
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclometalated gold(III) complexes: noticeable differences between (N,C) and (P,C) ligands in migratory insertion†
Jordi
Serra
a,
Pau
Font
a,
E. Daiann
Sosa Carrizo
 b,
Sonia
Mallet-Ladeira
c,
Stéphane
Massou
c,
Teodor
Parella
d,
Karinne
Miqueu
b,
Abderrahmane
Amgoune
e,
Xavi
Ribas
*a and
Didier
Bourissou
*e
b,
Sonia
Mallet-Ladeira
c,
Stéphane
Massou
c,
Teodor
Parella
d,
Karinne
Miqueu
b,
Abderrahmane
Amgoune
e,
Xavi
Ribas
*a and
Didier
Bourissou
*e
aQBIS-CAT Group, Institut de Química Computacional i Catàlisi (IQCC), Departament de Química, Universitat de Girona, Campus Montilivi, Girona, E-17003, Catalonia, Spain. E-mail: xavi.ribas@udg.edu
bCNRS/UNIV PAU & PAYS ADOUR, Institut des Sciences Analytiques et de Physico-Chimie pour l'Environnement et les Matériaux (IPREM, UMR 5254), Hélioparc, 2 Avenue du Président Angot, 64053 Pau Cedex 09, France
cInstitut de Chimie de Toulouse (FR 2599), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France
dServei de RMN, Facultat de Ciències, Universitat Autonòma de Barcelona, Campus UAB, Bellaterra E-08193, Catalonia, Spain
eCNRS, Université Paul Sabatier, Laboratoire Hétérochimie Fondamentale Appliquée (LHFA, UMR 5069), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France. E-mail: dbouriss@chimie.ups-tlse.fr
First published on 26th March 2018
Abstract
Gold(III) complexes are garnering increasing interest for opto-electronic, therapeutic and catalytic applications. But so far, very little is known about the factors controlling their reactivity and the very influence of the ancillary ligand. This article reports the first comprehensive study on this topic. The reactivity of a cationic (N,C) gold(III) complex, namely 1A, towards ethylene has been thoroughly studied and compared with that of the related (P,C) complex 1C. A cationic gold(III) complex 5A resulting from double insertion of ethylene was selectively obtained. Complex 5A was found to be remarkably stable. It was trapped with chloride and fully characterized. In marked contrast to that observed with 1C, no β-H elimination or linear-to-branched rearrangement of the alkyl chain occurred with 1A. The energy profile for the reactions of 1A with ethylene has been comprehensively investigated computationally, and the influence of the ancillary ligand has been precisely delineated. Because nitrogen is a weaker donor than carbon (and phosphorus), the (N,C) ligand is very electronically dissymmetric, much more than the (P,C) ligand. This makes the two reactive sites at gold quite different, which noticeably influences the competition between migratory insertion and β-H elimination, and actually changes the outcome of the olefin insertion at gold. This study provides valuable insight into the influence of ancillary ligands on gold(III) reactivity, something critical to further develop Au(III) and Au(I)/Au(III) catalysis.
Introduction
The past few years have witnessed spectacular development in gold(III) chemistry.1–4 Au(III) complexes display very interesting luminescence properties5,6 and biological activities,7,8 and increasing efforts are made to develop their opto-electronic and therapeutic applications. Au(III) complexes also show unique catalytic properties, for the electrophilic activation of π–CC bonds and carbonyl compounds, and as intermediates in Au(I)/Au(III) redox cycles.3,9–11Hard C- and N-based ligands occupy a forefront position in gold(III) chemistry.1,12 In particular, (N,C) cyclometalated complexes A, and (C,N,C) and (N,C,C) pincer complexes B and B′ offer a unique balance between stability and properties (Scheme 1).6,10c,d,13–15 Strikingly, soft P donors have also proved recently to efficiently stabilize gold(III) species.16 (P,C) cyclometalated gold(III) complexes C (which are readily available by chelate-assisted oxidative addition of C–X bonds)16a have been shown to display rich reactivity.16b–f Unprecedented elementary organometallic reactions such as migratory insertion of alkenes16b,c,e and β-hydride elimination16d have been evidenced, enlarging the portfolio of chemical transformations of gold complexes.2
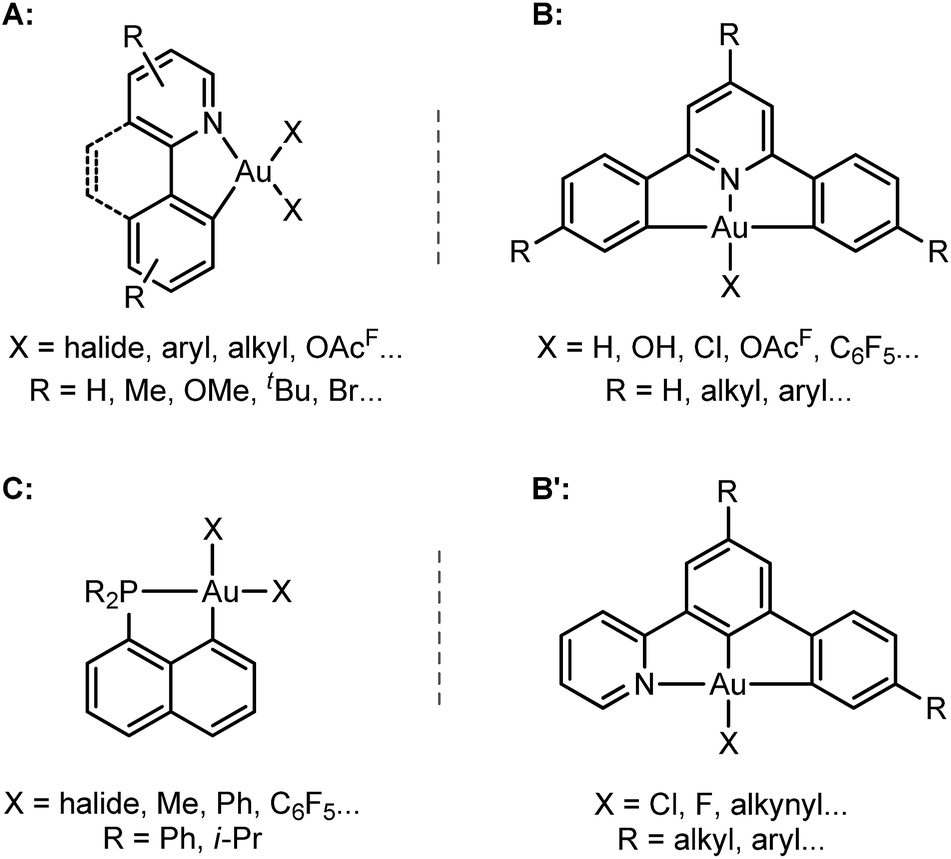 | ||
| Scheme 1 General structures of (N,C), (C,N,C), (N,C,C) and (P,C) gold(III) complexes A–C. | ||
To progress and develop further the chemistry of gold(III) complexes, it is critical to better understand how their properties and reactivity are influenced by ancillary ligands. It was precisely the focus of this work to bridge the gap between widespread (N,C) gold(III) complexes and recently introduced (P,C) gold(III) species, and to try to answer the following questions:
(i) Is the unprecedented reactivity recently evidenced with (P,C) gold(III) complexes specific to this ligand set or is it general? In other words, are (N,C) gold(III) complexes also prone to migratory insertion and β-H elimination?
(ii) What is the precise influence of the ancillary ligand on reactions with alkenes? Does it affect the kinetics and thermodynamics? Does it change the mechanism and/or fate of the reaction?
To this end, we studied a cationic tricoordinate (N,C) gold(III) complex, namely [(N,C)AuPh]+1A (Scheme 2), and we report here a detailed investigation of its reactivity towards ethylene. This study draws some analogies with the related [(P,C)AuPh]+ complex 1C,16e but also reveals noticeable differences. The weaker donicity of N versus P makes the (N,C) ligand much more electronically dissymmetric than the (P,C) ligand. As shown by detailed DFT investigations of the reaction profiles, these electronic properties noticeably influence the balance between the different reaction paths and eventually change the outcome of the ethylene insertion at gold. To the best of our knowledge, this is the first time that the impact of ancillary ligands on gold(III) reactivity is thoroughly explored.
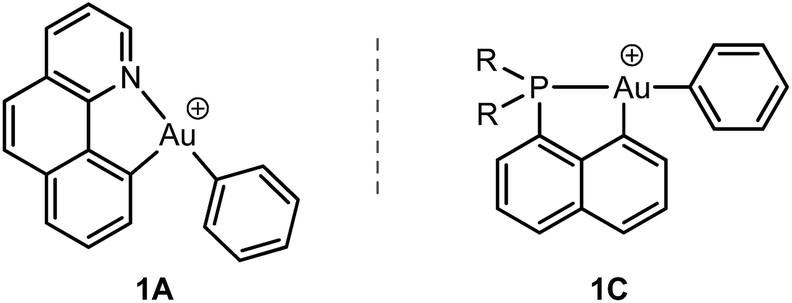 | ||
| Scheme 2 Structures of the (N,C) and (P,C) cationic gold(III) complexes studied and compared in this work. | ||
Results and discussion
Reaction of the [(N,C)AuPh]+ complex 1A with ethylene
Initially, we attempted the selective mono-arylation of the [(N,C)AuI2] complex 1, which was recently prepared via CAR–I oxidative addition to Au(I),13f following the same synthetic procedure as that described for the [(P,C)AuI2] complex.16e However, treatment of 1 with PhMgBr led to an intractable mixture of products. Hopefully, exchange of iodines at gold for more labile trifluoroacetate groups (complex 2) allowed for the synthesis of the desired [(N,C)AuBrPh] complex 3-Br(Scheme 3), applying the conditions reported by Tilset and co-workers.17 Complex 3-Br was isolated as a white powder in 59% yield after column chromatography and characterized by NMR spectroscopy and high-resolution mass spectrometry (HRMS).18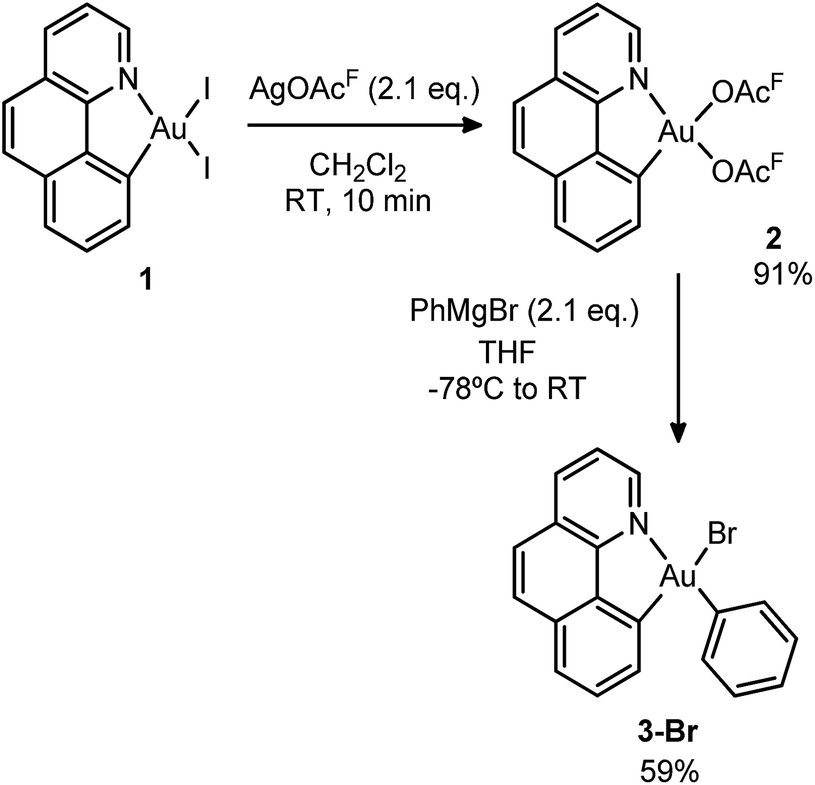 | ||
| Scheme 3 Synthesis of the Au(III) complex 3-Br, precursor to 1A. | ||
The cationic [(N,C)AuPh]+ complex 1A was then generated by abstracting the bromide at the Au(III) atom with AgSbF6 in dichloromethane under ethylene pressure (2 bar). No changes were observed within 72 h at room temperature, whereas upon heating at 40 °C for 18 h, complete conversion into a new species with aliphatic signals above δ 1.8 ppm was achieved (Scheme 4). Moreover, crystals formed spontaneously in small amounts inside the NMR tube, and X-ray diffraction analysis revealed double insertion of ethylene plus coordination of one molecule of adventitious water (Scheme 4). At this point, aiming at generating a more stable neutral four-coordinate gold(III) complex to allow for easier handling and characterization,16d,e we quenched the reaction crude with nBu4NCl. A white solid was isolated in 60% yield and characterized by multinuclear NMR spectroscopy and HRMS as 5A-Cl. The relative integration of the aromatic and aliphatic signals in the 1H NMR spectrum (13 versus 8H), and the peak at m/z 508.1346 corresponding to the mass of [(N,C)AuPh]+ augmented by two ethylene molecules, corroborated the double insertion into the Au–Ph bond. The solid-state structure of the complex 5A-Cl was also determined by X-ray crystallography.18 The gold atom sits in a square-planar environment formed by the (N,C) chelate ligand, the Cl atom and the (CH2)4Ph chain located cis to the aryl moiety.
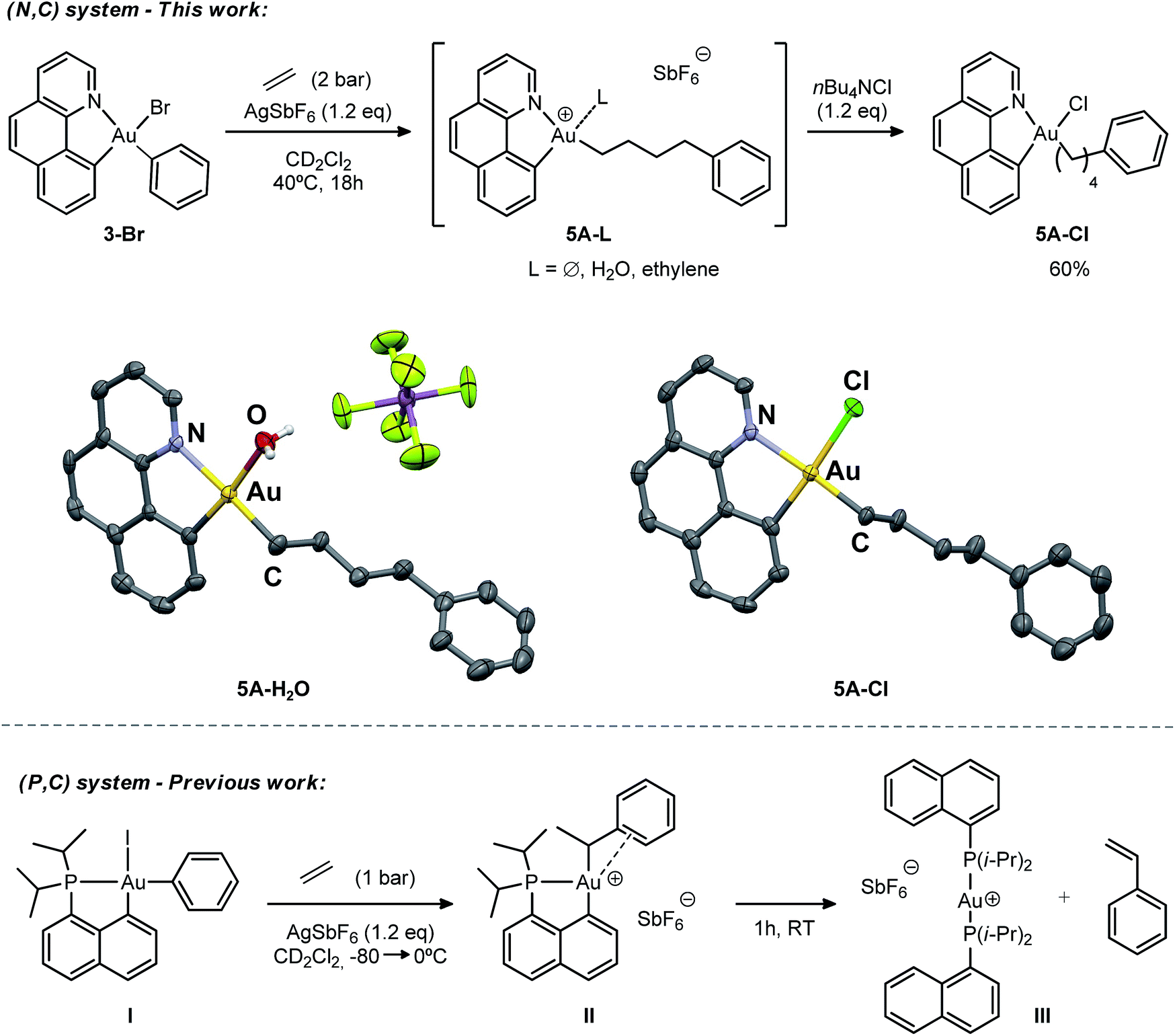 | ||
| Scheme 4 Reactions of the (N,C) and (P,C)-ligated gold(III) complexes 1A and 1C with ethylene, and the molecular view of 5A-H2O and 5A-Cl with thermal ellipsoids drawn at the 50% probability level (hydrogen atoms have been omitted for clarity). | ||
The stability of the cationic gold complex 5A generated under these conditions contrasts with the instability of 1A. In the absence of ethylene, the latter fully decomposes within 6 h in solution at room temperature into several side-products and metallic gold. At this stage, and without knowing the precise structure of 5A due to its ill-defined 1H NMR spectrum (the fourth coordination site at gold may be occupied by the SbF6 counter anion, the pendant Ph ring, an H2O or ethylene molecule), it is difficult to rationalize this difference.
The double addition of ethylene to complex 1A is reminiscent of the insertion of two norbornene (NB) molecules into the Au–Me bond of the (P,C)-ligated gold(III) dimethyl complex.16b,c In this process, the insertion of a second NB into the Au–Cnorbornyl bond gives access to a more thermodynamically stable complex, with the Au–Cnorbornyl bond at the trans position relative to phosphorus. In a likewise manner, the strong trans influence of the benzoquinolinic carbon in 1A triggers the migration of the alkyl group by insertion of a second ethylene molecule at the cis position relative to the aryl moiety (see DFT calculations below). In spite of the presence of a hydrogen atom at a β position and the tendency of cationic low-coordinate gold(III) alkyl complexes to undergo β-H elimination,16d complex 5A shows unusual thermal stability and no formation of styrene or higher olefins was detected during the course of the reaction. This is in stark contrast to the behaviour of the [(P,C)AuPh]+ complex 1C, where, after ethylene insertion, a β-H elimination and re-insertion sequence takes place leading to the gold(III)–arene complex II, which after 1 h at room temperature eventually evolves into the linear gold(I) complex III by reductive elimination, styrene release and ligand redistribution (Scheme 4, bottom).16e
D-labeling experiment
To assess the possible occurrence of β-H elimination in competition with ethylene insertion, a D-labeling experiment was carried out using trans-ethylene-d2 (Scheme 5). The reaction conditions have been adapted to enable the use of trans-ethylene-d2 which was supplied by Cluzeau Info Labo in a lecture bottle (1 atm, 95.2% D). Using an atmospheric pressure of ethylene, complete conversion was achieved after two ethylene refillings and 2 × 24 hours heating at 40 °C. The reaction was then repeated with trans-ethylene-d2 using the same experimental conditions.18 The reaction mixture was then trapped with nBu4NCl and analysed by NMR spectroscopy. The 1H NMR spectrum shows the expected integration and pattern for the Au(CHD)4Ph enchainment (Scheme 5): the two terminal CHD moieties resonate as doublets compared to triplets for the non-deuterated complex (5A-Cl). Consistently, the corresponding 13C NMR signals all appear as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplets in the 13C{1H} NMR spectrum. There is very little sign of H/D scrambling, if any, confirming that β-H elimination does not occur to a significant extent under these conditions and that ethylene insertion prevails.
:1:1 triplets in the 13C{1H} NMR spectrum. There is very little sign of H/D scrambling, if any, confirming that β-H elimination does not occur to a significant extent under these conditions and that ethylene insertion prevails.
 | ||
| Scheme 5 D-labeling experiment using trans-ethylene-d2 affording selectively the 5A-Cl-d4 product. Aliphatic 1H and 13C NMR signals of 5A-Cl (A) and 5A-Cl-d4 (B, C), as obtained by the analysis of the reaction crudes. In the case of the complex derived from trans-ethylene-d2, deuterium-decoupled (B) and deuterium-coupled (C) NMR analyses are shown. | ||
Synthesis and characterization of the [(N,C)Au(nBu)]+ complex 6A
To gain a better understanding of the reluctance of complex 5A to undergo β-H elimination, the analogous gold(III) n-butyl complex 6-Cl was synthesized and the behaviour of its cationic version was investigated (Scheme 6). The selective mono-alkylation of [(N,C)Au(OAcF)2] complex 2 was successfully accomplished using 2.1 equiv. of the Grignard reagent nBuMgCl under identical conditions to those applied for the preparation of the [(N,C)AuBrPh] complex 3-Br. After column chromatography, the target [(N,C)AuCl(nBu)] 6-Cl complex was isolated as a bench-top stable white powder in 73% yield and characterized by multinuclear NMR spectroscopy and high-resolution mass spectrometry.18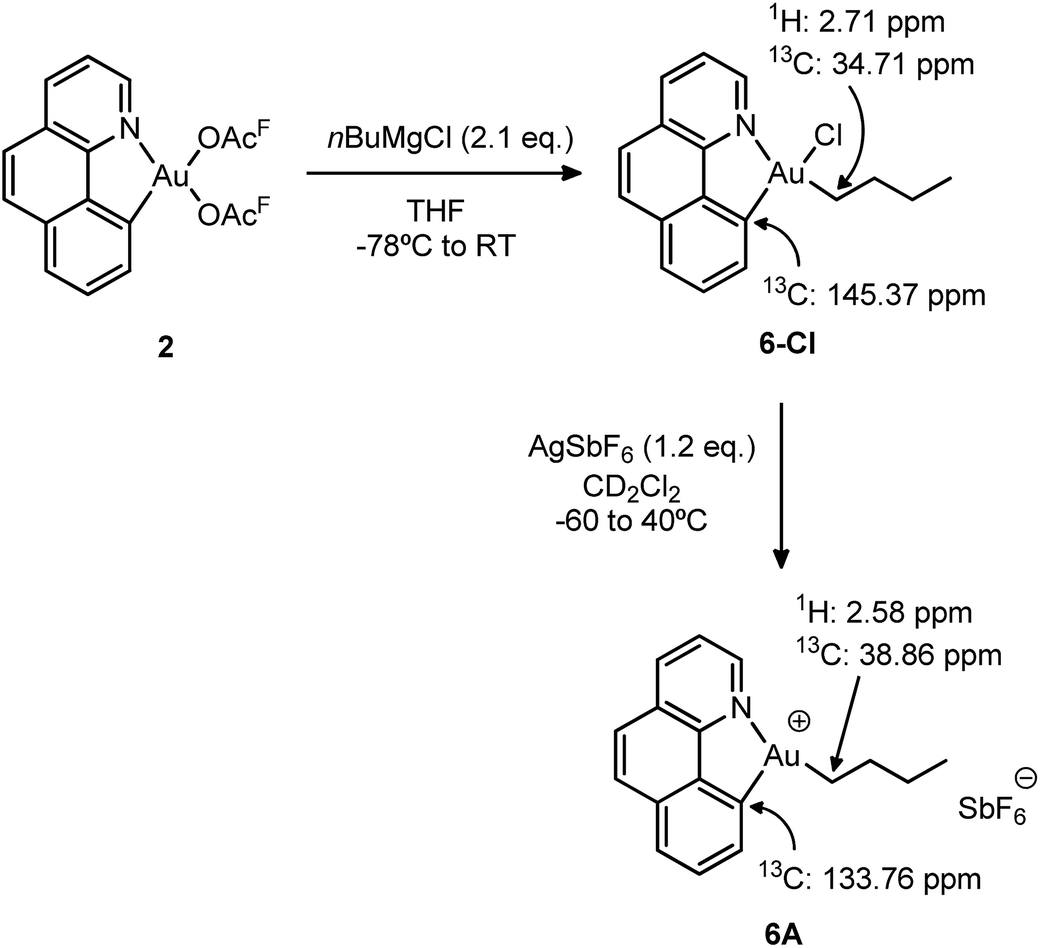 | ||
| Scheme 6 Synthesis of the n-butyl complexes 6-Cl and 6A and the most diagnostic 1H and 13C NMR shifts. | ||
We then proceeded with the generation of the corresponding cationic gold(III) complex 6A by chloride abstraction with AgSbF6 in CD2Cl2. As indicated by 1H NMR, it is instantaneously formed at −60 °C and it remains unchanged upon warming up to room temperature. No signs of degradation or β-H elimination (formation of butenes and butane) were observed by 1H and 13C NMR, even after heating at 40 °C for 24 h. In consequence, the cationic complex was fully characterized by NMR spectroscopy at room temperature without particular precautions. The 13C NMR data reveal a significant shift to high field of the aromatic carbon directly bound to gold upon cationization (from δ 145.37 ppm in the neutral complex 6-Cl to δ 133.76 ppm in the cationic complex 6A). On the other hand, the alkylic carbon of the n-butyl chain attached to gold shifts to low field with Δδ = 4.6 ppm. Overall, the salient thermal stability of the n-butyl cationic complex is in accordance with the lack of β-H elimination in the ethylene insertion reaction of [(N,C)AuPh]+ complex 5A.
In order to explain the double insertion of ethylene and selective formation of 5A, and to understand the different outcomes observed with the (N,C) and (P,C) complexes, a thorough DFT study was performed.
Computational investigation of the energy profile for the reaction of 1A with ethylene
DFT calculations were carried out at the B3PW91/SDD + f(Au),6-31G**(other atoms) level of theory. Solvent effects (DCM) were taken into account by means of a polarizable continuum model (PCM).18 Two energy minima corresponding to the cis and trans isomers of 1A were found on the potential energy surface (PES) (Fig. 1). By convention, cis and trans refer to the relative position of the phenyl (or alkyl group) at gold with respect to the carbon atom of the benzoquinolinic ligand. The cis isomer 1A is much more thermodynamically stable (by 36.2 kcal mol−1), in line with the detrimental trans arrangement of the phenyl and benzoquinolinic carbon atoms in t-1A.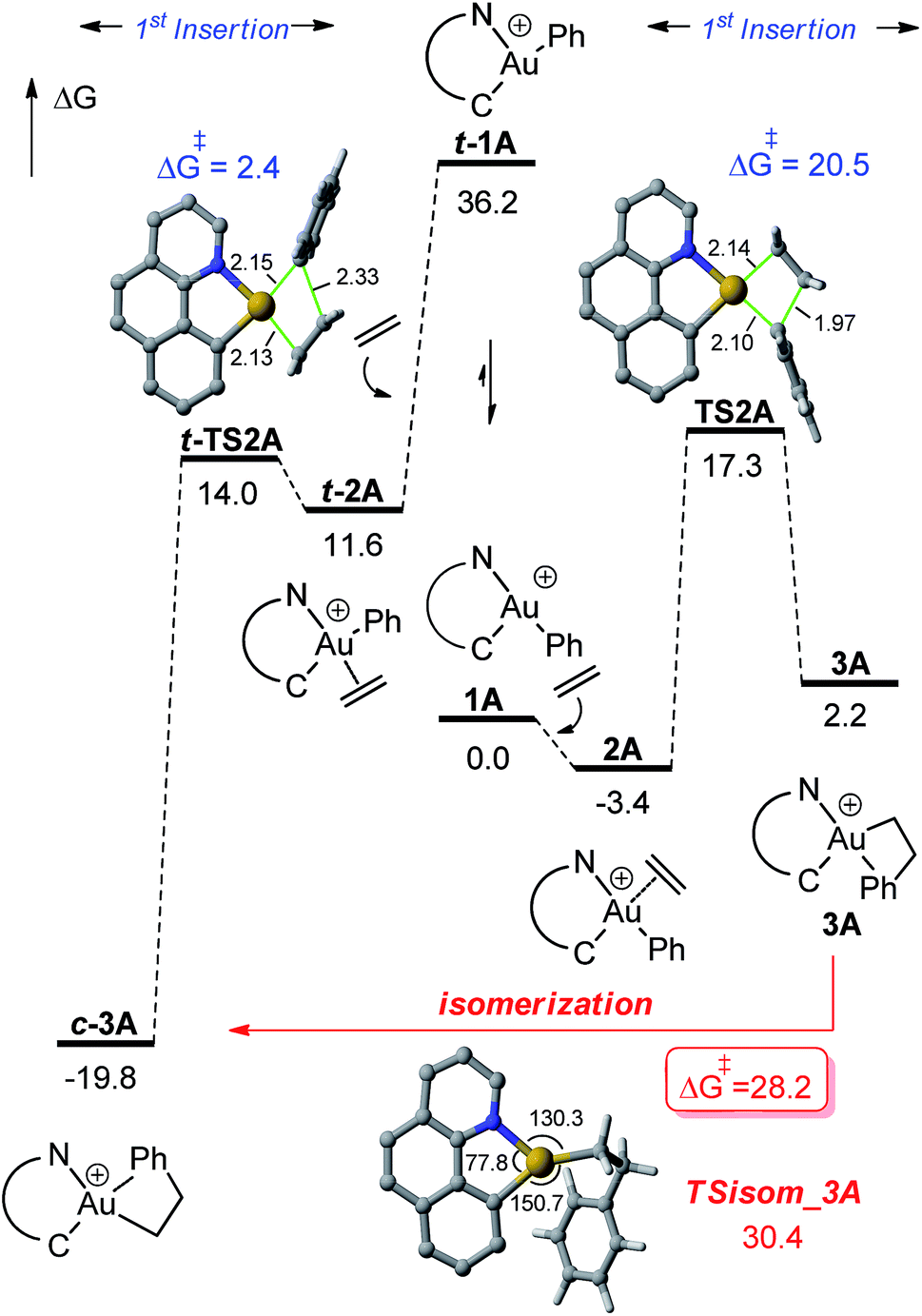 | ||
| Fig. 1 Energy profile (ΔG in kcal mol−1) computed at the PCM(dichloromethane)-B3PW91/SDD + f(Au)/6-31G**(other atoms) level of theory for the insertion of ethylene into the Au–Ph bond of 1A (cis and trans isomers). | ||
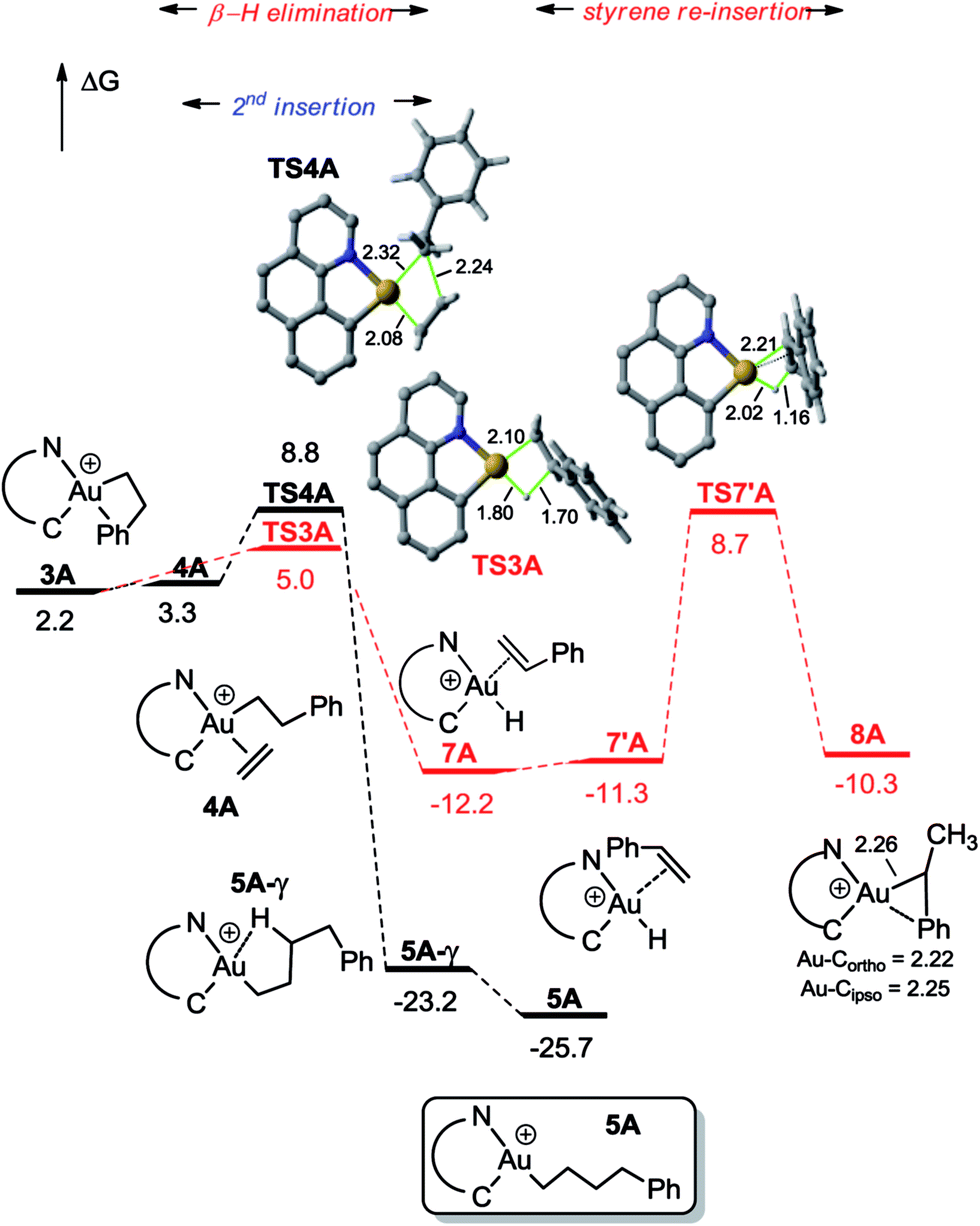 | ||
| Fig. 2 Energy profile (ΔG in kcal mol−1) computed at the PCM(dichloromethane)-B3PW91/SDD + f(Au)/6-31G**(other atoms) level of theory for the second ethylene insertion and β-H elimination from 3A. | ||
For the sake of completeness, β-H elimination, γ-H transfer and third ethylene insertion into 5A, as well as second ethylene insertion into 8A have been considered but all these transformations were found to be kinetically or thermodynamically disfavoured (Fig. S7 and S9–S11†).18 The same holds true for the second ethylene insertion and β-H elimination from c-3A, the more stable isomer of the first ethylene insertion. It is in particular worth noting that if the activation barrier for β-H elimination from 5A is not prohibitively high (19.0 kcal mol−1), the reaction is strongly endergonic (ΔG 18.5 kcal mol−1) due to the unfavourable trans arrangement of the resulting gold hydride olefin complex. This probably explains the stability of the [(N,C)Au(nBu)]+ complex 6A noticed experimentally. The activation barrier for the third ethylene insertion is ca. 17 kcal mol−1 higher than that of the first insertion, in line with the higher reactivity of the Au–Csp2vs. Au–Csp3 bond, as previously observed with the [(P,C)Ph]+ and [(P,C)AuMe]+ complexes.16d,e
Comparison of the (N,C) and (P,C) complexes
The above discussion has pointed out noticeable differences between the (N,C) and (P,C)-ligated gold complexes which are mainly associated with the weaker donor character of N versus P. As a result, the (N,C) ligand induces significant electronic dissymmetry on gold compared to the (P,C) ligand, and the two reaction sites trans to N or C (cf. square-planar geometry of the Au(III) complexes) behave quite differently, as previously noticed by Tilset, Nova and co-workers for the tolylpyridine (tpy) ligand.13c,dTo confirm and highlight the difference in the electronic properties of the ligand, calculations were performed on the naked [(N,C)Au]2+ and [(P,C)Au]2+ fragments (Fig. 3).18 In both cases, the LUMO corresponds to the vacant orbital at Au at the trans position to the N or P [main contribution of the σ*(Au–N)/(Au–P) orbitals]. It is significantly lower in energy (by 1 eV) with the (N,C) ligand. In contrast, the LUMO + 1 orbitals of the two fragments, which correspond to the vacant orbitals at Au at the trans position to the aryl moiety, lie close in energy. Thus, the (N,C) ligand induces a significantly larger LUMO–LUMO + 1 gap than the (P,C) ligand and hence a stronger electronic dissymmetry of the coordination sphere at gold. Note also that the weaker donicity of the (N,C) ligand results in a significantly more electrophilic gold center than with the (P,C) ligand: the LUMO is noticeably lower in energy and the Bader atomic charge at gold is much higher (1.05 versus 0.58).
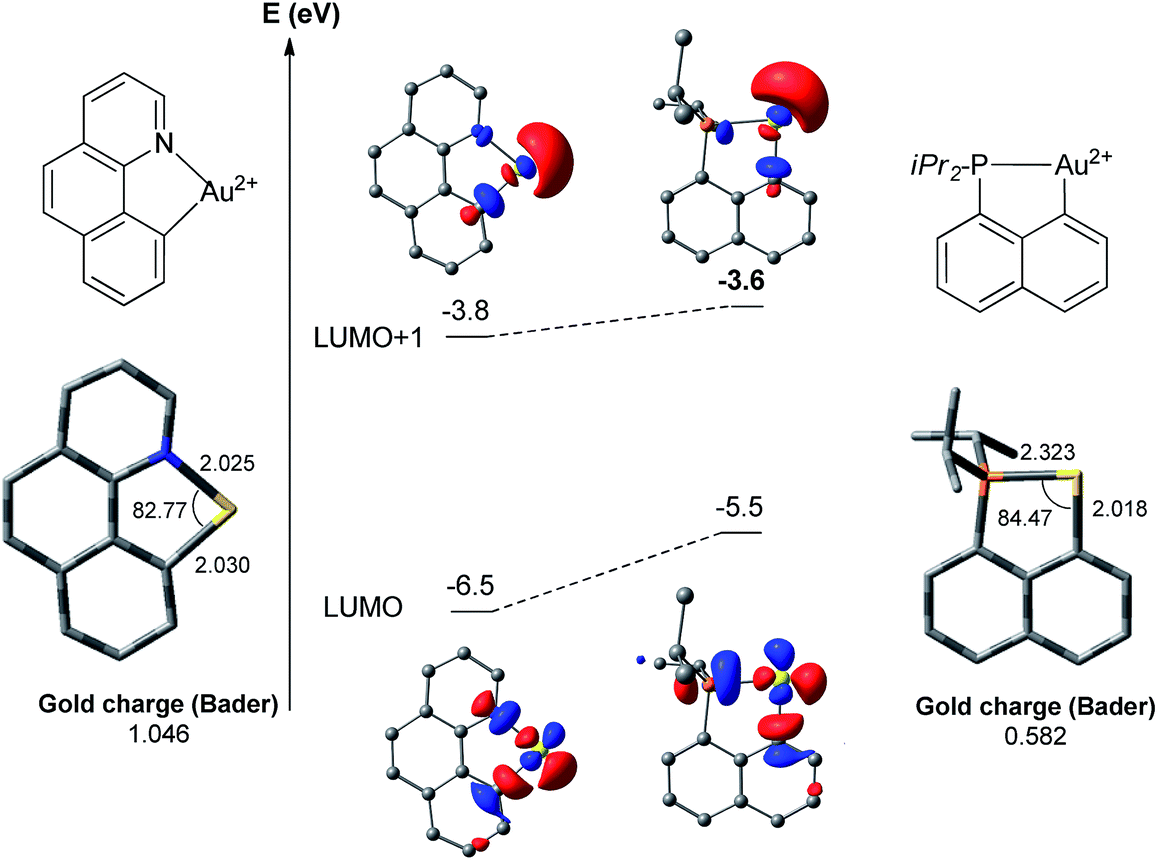 | ||
| Fig. 3 Optimized structures (with selected geometrical data) and the lowest energy molecular orbitals of the [(N,C)Au]2+ and [(P,C)Au]2+ fragments computed at the PCM(dichloromethane)-B3PW91/SDD + f(Au)/6-31G**(other atoms) level of theory. | ||
To further compare the two systems and pinpoint the differences, calculations on the [(P,C)AuPh]+ complex 1C were re-performed16e at the same level of theory as for the [(N,C)AuPh]+ complex 1A and completed to include the second ethylene insertion (Fig. S12†).18 The difference between the (N,C) and (P,C) ligands is spectacular in the energy gap between the cis and trans isomers of 1A and 1C (as well as of related species). As mentioned above, the cis form of 1A is more stable than the trans isomer by 36.2 kcal mol−1 (ΔG). The energy gap falls down to 7.1 kcal mol−1 for 1C. The difference is also striking in the first ethylene insertion as well as in the competition between second ethylene insertion and β-H elimination. With the (N,C) ligand, the first ethylene insertion is slightly uphill in energy (ΔG +2.2 kcal mol−1) while the second insertion is strongly exergonic (ΔG −27.9 kcal mol−1) and favoured over β-H elimination. In contrast, ethylene insertion into the (P and C) complex 1C is downhill in energy (ΔG −10.0 kcal mol−1) but the second ethylene insertion involves a high activation barrier (21.6 kcal mol−1versus 15.1 kcal mol−1 for the first insertion) and does not compete with β-H elimination/re-insertion leading to the branched product II. In addition, reductive elimination of the gold hydride 7A (Csp2–H coupling) was found to be significantly more demanding energetically than that of the corresponding (P,C) complex 7C (the activation barrier is about 10 kcal mol−1 higher in energy, see Fig. S14†),18 in line with the stability of 5A and fast decomposition of II (see Scheme 4).
Conclusions
To sum up, the novel cyclometalated Au(III) complex [(N,C)AuPh]+1A was shown to react with ethylene to selectively give a double insertion product (5A), without β-H elimination nor rearrangement of the linear (CH2)4Ph chain. Trapping with chloride affords complex 5A-Cl which has been fully characterized, including by X-ray diffraction. The energy profile for the reactions of 1A with ethylene has been thoroughly investigated computationally, considering competitive paths, and the influence of the ancillary ligand has been delineated by comparing the (N,C) and (P,C) complexes 1A and 1C.Because of the weak donor character of nitrogen (compared with carbon and even phosphorus), the benzoquinoline ligand is very electronically dissymmetric. As a result, the two reactive sites at gold are quite different. This situation contrasts with the more symmetric nature of the (P,C) chelate and explains the peculiar reactivity of 1A. The electronic dissymmetry of the (N,C) ligand explains why β-H elimination is not observed upon reaction of 1A with ethylene, neither upon cationisation of 6-Cl. The activation barrier for β-H elimination is not prohibitively high, but the reaction is uphill in energy due to the unfavourable position of the hydride (trans to the carbon atom of the ligand) in the resulting complex.
The detailed mechanistic study of migratory insertion and β-H elimination has revealed that the properties of the ancillary ligand, and in particular the electronic dissymmetry that the (N,C) ligand induces at gold play a prominent role in reactivity. This situation is somewhat reminiscent of that arising with phosphine–sulfonate bidentate ligands which, thanks to their strong electronic dissymmetry, bestow unique properties to Pd(II) complexes towards the coordination–insertion polymerization of ethylene and polar olefins.23
This study reveals that (N,C) Au(III) complexes possess rich reactivity. It generalizes migratory insertion of alkenes at gold, an elementary process unknown with gold until very recently, but involved in many synthetically useful transformations (such as the Mizoroki–Heck reaction, the oligomerization/polymerization of olefins, etc.). The ancillary ligand markedly influences the outcome of the reaction and thus, modulation of its structure paves the way for tuning and optimizing the properties of gold(III) complexes.
Accordingly, future work will seek to take advantage of well-defined (N,C) and (P,C) gold(III) complexes in catalysis and to expand further the variety of chelating ligands.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Centre National de la Recherche Scientifique and the Université de Toulouse, the European Research Council (Starting Grant Project ERC-2011-StG-277801), Spanish MINECO (CTQ2016-77989-P and CTQ2015-64436-P) and Generalitat de Catalunya (2017 SGR 264) is gratefully acknowledged. We thank Umicore AG & Co for a generous gift of gold precursors. UPPA, MCIA (Mésocentre de Calcul Intensif Aquitain) and CINES under allocation 2017 (A002080045) made by Grand Equipement National de Calcul Intensif (GENCI) are acknowledged for computational facilities. Isabelle Favier and Charlie Blons (LHFA) are gratefully acknowledged for their assistance in setting up the reaction with ethylene-d2. E. Daiann Sosa-Carrizo thanks CDAPP for funding part of his post-doctoral contract. J. S. and P. F. thank the GDRI HC3A for the attribution of mobility grants and UdG for a PhD grant. X. R. thanks ICREA-Acadèmia award. We also thank STR from UdG for technical support.Notes and references
- R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994–2015 CrossRef CAS PubMed.
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Arabian J. Sci. Eng., 2012, 37, 1187–1225 CrossRef CAS.
- D. A. Roşca, J. A. Wright and M. Bochmann, Dalton Trans., 2015, 44, 20785–20807 RSC.
- For reviews, see: (a) V. W. W. Yam and E. C. C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC; (b) C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409–12420 RSC; (c) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- For selected recent examples, see: (a) W. P. To, G. S. M. Tong, W. Lu, C. Ma, J. Liu, A. L. F. Chow and C. M. Che, Angew. Chem., Int. Ed., 2012, 51, 2654–2657 CrossRef CAS PubMed; (b) W. P. To, K. T. Chan, G. S. M. Tong, C. Ma, W. M. Kwok, X. Guan, K. H. Low and C. M. Che, Angew. Chem., Int. Ed., 2013, 52, 6648–6652 CrossRef CAS PubMed; (c) C. Y. Sun, W. P. To, X. L. Wang, K. T. Chan, Z. M. Su and C. M. Che, Chem. Sci., 2015, 6, 7105–7111 RSC; (d) M. C. Tang, D. P. K. Tsang, Y. C. Wong, M. Y. Chan, K. M. C. Wong and V. W. W. Yam, J. Am. Chem. Soc., 2014, 136, 17861–17868 CrossRef CAS PubMed; (e) V. K. M. Au, D. Wu and V. W. W. Yam, J. Am. Chem. Soc., 2015, 137, 4654–4657 CrossRef CAS PubMed; (f) E. Y. H. Hong, C. T. Poon and V. W. W. Yam, J. Am. Chem. Soc., 2016, 138, 6368–6371 CrossRef CAS PubMed; (g) M. C. Tang, C. H. Lee, S. L. Lai, M. Ng, M. Y. Chan and V. W. W. Yam, J. Am. Chem. Soc., 2017, 139, 9341–9349 CrossRef CAS PubMed; (h) R. Kumar, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 14287–14290 CrossRef CAS PubMed; (i) L. Currie, J. Fernandez-Cestau, L. Rocchigiani, B. Bertrand, S. J. Lancaster, D. L. Hughes, H. Duckworth, S. T. E. Jones, D. Credgington, T. J. Penfold and M. Bochmann, Chem.–Eur. J., 2017, 23, 105–113 CrossRef CAS PubMed; (j) A. Szentkuti, M. Bachmann, J. A. Garg, O. Blacque and K. Venkatesan, Chem.–Eur. J., 2014, 20, 2585–2596 CrossRef CAS PubMed.
- For reviews, see: (a) T. Zou, C. T. Lum, C. N. Lok, J. J. Zhang and C. M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC; (b) S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CrossRef CAS; (c) C. M. Che and R. W. Y. Sun, Chem. Commun., 2011, 47, 9554–9560 RSC; (d) I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS.
- For selected recent examples, see: (a) D. Hu, Y. Liu, Y. T. Lai, K. C. Tong, Y. M. Fung, C. N. Lok and C. M. Che, Angew. Chem., Int. Ed., 2016, 55, 1387–1391 CrossRef CAS PubMed; (b) C. Y. S. Chung, S. K. Fung, K. C. Tong and P. K. Wan, Chem. Sci., 2017, 8, 1942–1953 RSC; (c) S. K. Fung, T. Zou, B. Cao, P. Y. Lee, Y. M. E. Fung, D. Hu, C. N. Lok and C. M. Che, Angew. Chem., Int. Ed., 2017, 56, 3892–3896 CrossRef CAS PubMed; (d) K. Tsubokura, K. K. H. Vong, A. R. Pradipta, A. Ogura, S. Urano, T. Tahara, S. Nozaki, H. Onoe, Y. Nakao, R. Sibgatullina, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3579–3584 CrossRef CAS PubMed.
- For reviews, see: (a) C. Nevado and T. D. Haro, in New Strategies in Chemical Synthesis and Catalysis, Wiley–VCH Verlag GmbH & Co. KGaA, 2012, pp. 247–272 Search PubMed; (b) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910–1925 RSC; (c) A. S. K. Hashmi, Gold-Catalyzed Organic Reactions, in Inventing Reactions, ed. GooßenL. J., Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 143–164 Search PubMed; (d) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed.
- For selected recent examples, see: (a) C. Y. Wu, T. Horibe, C. B. Jacobsen and F. D. Toste, Nature, 2015, 517, 449–454 CrossRef CAS PubMed; (b) P. T. Bohan and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 11016–11019 CrossRef CAS PubMed; (c) J. Serra, C. J. Whiteoak, F. Acuna-Pares, M. Font, J. M. Luis, J. Lloret-Fillol and X. Ribas, J. Am. Chem. Soc., 2015, 137, 13389–13397 CrossRef CAS PubMed; (d) M. Hofer, A. Genoux, R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1021–1025 CrossRef CAS PubMed; (e) S. Witzel, J. Xie, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2017, 359, 1522–1528 CrossRef CAS; (f) M. J. Harper, E. J. Emmett, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2017, 139, 12386–12389 CrossRef CAS PubMed; (g) A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef PubMed.
- The propensity of gold(III) to undergo extraordinarily fast reductive elimination has also been used recently to develop an efficient way to form C(sp3)–CF3 bonds and prepare 18F–radiolabeled compounds, see: (a) W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159–164 CrossRef CAS PubMed; (b) M. D. Levin, T. Q. Chen, M. E. Neubig, C. M. Hong, C. A. Theulier, I. J. Kobylianskii, M. Janabi, J. P. O'Neil and F. D. Toste, Science, 2017, 356, 1272–1276 CrossRef CAS PubMed.
- W. Henderson, The Chemistry of Cyclometallated Gold(III) Complexes with C,N–Donor Ligands, in Adv. Organomet. Chem., ed. West R. and Hill A. F., Academic Press, 2006, Vol. 54, pp. 207–265 Search PubMed.
- For (N,C) gold(III) complexes, see: (a) E. C. Constable, R. P. G. Henney and T. A. Leese, J. Organomet. Chem., 1989, 361, 277–282 CrossRef CAS; (b) E. C. Constable and T. A. Leese, J. Organomet. Chem., 1989, 363, 419–424 CrossRef CAS; (c) E. Langseth, A. Nova, E. A. Tråseth, F. Rise, S. Øien, R. H. Heyn and M. Tilset, J. Am. Chem. Soc., 2014, 136, 10104–10115 CrossRef CAS PubMed; (d) M. S. M. Holmsen, A. Nova, D. Balcells, E. Langseth, S. Øien-Ødegaard, R. H. Heyn, M. Tilset and G. Laurency, ACS Catal., 2017, 7, 5023–5034 CrossRef CAS; (e) R. Kumar, A. Linden and C. Nevado, J. Am. Chem. Soc., 2016, 138, 13790–13793 CrossRef CAS PubMed; (f) J. Serra, T. Parella and X. Ribas, Chem. Sci., 2017, 8, 946–952 RSC; (g) A. Maity, A. N. Sulicz, N. Deligonul, M. Zeller, A. D. Hunter and T. G. Gray, Chem. Sci., 2015, 6, 981–986 RSC; (h) L. Rocchigiani, J. Fernandez-Cestau, G. Agonigi, I. Chambrier, P. H. M. Budzelaar and M. Bochmann, Angew. Chem., Int. Ed., 2017, 56, 13861–13865 CrossRef CAS PubMed.
- For (C,N,C) and (N,C,C) gold(III) complexes, see: (a) D. A. Roşca, D. A. Smith and M. Bochmann, Chem. Commun., 2012, 48, 7247–7249 RSC; (b) D. A. Roşca, D. A. Smith, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2012, 51, 10643–10646 CrossRef PubMed; (c) N. Savjani, D. A. Roşca, M. Schormann and M. Bochmann, Angew. Chem., Int. Ed., 2013, 52, 874–877 CrossRef CAS PubMed; (d) D. A. Roşca, D. A. Smith, D. L. Hughes and M. Bochmann, Nat. Commun., 2013, 4, 2167 Search PubMed; (e) D. A. Roşca, J. Fernandez-Cestau, J. Morris, J. A. Wright and M. Bochmann, Sci. Adv., 2015, 1, e1500761 Search PubMed; (f) R. Kumar, J. P. Krieger, E. Gómez-Bengoa, T. Fox, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 12862–12865 CrossRef CAS PubMed.
- For examples of gold(III) complexes featuring chelating O-based ligands, see: (a) A. S. K. Hashmi, J. P. Weyrauch, M. Rudolph and E. Kurpejović, Angew. Chem., Int. Ed., 2004, 43, 6545–6547 CrossRef CAS PubMed; (b) P. Oña-Burgos, I. Fernández, L. Roces, L. Torre-Fernández, S. García-Granda and F. López-Ortiz, Organometallics, 2009, 28, 1739–1747 CrossRef; (c) M. W. Johnson, A. G. DiPasquale, R. G. Bergman and F. D. Toste, Organometallics, 2014, 33, 4169–4172 CrossRef CAS PubMed; (d) J. F. Cui, H. M. Ko, K. P. Shing, J. R. Deng, N. C. H. Lai and M. K. Wong, Angew. Chem., Int. Ed., 2017, 56, 3074–3079 CrossRef CAS PubMed.
- For naphthyl-bridged (P,C) cyclometalated complexes, see: (a) J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778–1781 CrossRef CAS PubMed; (b) F. Rekhroukh, R. Brousses, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 1266–1269 CrossRef CAS PubMed; (c) F. Rekhroukh, L. Estevez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Organometallics, 2016, 35, 995–1001 CrossRef CAS; (d) F. Rekhroukh, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 11920–11929 CrossRef CAS PubMed; (e) F. Rekhroukh, C. Blons, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Chem. Sci., 2017, 8, 4539–4545 RSC; (f) H. Kawai, W. J. Wolf, A. G. DiPasquale, M. S. Winston and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 587–593 CrossRef CAS PubMed.
- E. Langseth, C. H. Görbitz, R. H. Heyn and M. Tilset, Organometallics, 2012, 31, 6567–6571 CrossRef CAS.
- See ESI.†.
- D. Balcells, O. Eisenstein, M. Tilset and A. Nova, Dalton Trans., 2016, 45, 5504–5513 RSC.
- Another minimum with ethylene oriented parallel to the coordination plane of gold was also located on the potential energy surface. It is less stable by 4.2 kcal mol−1.18.
- For π–arene interactions in Au(III) complexes, see ref. 16e and L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann, Chem. Commun., 2017, 53, 4358–4361 RSC.
- A γ-CH agostic complex has been characterized spectroscopically upon insertion of NB from the [(P,C)AuMe]+ complex: F. Rekhroukh, L. Estévez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2016, 55, 3414–3418 CrossRef CAS PubMed.
- (a) S. Noda, A. Nakamura, T. Kochi, L. W. Chung, K. Morokuma and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14088–14100 CrossRef CAS PubMed; (b) K. Nozaki, S. Kusumoto, S. Noda, T. Kochi, L. W. Chung and K. Morokuma, J. Am. Chem. Soc., 2010, 132, 16030–16042 CrossRef CAS PubMed; (c) Y. Mitsushige, B. P. Carrow, S. Ito and K. Nozaki, Chem. Sci., 2016, 7, 737–744 RSC; (d) R. Nakano, L. W. Chung, Y. Watanabe, Y. Okuno, Y. Okumura, S. Ito, K. Morokuma and K. Nozaki, ACS Catal., 2016, 6, 6101–6113 CrossRef CAS; (e) D. Guironnet, L. Caporaso, B. Neuwald, I. Göttker-Schnetmann, L. Cavallo and S. Mecking, J. Am. Chem. Soc., 2010, 132, 4418–4426 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental conditions and procedures, analytical data, theoretical details (PDF), crystallographic data for compounds 5A-H2O and 5A-Cl, and DFT optimized structures (XYZ). CCDC 1574199 and 1574200. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04899h |
| This journal is © The Royal Society of Chemistry 2018 |