)) and crossover products (Mes(Bpin-d12) and (Mes-d9)Bpin) in a 1:1 ratio after quenching with DMAP, as the solvent cage effect should only play an insignificant role in the second step in path II due to the comparatively high free-energy barrier of this step. Alternatively, path III would lead to the observation of only the non-crossover products, as the H/D scrambling of complex 5 and its deuterium analog [IMe4B(H)2((Mes-d9)(Bpin-d12))][B(C6F5)4] was found to be much slower (less than 1% crossover product was observed after stirring for 30 min at room temperature).
Intriguingly, the observed ratios of non-crossover to crossover products imply that this reaction could proceed through both paths II and III with the latter favored at lower temperature (Table 1). When this reaction is conducted at −78 °C, the ratio of non-crossover to crossover products is 90:10.
| Entry | Temp. (K) | Non-crossover:crossover products |
k II/kIII |
|---|---|---|---|
| 1 | 195 | 90:10 |
0.24 |
| 2 | 213 | 87:13 |
0.36 |
| 3 | 233 | 82:18 |
0.54 |
| 4 | 253 | 80:20 |
0.69 |
| 5 | 273 | 76:24 |
0.91 |
| 6 | 283 | 73:27 |
1.18 |
Increasing the reaction temperature to 10 °C resulted in a decrease in the ratio to 73:27. Using the ratio of non-crossover to crossover products, we can estimate the ratio of the rate constants for path II and III, kII/kIII (for calculation details see ESI†), and thus approximate ΔΔH≠ and ΔΔS≠, which are estimated to be 1.9(1) kcal mol−1 and 6.9(4) cal mol−1 K−1, respectively. However, from the experimental data, we cannot tell whether this activation parameter difference corresponds to the rate-determining step (RDS) or a product-determining step after the RDS.
Theoretical studies
To further understand the reaction mechanism, the free-energy profile of this reaction was computed using density functional theory (DFT) calculations (M06-2X) (Fig. 3).30 First, H2 coordinates to the borenium center (B1) of 1 to form a dihydrogen complex 1·H2 through transition state TS1, in which the H–H bond is slightly activated and elongated from 0.737 to 0.817 Å. Subsequently, H2 is cleaved to form an intermediate IM1 through transition state TS2, which is the rate-limiting step. TS2 is 15.6 kcal mol−1 greater than free 1 and H2 in free energy, which is comparable to the experimental value of 16.3 kcal mol−1. In TS2, the H–H bond is substantially weakened and elongated to 1.040 Å. Both H atoms are closely bonded to the B1 atom with B1–H1 and B1–H2 bond distances of 1.269 and 1.244 Å, respectively. Unlike the four-centered transition state for σ bond metathesis,6 only a very weak interaction between B2 and the H2 atom (1.729 Å) is observed, which corresponds to a bond order of 0.11 according to natural bond orbital (NBO) analysis. The B1–B2 bond in TS2 is significantly weakened as well, and its distance is increased from 1.708 Å in 1 to 1.761 Å. In IM1, the H–H bond is completely cleaved. Concomitantly, a B1–H1 bond is observed with a bond length of 1.192 Å. The H2 atom forms a highly unsymmetrical three-center two-electron (3c2e) bond with B1 and B2, and the B1–H2 and B2–H2 bond distances are 1.245 and 1.469 Å, respectively. Interestingly, the ipso carbon atom of the mesityl moiety also forms a B–C–B 3c2e bond with B1 and B2, and the B1–C1 and B2–C1 bond distances are 1.708 and 1.765 Å, respectively. As a result, IM1 can be considered as an adduct of borenium [IMe4BH2]+ and MesBpin linked with two 3c2e bonds.31IM1 can rearrange to the final product 5 through transition state TS3 with a very low free-energy barrier of 0.5 kcal mol−1, which can be deemed as following path III in Scheme 4. On the other hand, IM1 can also undergo a reversible ligand exchange through TS3′ to afford 7 and HBpin, which are 8.6 kcal mol−1 less stable than 5 in free energy. The reaction from 7 and HBpin to 5 through IM1 needs to overcome a free-energy barrier of 14.7 kcal mol−1, which is in good agreement with our experimental value (15.9 kcal mol−1). This pathway corresponds to path II in Scheme 4. Therefore, these calculation results clearly show that the observation of the crossover experiment is not due to the existence of two H2 activation pathways. Instead, the observed crossover products originate from the ligand exchange of IM1 following the cleavage of the H–H bond, and the percentage of crossover product in the crossover experiment is determined by the free-energy difference between TS3 and TS3′. The calculated ΔΔH≠ and ΔΔS≠ values between TS3′ and TS3 are 4.7 kcal mol−1 and 5.6 cal mol−1 K−1, respectively, and are in relatively good agreement with the experimental values. We also calculated the KIE value for this reaction and the computed value of 0.81 agrees very well with the measured KIE value. | ||
| Fig. 3 The Gibbs free energy profile at 298 K for the reaction between 1 and H2 (H atoms (except H1 and H2) are omitted for clarity). | ||
To obtain insight into the H–H bond activation process, NBO population analysis along the intrinsic reaction coordinate (IRC) was carried out. As shown in Fig. 4, the electron populations32 of both hydrogen atoms are quite similar throughout the whole process, clearly indicating that the H–H bond is cleaved in a homolytic manner. During the process of coordination of H2 to 1 to form 1·H2, the electron populations of the H1 and H2 atoms decrease by 0.26 and 0.28 e, respectively. NBO occupation analysis revealed that these population changes are mainly due to the charge transfer from the H–H σ bond orbital to the empty p orbital of the B1 atom; the loss of the occupancy of the H–H σ bond orbital (0.60 e) matches very well with the increase of the occupancy of the p orbital of the B1 atom (0.62 e). This charge transfer results in an increase of the population of the B1 atom by 0.75 e, which induces polarization of the B–B σ bond towards the B1 atom and a decrease of the population of the B2 atom by 0.16 e. The contribution from the atomic orbitals of B1 to the B–B σ bonding orbital also increases from 52% in 1 to 59% in 1·H2. Such polarization is even larger than a known boryltitanium complex in which the boron atom contributes 54% of the Ti–B bond.33 This polarization of the B–B bond is in sharp contrast with the well-studied Lewis base coordinated B2pin2 complexes in which the B–B bond is polarized to the sp2 boron atom.33b,34 The IMe4 and mesityl ligands of the B1 atom in 1 are likely responsible for this counter-intuitive observation. Unlike the pinacol ligand, which can be an efficient electron-withdrawing group at an sp3 hybridized B atom,33b,34b neither IMe4 nor Mes can effectively draw electron density from the B1 atom when it receives electron charge from H2.
 | ||
| Fig. 4 Electron population change along the IRC of the reaction between 1 and H2. | ||
As the reaction proceeds from 1·H2 to TS2, the H–H σ* orbital gains 0.20 e, accompanied with a loss of 0.17 e from the B–B σ orbital, which is consistent with a charge transfer from the B–B σ orbital to the H–H σ* orbital. Due to the polarization of the B–B σ bond (the contribution from the atomic orbitals of B1 is 60%), this charge transfer predominately originates from the B1 atom. The back donation from the B1 atom, combined with the concomitant charge transfer from the H–H σ bond orbital to the empty p orbital of the B1 atom, ensures that both H atoms strongly coordinate to the B1 atom (the bond orders of B1–H1 and B1–H2 are 0.64 and 0.54 respectively). Furthermore, the polarization of the B–B bond results in an increase of positive charge on the Bpin moiety in TS2 (q = 0.21) compared to 1 (q = 0.04), which induces the migration of the electron-rich mesityl group from the B1 to B2 atom to form a B–C–B 3c2e bond in IM1.
Conclusions
In this work we have reported the synthesis of an NHC-coordinated borylborenium complex (1). When this borylborenium complex was treated with H2, addition of H2 to the borenium center was observed along with elimination of mesityl and Bpin substituents. The mechanistic studies revealed an unprecedented concurrent addition/elimination process in which polarization of the B–B bond upon coordination of H2 plays a critical role. Our discovery advances the understanding of dihydrogen activation involving high valent species, and may offer a new pathway for the activation of a broader range of element–element bonds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21372048, 21573044, 21672039) and the Shanghai Science and Technology Committee (16DZ2270100) is gratefully acknowledged. We also thank the High Performance Computer Center of Fudan University for the allocation of computing time. We thank Dr Yue-Jian Lin for their help in the crystallography study.Notes and references
- G. J. Kubas, Chem. Rev., 2007, 107, 4152–4205 CrossRef CAS PubMed.
- J. G. de Vries and C. J. Elsevier, The Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, 2007 Search PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed.
- R. Waterman, Organometallics, 2013, 32, 7249–7263 CrossRef CAS.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS PubMed.
- (a) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed; (c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed; (d) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- Addition of covalent bonded species, such as Cl2, to high valent metal complexes with non-innocent supporting ligands is known, see: K. J. Blackmore, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2005, 44, 5559–5561 CrossRef CAS PubMed.
- Z. Lu, Z. Cheng, Z. Chen, L. Weng, Z. H. Li and H. Wang, Angew. Chem., Int. Ed., 2011, 50, 12227–12231 CrossRef CAS PubMed.
- G. I. Nikonov, S. F. Vyboishchikov and O. G. Shirobokov, J. Am. Chem. Soc., 2012, 134, 5488–5491 CrossRef CAS PubMed.
- (a) M. Devillard, R. Brousses, K. Miqueu, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5722–5726 CrossRef CAS PubMed; (b) M. Devillard, S. Mallet-Ladeira, G. Bouhadir and D. Bourissou, Chem. Commun., 2016, 52, 8877–8880 RSC.
- For examples of the activation of H2 with borenium complexes in the presence of bases, see: (a) J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS PubMed; (b) E. R. Clark, A. Del Grosso and M. J. Ingleson, Chem.–Eur. J., 2013, 19, 2462–2466 CrossRef CAS PubMed; (c) P. Eisenberger, B. P. Bestvater, E. C. Keske and C. M. Crudden, Angew. Chem., Int. Ed., 2015, 54, 2467–2471 CrossRef CAS PubMed.
- Y. Wang, W. Chen, Z. Lu, Z. H. Li and H. Wang, Angew. Chem., Int. Ed., 2013, 52, 7496–7499 CrossRef CAS PubMed.
- D. P. Curran, A. Solovyev, M. M. Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed.
- For examples of borylboreniums see: (a) H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kramer, T. Kupfer, K. Radacki, E. Siedler, A. Trumpp, K. Wagner and C. Werner, J. Am. Chem. Soc., 2013, 135, 8702–8707 CrossRef CAS PubMed; (b) A. Prokofjevs, Angew. Chem., Int. Ed., 2015, 54, 13401–13405 CrossRef CAS PubMed.
- A. Solovyev, Q. Chu, S. J. Geib, L. Fensterbank, M. Malacria, E. Lacôte and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 15072–15080 CrossRef CAS PubMed.
- D. S. Laitat, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef PubMed.
- (a) C. Fan, L. G. Mercier, W. E. Piers, H. M. Tuononen and M. Parvez, J. Am. Chem. Soc., 2010, 132, 9604–9606 CrossRef CAS PubMed; (b) A. Y. Houghton, V. A. Karttunen, C. Fan, W. E. Piers and H. M. Tuononen, J. Am. Chem. Soc., 2013, 135, 941–947 CrossRef CAS PubMed.
- In the presence of Lewis bases, organoboranes are known to activate H2 even at −78 °C, for examples see (a) D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428–7432 CrossRef CAS PubMed; (b) P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433–7437 CrossRef CAS PubMed; (c) L. Greb, P. Oňa-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 51, 10164–10168 CrossRef CAS PubMed.
- C. C. Caries and S. Guccione, Organometallics, 2008, 27, 747–752 CrossRef.
- A. Prokofjevs, J. W. Kampf, A. Solovyev, D. P. Curran and E. Vedejs, J. Am. Chem. Soc., 2013, 135, 15686–15689 CrossRef CAS PubMed.
- Observation of no H/D scrambling in Mes group eliminates the potential mechanism involving hydrogenolysis of B–C bond and subsequent C–H borylation.
- For examples of heterolytic cleavage of H2 along B–E bonds see ref. 19 and S. J. Geier, T. M. Gilbert and D. W. Stephan, J. Am. Chem. Soc., 2008, 130, 12632–12633 CrossRef CAS PubMed.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS PubMed.
- E. v. Grotthuss, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067–14071 CrossRef PubMed.
- N. Tsukahara, H. Asakawa, K.-H. Lee, Z. Lin and M. Yamashita, J. Am. Chem. Soc., 2017, 139, 2593–2596 CrossRef CAS PubMed.
- B. E. Hauger, D. Gusev and K. G. Caulton, J. Am. Chem. Soc., 1994, 116, 208–214 CrossRef CAS.
- J. M. Camara and T. B. Rauchfuss, J. Am. Chem. Soc., 2011, 133, 8098–8101 CrossRef CAS PubMed.
- (a) Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101–194118 CrossRef PubMed; (b) Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 5121–5129 CrossRef CAS PubMed.
- A. Hübner, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2012, 51, 12514–12518 CrossRef PubMed.
- Electron population of an atom is the total number of electrons on the atom. A positive value represents an increase of population relative to that of 1 and H2.
- (a) T. Terabayashi, T. Kajiwara, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14162–14163 CrossRef CAS PubMed; (b) J. Cid, J. J. Carbó and E. Fernández, Chem.–Eur. J., 2012, 18, 12794–12802 CrossRef CAS PubMed.
- (a) K.-s. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253–7255 CrossRef CAS PubMed; (b) A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158–7161 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1543216 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04987k |
| This journal is © The Royal Society of Chemistry 2018 |