
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of the lipid bilayer on energy transfer kinetics in the photosynthetic protein LH2†
John I.
Ogren‡
a,
Ashley L.
Tong‡
a,
Samuel C.
Gordon
a,
Aurélia
Chenu
a,
Yue
Lu
b,
Robert E.
Blankenship
b,
Jianshu
Cao
a and
Gabriela S.
Schlau-Cohen
 *a
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: gssc@mit.edu
bDepartment of Biology and Chemistry, Washington University in St. Louis, St. Louis, MO 63130, USA
First published on 9th February 2018
Abstract
Photosynthetic purple bacteria convert solar energy to chemical energy with near unity quantum efficiency. The light-harvesting process begins with absorption of solar energy by an antenna protein called Light-Harvesting Complex 2 (LH2). Energy is subsequently transferred within LH2 and then through a network of additional light-harvesting proteins to a central location, termed the reaction center, where charge separation occurs. The energy transfer dynamics of LH2 are highly sensitive to intermolecular distances and relative organizations. As a result, minor structural perturbations can cause significant changes in these dynamics. Previous experiments have primarily been performed in two ways. One uses non-native samples where LH2 is solubilized in detergent, which can alter protein structure. The other uses complex membranes that contain multiple proteins within a large lipid area, which make it difficult to identify and distinguish perturbations caused by protein–protein interactions and lipid–protein interactions. Here, we introduce the use of the biochemical platform of model membrane discs to study the energy transfer dynamics of photosynthetic light-harvesting complexes in a near-native environment. We incorporate a single LH2 from Rhodobacter sphaeroides into membrane discs that provide a spectroscopically amenable sample in an environment more physiological than detergent but less complex than traditional membranes. This provides a simplified system to understand an individual protein and how the lipid–protein interaction affects energy transfer dynamics. We compare the energy transfer rates of detergent-solubilized LH2 with those of LH2 in membrane discs using transient absorption spectroscopy and transient absorption anisotropy. For one key energy transfer step in LH2, we observe a 30% enhancement of the rate for LH2 in membrane discs compared to that in detergent. Based on experimental results and theoretical modeling, we attribute this difference to tilting of the peripheral bacteriochlorophyll in the B800 band. These results highlight the importance of well-defined systems with near-native membrane conditions for physiologically-relevant measurements.
1 Introduction
Photosynthesis powers most life on Earth and is responsible for the generation of over 100 gigatons of biomass on an annual basis.1,2 Purple non-sulfur bacteria are an important model system for understanding the mechanism of photosynthesis. These systems, and in particular, the photosynthetic apparatus of Rhodobacter sphaeroides (R. sphaeroides) are well studied and have provided deep insight into the photosynthetic machinery as well as biological energy transfer.3,4 In bacterial photosynthesis, Light-Harvesting Complex 2 (LH2) acts as the primary antenna protein, absorbing photons and initiating the photosynthetic process. The absorbed energy then migrates via both intra-protein and inter-protein energy transfer to reach Light-Harvesting Complex 1 (LH1), which funnels the energy to the site of charge separation, the Reaction Center (RC). Although vital to photosynthetic biomass production, the in vivo dynamics of the initial energy transfer steps within this network have not been determined due to the intrinsic difficulties associated with production, purification, and sample preparation of membrane proteins. Specifically, producing spectroscopically amenable samples that mimic the in vivo membrane architecture and environment has proven challenging.LH2 (Fig. 1A) is a nonamer of heterodimers.5,6 Each heterodimer consists of an α subunit and a β subunit, both containing a transmembrane alpha helix. The assembly of heterodimers produces a symmetrical, hollow, cylindrical structure. Each heterodimer contains three non-covalently bound bacteriochlorophyll a (Bchla) and one carotenoid. Two of the Bchla are held parallel to the normal of the membrane and form a pigment ring that absorbs at 850 nm (B850 band, red) and the third is held perpendicular to the membrane normal forming a pigment ring absorbing at 800 nm (B800 band, blue). Energy transfer occurs within the B800 band, within the B850 band, and from the B800 to B850 band. The B850 band has a higher lying set of excited states known as B850* that absorb near 800 nm, overlapping with the B800 band.7–23 The overall bandwidth of the B850 excitons has been shown to extend from ∼760 nm to ∼865 nm at low temperatures.7 The absorption spectra of these LH2 bands can be seen in Fig. 1B.
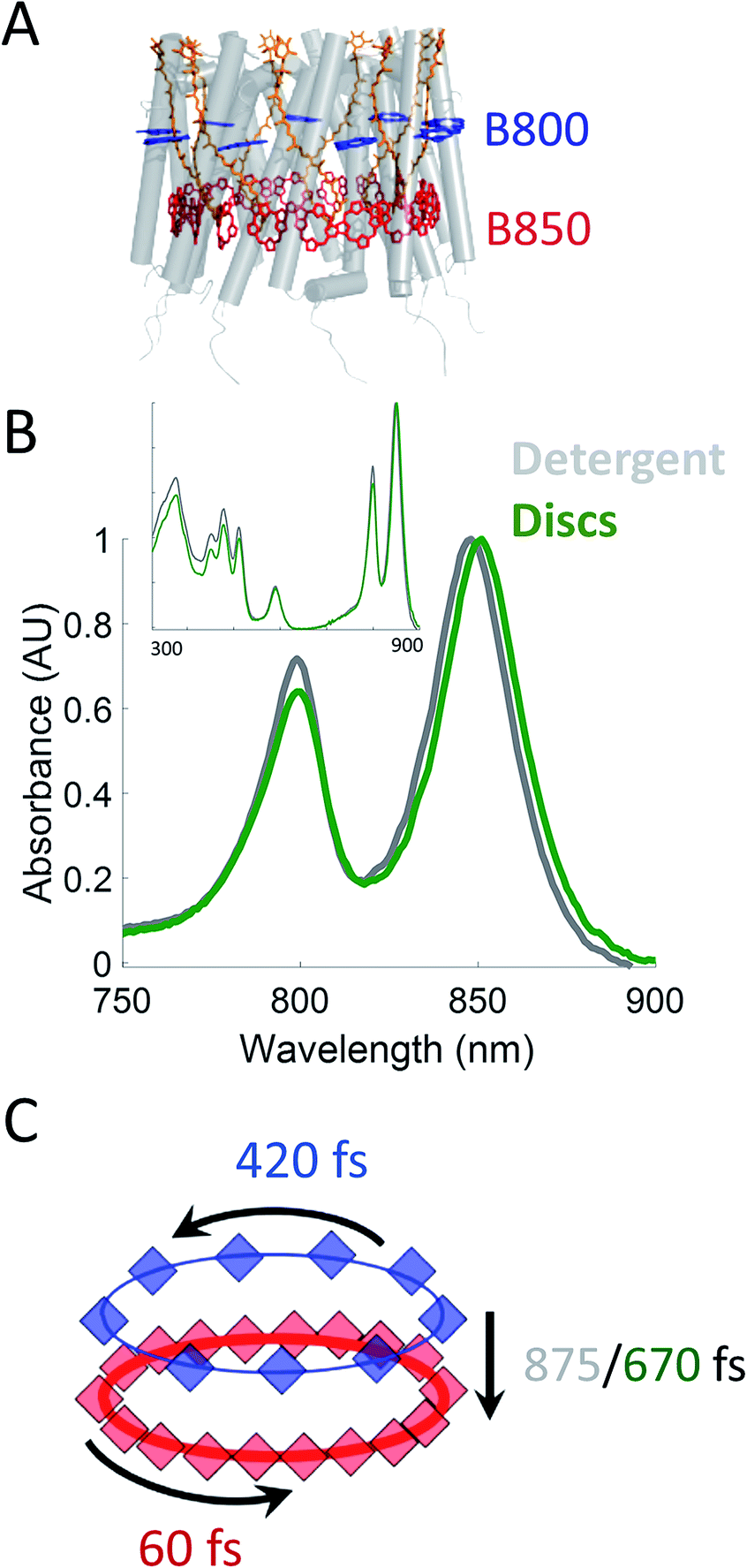 | ||
| Fig. 1 (A) Structural model (PDB 2FKW) of LH2 showing the alpha helical backbone (grey), the carotenoids (orange), the B800 band (blue), and the B850 band (red). (B) UV-VIS linear absorption spectra for LH2 in LDAO detergent (grey) and solubilized in DMPC membrane discs (green) in the B800/B850 region. The data is normalized to the B850 peak on the wavelength scale and shows a peak shift of the B850 band from 849 nm to 852 nm for LH2 in detergent and discs, respectively. Insert: the full spectrum of LH2 in detergent and discs from 250 to 900 nm showing the nearly identical structure of the two LH2 samples, independent of solubilization condition. (C) Time constants for energy transfer as measured by transient absorption spectroscopy on both the detergent solubilized LH2 and the membrane disc embedded LH2. Energy transfer within the B800 and B850 bands is similar for both samples (blue and red, respectively) but energy transfer between bands indicates structural differences induced by the membrane condition (detergent – grey; membrane discs – green). | ||
To date, there have been a significant number of studies using spectroscopic methods to determine energy transfer rates within LH2 and other components of the photosynthetic apparatus of purple non-sulfur bacteria.9,13,17,18,24–33 They have employed three predominant methodologies to solve the problems of protein production and purification as well as appropriate sample preparation. The first relies on detergent micelle formation surrounding the hydrophobic portion of the protein to produce a soluble protein–detergent complex.34,35 Although effective in solubilizing membrane proteins, the harsh conditions of detergents can produce drastic changes to protein structure, including loss or change of function as well as denaturation.36–38 The second method reconstitutes membrane proteins into lipid vesicles in vitro.14,39–41 Although this provides a hospitable membrane environment, these systems are heterogeneous in size, have varied and uncontrolled local membrane curvature, and are highly scattering, which can overwhelm spectroscopic signals. Furthermore, membrane vesicles often incorporate multiple proteins, which have been shown to introduce effects from protein–protein interactions that alter the dynamics in photosynthetic light-harvesting complexes.42 These effects illustrate that benchmarking the dynamics of individual light-harvesting complexes in the membrane environment requires a system with isolated proteins. The third method uses LH2-only chromatophores, membrane sections, or live cells.9,37,41,43 Similar to vesicles, this provides a hospitable membrane environment while also including effects from protein–protein interactions. Furthermore, the membrane morphology and composition can be heterogeneous or even unknown.44 Thus, the intrinsic energy transfer dynamics of LH2 in the membrane environment, without the effects of protein–protein interactions, have not yet been determined.
One emerging platform, membrane discs commonly known as ”nanodiscs”, overcomes the limitations of the sample preparation methods commonly employed by the spectroscopic community. These membrane discs provide a simplified near-native environment to solubilize individual membrane proteins.45–49 They are biochemically produced from subcomponents and are comprised of an amphiphilic belting protein, termed membrane scaffolding protein (MSP), a mixture of lipids used to form a bilayer, and the target membrane protein of interest.47,50,51 When mixed in precise stoichiometric ratios, these components self-assemble such that the MSP surrounds a lipid bilayer with the target membrane protein embedded.51 The resultant discs exhibit remarkably homogeneous diameters that can be straightforwardly characterized.50 The type of lipids used to form the discs can be selected for length and head group composition to best mimic the native environment. The lipid bilayer produced within the discs forms a natively flat landscape versus an irregularly curved environment as in the membrane vesicles.49 Furthermore, because the size of these discs is still small relative to the wavelengths of UV through near-IR light (typically 10 to 30 nm), these discs produce very little scattering, making the membrane proteins embedded within this platform amenable to spectroscopic studies.52 Finally, the number of proteins embedded can be controlled by the ratio of target protein to belting protein and the choice of belting protein, which determines disc size. Using a small disc size and substoichiometric ratios of target protein to belting protein allows a single protein to be incorporated into the membrane discs. Therefore, the membrane discs provide unprecedented control over and knowledge of the molecular and morphological properties. Specifically, this control over membrane disc composition allows the energy transfer dynamics of individual proteins to be explored without the complexity of protein–protein interactions.
In this work, we incorporate individual LH2s from R. sphaeroides into membrane discs to achieve both a spectroscopically viable sample and a simplified near-native membrane.47,53,54 To compare the effect of the near-native environment to the detergent-solubilized environment, we determine the intra-protein energy transfer rates using transient absorption spectroscopy and transient absorption anisotropy measurements for LH2 in both preparations. We find (Fig. 1C) that the intra-band energy transfer dynamics remain similar as a function of solubilization environment, at 420 fs and 60 fs within the B800 and the B850 bands, respectively. In contrast, we find a striking difference between the two solubilization conditions in the inter-band energy transfer rate. For this B800 to B850 energy transfer, the detergent solubilized and disc-embedded systems show rates of 875 and 670 fs, respectively, indicating a change in local environment of the pigments. Through these results, we demonstrate that membrane discs provide a new platform to explore the physiological dynamics of photosynthetic light-harvesting complexes.
2 Results and discussion
2.1 LH2 in membrane discs
As shown in Fig. 2, we have successfully expressed, assembled, and loaded a single LH2 into two types of membrane discs, one made from DMPC lipids and the other from POPC lipids (see ESI Fig. S4† for POPC). LH2 has also recently been inserted into discs made from DMPC for labeling-based studies.55 TEM characterization of the loaded discs demonstrates disc formation and that the discs contain only a single LH2 (Fig. 2A). Analysis of the TEM images showed a diameter of 20 ± 5 nm for >100 purified disc particles (Fig. 2B). The 10 nm diameter of LH2 prevents loading of two LH2 in discs of this size. Discs constructed with MSP1E3D1 exhibit a bimodal distribution of diameter, as characterized for empty discs in ESI Fig. S5.† The larger size of LH2 biases the bimodal distribution towards the observed larger disc diameter. Loaded discs are purified by size exclusion chromatography (Fig. 2C). Peak 2 in the trace contains LH2 discs. Loading was confirmed by SDS-PAGE on the fraction containing LH2, which showed bands for both LH2 and MSP1E3D1 (Fig. 2D).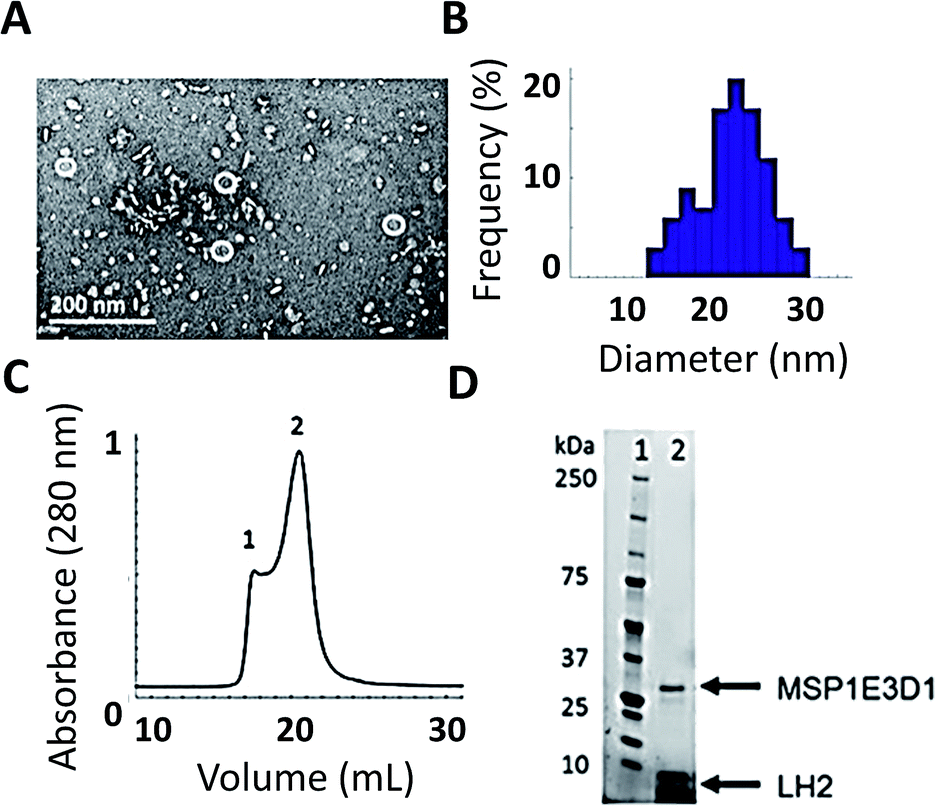 | ||
| Fig. 2 Successful incorporation of LH2 into membrane discs. (A) TEM image of FPLC purified LH2 discs. (B) TEM size distribution of >100 LH2 discs. Some 12 ± 2 nm empty discs remain after purification. LH2 discs are 20 ± 5 nm. (C) Size-exclusion chromatography from FPLC of LH2 discs. Peak 1 contains larger aggregates and peak 2 contains LH2 discs. (D) SDS-PAGE of peak 2 (C) from FPLC showing both the belting protein and LH2. | ||
The linear absorption spectra (Fig. 1B) show that the LH2 complexes maintain integrity within the disc. The same peaks are observed in detergent-solubilized and disc-embedded LH2, demonstrating that the structure of the protein is robust to the disc assembly process. However, the linear absorption spectra of LH2 in detergent and in discs shows a consistent shift of the B850 absorption peak from 849 nm in detergent to 852 nm in membrane discs, as previously reported.41,56 Pressure-dependent spectral shifts have also been previously reported.57 The electronic structure of the pigments is highly sensitive to the protein environment and so structural perturbations can alter the absorption spectra. The complex and varied interactions of membrane proteins with detergents are both protein and detergent specific. As a result, it is difficult to identify the molecular origin of spectral shifts that depend on solubilization environments. The 3 nm shift highlights that the local environment of the B850 pigments changes due to solubilization environment.
Time-correlated single-photon counting (TCSPC) was used to determine the fluorescence kinetics of detergent-solubilized and disc-embedded LH2 (ESI Fig. S9 and Table S3†). All four samples exhibit mono-exponential decays with a time constant of ∼1 ns, consistent with previous work.41 Fluorescence decays of detergent solubilized and membrane-embedded LH2 have previously been shown to have a mono-exponential decay and bi-exponential decay, respectively, where the bi-exponential decay exhibits shorter time constants.41,44 The shorter time constants are attributed to protein–protein energy transfer enabling exciton–exciton annihilation, which does not occur in the membrane discs. The mono-exponential decays observed here, therefore, are consistent with previous work and illustrate the utility of membrane discs as model systems for energy transfer kinetics of single LH2s in a near-physiological environment.
2.2 Transient absorption spectroscopy and transient absorption anisotropy
To probe the effect of solubilization environment on LH2, the energy transfer kinetics were measured in two detergents, LDAO and β-OG, and two lipid compositions of the membrane, DMPC and POPC. Detergents that have been shown to solubilize LH2 while maintaining the integrity of the complex are LDAO, DDM, and β-OG. LDAO and DDM are the most commonly used detergents, while β-OG is more rarely used. However, the detergent tails of LDAO and DDM are the same length, while the tail of β-OG is significantly shorter.5,43,58 Specifically, the hydrocarbon chain of β-OG is six carbons shorter than that of LDAO. β-OG also has a much bulkier head group than LDAO (ESI Fig. S8†). Therefore, LDAO and β-OG were selected to provide two distinct detergent tail lengths. The hydrocarbon chain of POPC is two carbons longer than that of DMPC, although both are similar lengths to the majority of native lipids in R. sphaeroides.Fig. 3A presents 800 nm pump–850 nm probe transient absorption spectra recorded with the pulse polarization set to the magic angle (MA = 54.7°) for all four samples. This pulse combination directly probes the time evolution of the population of states at 850 nm (in the B850 ring) after initial excitation of states at 800 nm (in the B800 ring). The spectra are fit to a sum of three exponentials, the results of which are shown in Table 1. The spectrum of LH2 in β-OG collapses to a sum of two exponentials, indicating different energy transfer pathways than the other three samples. For the three component spectra, the fast decay component is assigned to B850* to B850 and decays over the course of the first 0.5 ps, in line with previous experimental and theoretical work.12 The second decay is assigned to the transfer of population between the B800 and B850 Qy excited states (lowest lying energy states), directly reporting on inter-band energy transfer dynamics. According to previous work, transfer from B800 to B850 occurs directly and via B850* with approximately the same timescale.12 A 30% increase in the timescale of this decay component was observed in membrane discs, revealing a difference in B800 to B850 energy transfer between detergent-solubilized and membrane-reconstituted LH2 due to one or both of the energy transfer routes. Longer timescale processes, i.e. vibrational relaxation (Stokes' shift) and the decay back to the ground state, are collectively fit by the third time component of picoseconds.
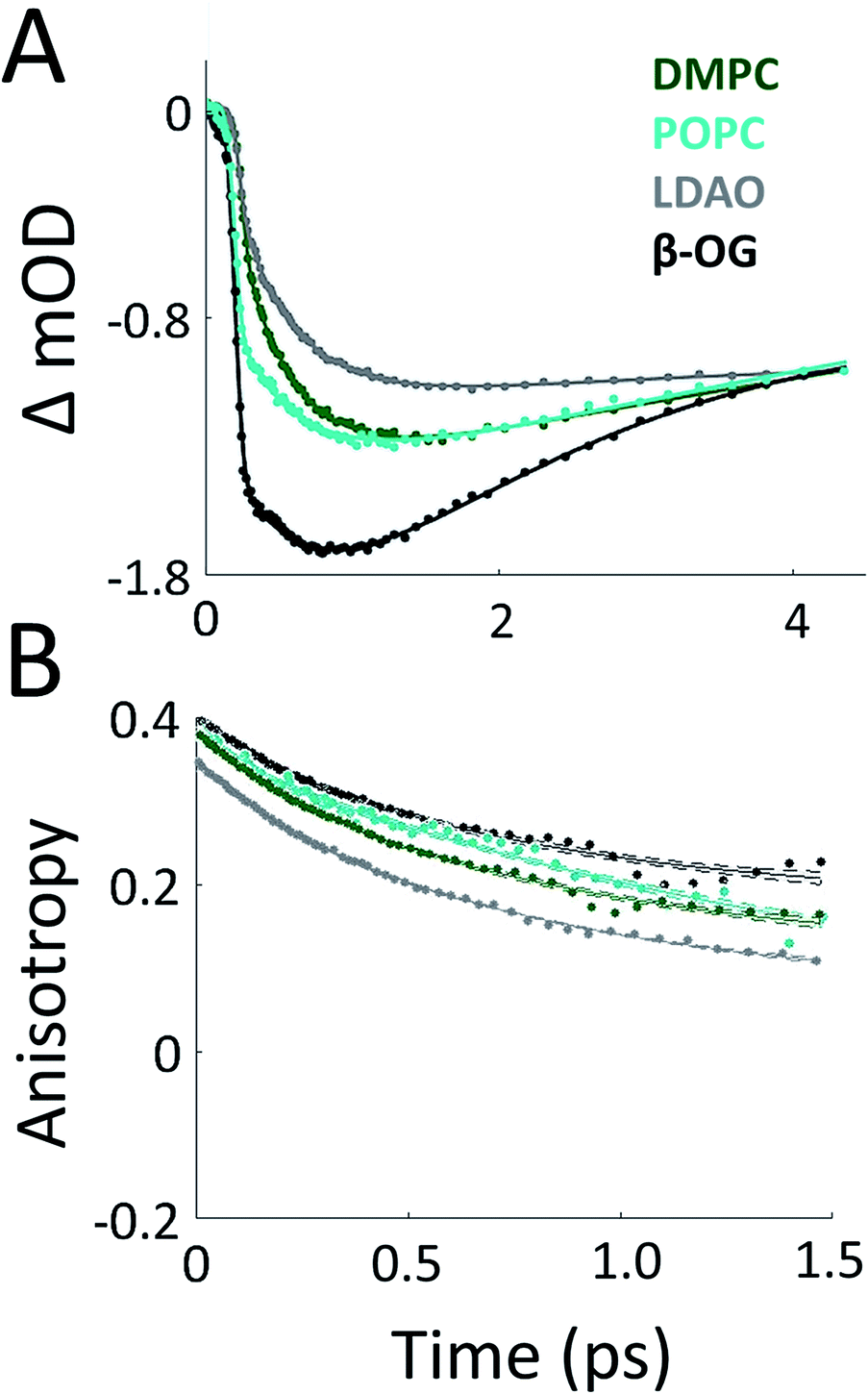 | ||
| Fig. 3 Transient absorption data of LH2 in different solubilization environments. (A) Magic angle 800 nm pump–850 nm probe transient absorption spectra for LH2 in DMPC membrane discs (green), POPC membrane discs (teal), LDAO detergent (grey), and β-OG detergent (black). (B) 800 nm pump–800 nm probe anisotropy for the four LH2 solubilization conditions. Data is shown as points and exponential fits are overlaid as lines with 95% confidence intervals indicated by the shaded region around each line. | ||
| 800 pump–850 probe | 800 pump–800 probe | |||
|---|---|---|---|---|
| τ | MA | τ | Anisotropy | |
| β-OG | 400 fs | 24% | 425 fs | 49% |
| 5 ps | −76% | >5 ns | 51% | |
| LDAO | 280 fs | 19% | 420 fs | 58% |
| 875 fs | 36% | >5 ns | 42% | |
| >10 ps | −45% | |||
| DMPC discs | 325 fs | 8% | 419 fs | 58% |
| 670 fs | 32% | >5 ns | 42% | |
| >10 ps | −60% | |||
| POPC discs | 325 fs | 16% | 413 fs | 43% |
| 670 fs | 15% | >5 ns | 57% | |
| 9 ps | −69% | |||
While the rate of energy transfer from B800 to B850 changes with local environment, the dynamics of energy transfer within both the B800 and B850 rings do not. Transient absorption anisotropy measurements decay due to the orientational change of the excitation as it transfers within the band. Thus, the experiment identifies and quantifies energy migration around the ring.
The 800 nm anisotropy (Fig. 3B) is initially fit to a sum of two exponential decays. The long time component (>5 ns) is then fixed as an offset to improve the fit for the short time decay to produce the timescales shown in Table 1. For all four LH2 solubilization conditions, these components are similar in time and relative weight. The faster decay component of ∼420 fs corresponds to energy transfer within the B800 ring. The 850 nm anisotropy is fit in the same manner as the 800 nm anisotropy with an additional decay component. The results are shown in ESI Fig. S6 and Table S1.† The data show a rapid decay component corresponding to energy transfer within the B850 ring of ∼55 or ∼65 fs for LH2 solubilized in LDAO and in DMPC discs, respectively. The energy transfer dynamics within the B850 ring are much faster because the pigments are much more strongly coupled.25 In addition to the <1 ps energy transfer dynamics discussed here, in all data sets there is a long (nanoseconds) component that arises from excited state decay and in the B850 anisotropy there is a picoseconds component that arises from vibrational relaxation.
The energy transfer rates are similar not only in DMPC and POPC discs, as observed here, but are also consistent with previously published experiments for LH2 in LH1-knockout R. sphaeroides.26 Collectively, these results suggest that protein–protein interactions are not impacting the photophysics, in contrast to green plants.42 Furthermore, the similarity in energy transfer rates illustrates that a membrane maintains a similar local environment for LH2 regardless of lipid composition. This is likely due to the structured assembly of a membrane where the acyl chains predominantly interact with the protein in a side-by-side orientation, thus decreasing lipid–pigment interactions. The additional two carbon chain length of the POPC versus DMPC (see ESI Fig. S8†) does not have a significant effect on energy transfer between the B800 and B850 bands. This result excludes the possibility that the membrane height is the driving factor behind the increase in the rate of energy transfer in the disc samples. Interestingly, although DMPC discs are in a structured gel phase while POPC discs are in a liquid crystalline phase at the temperature of our experiments, there is no change to the energy transfer rates. This suggests that membrane phase has little effect on the energy transfer rates, and thus on the protein structure. Furthermore, these results highlight that energy transfer dynamics are robust to the dynamic lipid composition of the purple bacterial membrane.
To determine the molecular origin of the effect of solubilization environment, the energy transfer rates within the B800 band and from B800 to the B850 band were calculated using generalized Förster theory as a function of tilt of the Bchla in the B800 band (see ESI 5†).60 Because the Bchla in the B800 band protrude from the protein scaffold and interact with the lipid or detergent used for solubilization, they are much more susceptible to perturbation due to solubilization environment than the Bchla in the B850 band. Fig. 4 shows the calculated relative energy transfer rates within the B800 band and between the B800 and B850 band as a function of the tilt of the Bchlas in the B800 band. These calculations determine relative rates, and so take into account all the experimental energy transfer steps, including those via the B850* states. The rate of energy transfer from B800 to B850 is much more sensitive (purple) than the rate within the B800 ring (blue). Flattening the B800 Bchlas by 2° causes an enhancement of the B800–B850 rate by more than 30%, while the B800 rate remains almost constant with less than 1% enhancement. The sharp dependence of the B800–B850 relative rate on the tilt angle arises from sensitivity to the distance between the coupled dipoles, and hence on how the tilt is simulated. It can become smoother by including disorder.61 These theoretical predictions are consistent with our experimental results, which find a change in B800 to B850 energy transfer but similar energy transfer rates within the B800 band in the anisotropy measurements at 800 nm as a function of solubilization environment (Table 1). This model would also predict similar energy transfer rates within the B850 band as a function of solubilization environment as observed in the short time component of the anisotropy measurements at 850 nm (ESI Table S1†). The small differences in anisotropy decay are changes on timescales comparable to the pulse duration and thus are not as reliable as the other measurements. Essentially, the B800 Bchla tilt towards the orientation of the B850 Bchla, which increases the inter-band coupling, speeding up the overall B800 to B850 energy transfer step including contributions from both B800 to B850 and B800 to B850*. Because the B800 Bchla tilt together, their orientation relative to each other is largely unchanged, which leaves the intra-band coupling the same.
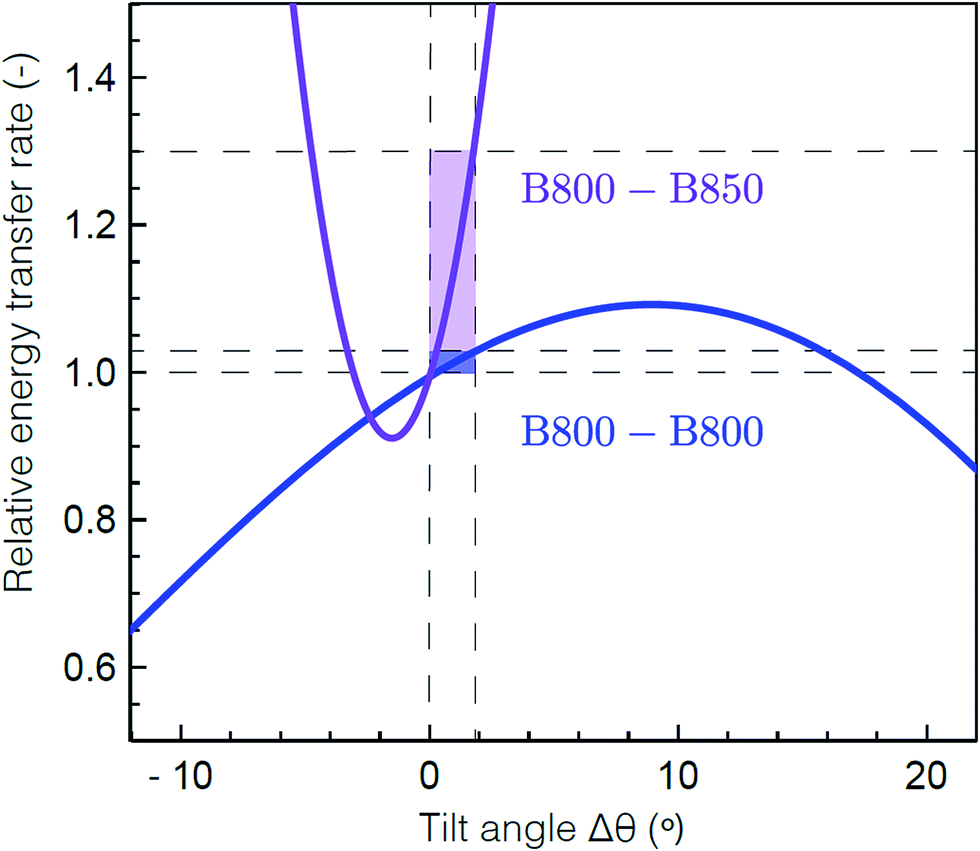 | ||
| Fig. 4 Theoretical energy transfer rates within LH2. The energy transfer rates within B800 (blue) and between B800 and B850 (purple) as a function of a tilt in the orientation of the B800 transition dipole moments relative to the original structure.59 A positive tilt results in a steady increase of the B800 rate from the original geometry, up to a maximum obtained for a tilt of Δθ = 9° corresponding to a flat B800 ring. The shaded domains illustrate the range for which the B800 rate increases by 1% (blue) and the corresponding, drastic change in the inter-ring transfer rate (purple). Flattening the B800 BChls by 2° enhances the B800–B850 rate by more than 30%. The presented B800–B850 rate displays the average rate from one B800 BChl to the six nearest B850 BChls. | ||
While the microscopic origin of the perturbation cannot be definitively determined, three possibilities are hydrophobic mismatch, lateral membrane pressure, or direct interaction of solubilizing environment with pigments. We consider these three physical processes. Hydrophobic mismatch occurs when the height of the membrane or detergent used does not match the hydrophobic region of the protein. In vivo, the hydrophilic headgroups of the bilayer interact with the hydrophilic protein regions, which are the N- and C-terminal regions of the β subunit in the case of LH2. The hydrophobic acyl chain region of the bilayer associates with the hydrophobic protein regions, which are the center of transmembrane alpha helices. The in vivo membrane height surrounding LH2 in R. sphaeroides is measured to be between 40 and 45 Å.62 To properly emulate the native environment, the solubilizing membrane or detergent should span a similar distance and have the hydrophilic and hydrophobic regions properly matched to the corresponding regions of the membrane protein.43,63 Hydrated DMPC and POPC (see ESI Fig. S8†) bilayers measure ∼44 Å in thickness, have a phosphatidylcholine headgroup, and a two acyl chain tail, which produce an environment that matches the native conditions.64 LDAO is an intermediate detergent with a weakly polar headgroup and a tail with 12 carbons.65 Although advantageous for solubilization, purification, and crystallization, LDAO produces micelles with a single acyl chain length of ∼15–16 Å.66,67 This reduced lipid height means that LDAO is shorter than the hydrophobic region of native LH2, producing a hydrophobic mismatch of ∼7 Å as measured from the crystal structure.59,65 Hydrophobic mismatch has been shown to alter membrane protein function, and thus is a likely candidate to change the tilt of the Bchla in the B800 band.68,69 Hydrophobic mismatch would also explain the greater perturbation of β-OG due to its even shorter hydrocarbon tail and bulkier head group.
The lateral membrane pressure profile is a second possible cause of the changing tilt of the B800 band Bchlas. A blue shift of the B850 band has been previously observed with increasing pressure.57 Here, we observe the same shift in moving from membrane solubilized to detergent solubilized LH2, as shown in Fig. 1B. In addition, only a very small shift is observed in the B800 band in either the membrane solubilized LH2 or the pressure-dependent absorption spectra. These similarities suggest that pressure may be the physical origin of the differences observed here. Furthermore, LH2 can induce curvature to the membrane that changes the lateral membrane pressure profile, as seen in previous work.70 Although difficult to measure in situ, the lateral pressure profile for membrane bilayers has been extensively studied using computational methods, which have included the effects on integral membrane proteins.71–74 In a lipid bilayer, such as the native environment or the lipid discs used in this work, the lateral stress profile has a positive (inward toward the membrane protein) pressure at the furthest extent due to the electrostatic interactions of the headgroups, a negative pressure due to the interfacial tension at the polar–apolar interface, and a positive pressure in the acyl chain region due to the repulsion between the hydrocarbon chains.75 In a protein–detergent system, it has been suggested that single chain, small headgroup detergents increase the pressure in the headgroup region while decreasing the pressure in the acyl chain relative to the bilayer system.76 The LDAO headgroup (see ESI Fig. S8†) is an amine oxide zwitterionic group which is small (2.8 nm2) compared to the phosphatidylcholine headgroup of the DMPC (5.7 nm2) and POPC (6.9 nm2).51 The β-OG headgroup is a glucoside that is nonionic, polar, and similar in size (∼5.1 nm2) compared to DMPC or POPC but is much bulkier due to its ring structure. In combination, LDAO's small headgroup, β-OG's bulkier headgroup, and the single acyl chain of detergents produce a very different intra-membrane region and thus a very different lateral membrane pressure profile than the DMPC or POPC discs.77
Finally, direct intermolecular interactions of single chain detergent molecules with pigments rather than the protein scaffold is a third possible cause of the changing tilt of the B800 band Bchlas. Similar effects have previously been reported in other photosynthetic systems (LHCII from Pisum sativum, peas) and perturbations in protein–pigment interactions due to detergent have been shown to alter the excited state lifetimes of LH2.78,79
Our data reveal that varying the solubilization environment alters the structure of LH2 in such a way as to increase the B800–B850 energy transfer rate. The energy transfer rates within individual proteins also determine the pathways of energy transfer between proteins to reach the reaction center. Energy transfer from one LH2 to another via the B850 rings is thought to occur on a 1–5 ps timescale.80 Currently, inter-protein energy transfer kinetics have been done in model systems that have multiple proteins incorporated and have multiple other processes occurring that may influence those kinetics. Because the B800 ring protrudes from the protein, energy transfer from one LH2 to another via the B800 rings may occur even faster than via the B850 rings. If the B800 to B850 energy transfer time were 875 fs, energy transfer from one LH2 to another via the B800 rings would be likely. However, we have determined the energy transfer time from B800 to B850 to be 670 fs in a near-native membrane environment, which means most energy transfer from one LH2 to another occurs via the B850 rings. While the timescales observed here suggest B850 to B850 transfer, further experiments are required to definitively determine the pathway of energy flow to the reaction center. The use of model membranes, as introduced here for studies of energy transfer in photosynthetic light harvesting, could be expanded to look at protein-to-protein energy transfer processes, such as LH2 to LH1.
3 Conclusion
Here, we have introduced the use of membrane discs as a tool to study the energy transfer dynamics of photosynthetic light-harvesting complexes in a near-native environment and demonstrated the utility of this approach for LH2. In contrast to traditional sample preparation methods, membrane discs allow the dynamics of individual proteins to be explored free from both non-physiological effects in detergent solubilization and the complications of protein–protein interactions. In LH2, one important energy transfer step, from the B800 to the B850 band, was found to be 30% faster in the membrane environment than in detergent, the typical experimental environment. Our experimental and theoretical results suggest that this change in dynamics is caused by an environment-induced tilt of the Bchla in the B800 band. Future structural studies may provide experimental evidence for this proposal. While X-ray crystallography relies on detergent-solubilized protein, nuclear magnetic resonance or circular dichroism could confirm the tilt of the B800 BChla as the structural change upon membrane incorporation, similar to approaches used to observe other structural changes.53,81–84Overall, our results highlight that detergent solubilization introduces non-physiological effects on dynamics in membrane proteins. The impact of detergent solubilization observed here suggests that detergent solubilization may similarly perturb the behavior of other membrane proteins, such as enzymes, photoreceptors, and ion channels. Furthermore, these results open the possibility to manipulate the membrane environment to produce systems with desired activity or functionality.
4 Experimental
4.1 Purification of MSP1E3D1
Over-expression of disc belting protein MSP1E3D1 was adapted from previously reported protocols.50,51 The plasmid for the belting protein MSP1E3D1 was acquired from Addgene (plasmid #20066) containing a 6X histidine tag and kanamyacin antibiotic resistance. MSP1E3D1 was transformed into One Shot BL21 Star (DE3) Chemically Competent E. coli (ThermoFisher Scientific #C601003) and glycerol stocks were made by flash freezing in liquid nitrogen and storing at −80 °C. Over-expression was carried out on a shaker at 37 °C. A 10 mL starter culture of Luria Bertani Broth was inoculated with the glycerol stock and grown overnight. The starter culture was used to inoculate 1 L of terrific broth. Protein over-expression was induced between 0.6 and 0.8 OD (at 600 nm) with a final concentration of 1 mM isopropyl β-D-1-thiogalactopyranoside. Cells were allowed to produce protein for five hours. The cell pellet was collected by centrifuging at 3k rpm at 4 °C for 20 minutes. The supernatant was discarded and the cell pellet was resuspended in 25 mL of 20 mM Tris, 150 mM NaCl, pH 7.4. Phenylmethylsulfonyl fluoride was added to a final concentration of 1 mM. Triton X-100 was added to 0.1% v/v. DNase I (New England Biolabs) was added (300 units) with a final concentration of 2.5 mM for magnesium chloride and 0.5 mM calcium chloride. Solution was homogenized in a tissue homogenizer. Solution was probe sonicated on ice for 3 minutes at 30% power for 30 seconds on and 30 seconds off. Solution was centrifuged at 4k rpm at 4 °C for one hour to remove cellular debris. The supernatant lysate was loaded onto a 25 mL Ni-NTA chromatography column and allowed to equilibrate with the beads on a nutating mixer at 4 °C overnight. Protein purification was carried out by previously established protocols.50,51 Protein mass was verified by reverse-phase liquid-chromatography mass-spectrometry (LC-MS). Purity was determined by denaturing gel electrophoresis (SDS-PAGE). To ensure there was no nucleic acid contamination, the 260/280 nm ratio was inspected by UV-VIS. Protein was concentrated to ∼500 μM and aliquoted into 50 μL aliquots, flash frozen in liquid nitrogen, and stored at −80 °C for later experiments.4.2 Lipid preparation
1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) were purchased from Avanti Polar lipids (#850345C and #850457, respectively). Chloroform was evaporated under a gentle stream of argon gas and desiccated overnight under vacuum. Lipid was resuspended in 20 mM Tris, 150 mM NaCl, 200 mM sodium cholate, pH 7.4 and sonified in a Branson Ultrasonic Bath until solution was clear. The concentration of lipid was determined by a phosphorus assay as previously reported.85 Lipids were degassed of oxygen to prevent oxidation by bubbling in nitrogen, aliquoting, freezing in liquid nitrogen, and then storing at −80 °C for later experiments.4.3 LH2 preparation
Production and purification of LH2 were performed similarly to the methodology used by Cogdell et.al.86 Wild type R. sphaeroides ATCC2.4.1 was provided by the Blankenship lab and single colonies were used to inoculate 1.6 L of growth media (1% w/w tryptone, 0.5% w/w yeast extract, 4 mM NaCl, 0.5 mM CaCl2, and 0.8 mM MgSO4). The culture was grown anaerobically for 3 days at approximately 30 °C with constant illumination from a 100 W incandescent light bulb. Cells were pelleted at 4k rpm at 4 °C and the supernatant was discarded. The pellet was resuspended in sonication buffer (20 mM Tris, 100 mM NaCl, 1 mM MgSO4, pH 7.5), and the cells were lysed using ten 1 minute on–1 minute off probe sonication cycles with the sample in an ice bath. The lysate was centrifuged for 8 hours at 28k rpm and the pellet resuspended in a minimal volume of solubilization buffer (20 mM Tris, pH 7.5). Linear absorption at 850 nm was used to quantify the concentration of LH2 and the concentration was adjusted with solubilization buffer to approximately 100 OD at 850 nm.87 Lauryldimethylamine oxide (LDAO, Sigma Aldrich) was added, dropwise, while stirring, up to 1.5% (w/w) and the sample was homogenized on ice using a plunger homogenizer. Once homogenized, the sample was diluted with solubilization buffer to 0.1% LDAO and centrifuged for 4 hours at 28k rpm.The supernatant, containing the detergent solubilized LH2, was loaded onto an ion exchange chromatography column (HiPrep DEAE FF 16/10, GE HealthSciences) and eluted with 20 mM Tris, 0.1% LDAO, pH 8 with a linear gradient from 0 to 400 mM NaCl over 18 column volumes (360 mL) at 4 °C collecting 4 mL fractions. Fractions were combined based on their UV-VIS spectrum specifically separating the LH2 from reaction center and the fractions were concentrated to <5 mL with 30 kDa filters and centrifugation at 4k rpm. The concentrated LH2 was loaded onto a gel filtration column (HiPrep Sephacryl S-200 HR, GE HealthSciences) and eluted with 20 mM Tris, 100 mM NaCl, 0.1% LDAO, pH 8 buffer at 4 °C collecting 1 mL fractions. Fractions were selected based on LH2 content, purity was determined by SDS-PAGE, and LH2 was concentrated to approximately 20 uM. 0.01% sodium azide was added to purified LH2, aliquoted to 100 μL volumes, flash frozen with liquid nitrogen, and stored at −80 °C.
4.4 LH2 disc assembly
Loaded discs were produced using a construct from the common apolipoprotein ApoA1, MSP1E3D1.45,50 MSP1E3D1, DMPC/POPC, and LH2 from R. sphaeroides were mixed together at ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :131:0.125. These ratios were selected to yield discs that were 1/4 filled with LH2 to prevent contamination of LH2 not embedded in the disc or multiply embedded proteins. The reaction was allowed to incubate on a rocker for 1 h at room temperature for DMPC and 4 °C for POPC. Bio-Beads SM-2 Resin was added to 2/3 the volume of the reaction. Bio-Beads were allowed to incubate with the reaction on a rocker for 1 h at room temperature for DMPC and 4 °C for POPC and then overnight at 4 °C for both to ensure efficient detergent removal. Bio-Beads were removed through centrifugation. The reaction was purified by the 6× histidine tag of the belting protein on a 1 mL Ni-NTA column to remove any LH2 not incorporated into discs. The reaction was allowed to equilibrate with the beads for 1 h at 4 °C on a nutating mixer. Column flow-through was collected and the column was washed with 1 mL of 40 mM Tris, 300 mM NaCl, 20 mM imidazole, pH 8.0 three times. Discs were eluted with 40 mM Tris, 300 mM NaCl, 400 mM NaCl, pH 8.0. Fractions containing discs, as determined by SDS-PAGE, were dialyzed against 20 mM Tris, 100 mM NaCl, 0.5 mM EDTA, pH 7.4 to remove imidazole. Determined by SDS-PAGE, no excess LH2 was present in the flow-through or washes, indicating 100% incorporation of LH2 into discs.
:131:0.125. These ratios were selected to yield discs that were 1/4 filled with LH2 to prevent contamination of LH2 not embedded in the disc or multiply embedded proteins. The reaction was allowed to incubate on a rocker for 1 h at room temperature for DMPC and 4 °C for POPC. Bio-Beads SM-2 Resin was added to 2/3 the volume of the reaction. Bio-Beads were allowed to incubate with the reaction on a rocker for 1 h at room temperature for DMPC and 4 °C for POPC and then overnight at 4 °C for both to ensure efficient detergent removal. Bio-Beads were removed through centrifugation. The reaction was purified by the 6× histidine tag of the belting protein on a 1 mL Ni-NTA column to remove any LH2 not incorporated into discs. The reaction was allowed to equilibrate with the beads for 1 h at 4 °C on a nutating mixer. Column flow-through was collected and the column was washed with 1 mL of 40 mM Tris, 300 mM NaCl, 20 mM imidazole, pH 8.0 three times. Discs were eluted with 40 mM Tris, 300 mM NaCl, 400 mM NaCl, pH 8.0. Fractions containing discs, as determined by SDS-PAGE, were dialyzed against 20 mM Tris, 100 mM NaCl, 0.5 mM EDTA, pH 7.4 to remove imidazole. Determined by SDS-PAGE, no excess LH2 was present in the flow-through or washes, indicating 100% incorporation of LH2 into discs.
LH2 discs were further purified by fast protein liquid chromatography (FPLC) with a BioLogic DuoFlow (Bio-Rad) on a Superdex 200 Increase 10/300 GL (GE Healthcare Lifesciences) at a flow rate of 0.75 mL min−1 with 20 mM Tris, 150 mM NaCl, pH 7.4. Fractions of main peaks were collected and analyzed by SDS-PAGE, linear absorption, and transmission electron microscopy (TEM) to identify the peak containing LH2 discs. The optimal lipid ratio was determined experimentally by maximizing the LH2 disc FPLC peak, which was found from characterization.
TEM samples were prepared on 400-mesh Cu–carbon coated films (Electron Microscopy Sciences) that were negatively glow-discharged. 5 μL of sample with appropriate dilution (1:20 after FPLC) was added to grid for 1 minute. Excess sample was removed with a Kimwipe from the edge to prevent deposition of fibers onto the grid. 5 μL of 2% uranyl acetate in water was added for 30 seconds. Excess stain was removed similarly to the sample. Samples were allowed to air dry for at least 1 hour. Samples were imaged on a FEI Tecnai (G2 Spirit TWIN) at 120 kV. The distribution of disc sizes was analyzed using more than 100 particles by ImageJ software (https://imagej.nih.gov).
4.5 Sample preparation for ultrafast spectroscopy
After purification, the LH2 discs were diluted to a final volume concentration of ∼500 nM corresponding to a visible absorption of ∼1.5 OD at 850 nm in 20 mM Tris, 150 mM NaCl, pH 7.4. For detergent solubilized samples, LH2 in LDAO was taken from stocks and diluted to a similar concentration in 20 mM Tris, 150 mM NaCl, pH 7.4, 0.1% LDAO. LH2 in LDAO was buffer exchanged by centrifugation into 0.75% β-OG over the course of 12 hours with an exchange every 20 minutes. The linear absorption was measured for both the flow through and the sample during buffer exchange to ensure there was no loss of the Bchls in the B800 band by monitoring for the appearance of a Bchl peak at 775 nm. A peristaltic pump flowed the sample being interrogated through a 1 mm path length flow cell (Starna) at >4 mL s−1, which ensures a new sample of protein for every laser shot. During acquisition, samples were stored on ice. Linear absorbance spectra were acquired pre and post-acquisition to monitor for degradation.4.6 Transient absorption and transient anisotropy measurements
The transient absorption apparatus is described in detail in the ESI.† Transient absorption measurements were performed for 800 nm pump–850 nm probe for all four samples. The 800 nm pulse was the direct output of the regenerative amplifier system that operates at a 5 kHz repetition rate. At the sample position the pulse duration was measured by SHG-FROG to be <45 fs with ∼30 nm bandwidth FWHM. White light supercontinuum was generated by focusing a portion of the regenerative amplifier output through argon.88,89 The white light was compressed and spectrally filtered in a prism compressor and the resulting pulse was centered at 850 nm with a bandwidth of ∼35 nm, <45 fs pulse duration. The power was adjusted such that the pump was ∼50 nJ per pulse and the probe was ∼2 nJ per pulse using neutral density filters and a waveplate–polarizer pair that also served to set the relative polarizations between the pump and probe. In the anisotropy experiments, simultaneously collected parallel (V–V) and perpendicular (V–H) transient absorption data were used to compute the numerator (N) and the denominator (D) of the anisotropy given by N = ΔAV–V − ΔAV–H and D = ΔAV–V + 2ΔAV–H. These functions were globally fit to extract the anisotropy decay rates and relative weight of each decay component.90 For transient anisotropy measurements with 800 nm pump–800 nm probe and 850 nm pump–850 nm probe, the probe path polarizer was set to 45° relative to the pump path polarizer. The transmitted probe pulse was then split into vertical and horizontal components with a polarizing beamsplitter and the individual components were detected on photodiodes with each output sent to separate lock-in amplifiers and detected with two GPIB channels.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based on work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences under Award Number DE-SC0018097 (to G. S. S.-C.). The authors also thank the Singapore-MIT alliance for Research and Technology for funding and the MIT Center for Materials Science and Engineering Shared Experimental Facilities for providing the TEM instrument.References
- J. Whitmarsh and J. Govindjee, Concepts in Photobiology: Photosynthesis and Photomorphogenesis, 1999, pp. 11–51 Search PubMed.
- R. E. Blankenship, Photosynth. Res., 1992, 33, 91–111 CrossRef CAS PubMed.
- R. van Grondelle, J. P. Dekker, T. Gillbro and V. Sundstrom, Biochim. Biophys. Acta, Bioenerg., 1994, 1187, 1–65 CrossRef.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 19–205 CrossRef.
- G. McDermott, S. Prince, A. Freer and A. Hawthornthwaite-Lawless, et al. , Nature, 1995, 374, 517 CrossRef CAS.
- T. Walz, S. J. Jamieson, C. M. Bowers, P. A. Bullough and C. N. Hunter, J. Mol. Biol., 1998, 282, 833–845 CrossRef CAS PubMed.
- K. Timpmann, G. Trinkunas, J. D. Olsen, C. N. Hunter and A. Freiberg, Chem. Phys. Lett., 2004, 398, 384–388 CrossRef CAS.
- M. Koolhaas, R. Frese, G. Fowler, T. Bibby, S. Georgakopoulou, G. Van der Zwan, C. Hunter and R. Van Grondelle, Biochemistry, 1998, 37, 4693–4698 CrossRef CAS PubMed.
- P. D. Dahlberg, G. J. Norris, C. Wang, S. Viswanathan, V. P. Singh and G. S. Engel, J. Chem. Phys., 2015, 143(10), 101101 CrossRef PubMed.
- H.-M. Wu, S. Savikhin, N. Reddy, R. Jankowiak, R. Cogdell, W. Struve and G. Small, J. Phys. Chem., 1996, 100, 12022–12033 CrossRef CAS.
- M. Rätsep, C. N. Hunter, J. D. Olsen and A. Freiberg, Photosynth. Res., 2005, 86, 37–48 CrossRef PubMed.
- V. Novoderezhkin, M. Wendling and R. Van Grondelle, J. Phys. Chem. B, 2003, 107, 11534–11548 CrossRef CAS.
- J. Trautman, A. Shreve, C. Violette, H. A. Frank, T. Owens and A. Albrecht, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 215–219 CrossRef CAS.
- A. F. Fidler, V. P. Singh, P. D. Long, P. D. Dahlberg and G. S. Engel, J. Chem. Phys., 2013, 139, 10B614_1 CrossRef PubMed.
- H. Van der Laan, T. Schmidt, R. Visschers, K. Visscher, R. Van Grondelle and S. Völker, Chem. Phys. Lett., 1990, 170, 231–238 CrossRef CAS.
- R. Monshouwer, I. O. de Zarate, F. van Mourik and R. van Grondelle, Chem. Phys. Lett., 1995, 246, 341–346 CrossRef CAS.
- S. Hess, E. Akesson, R. Cogdell, T. Pullerits and V. Sundström, Biophys. J., 1995, 69, 2211–2225 CrossRef CAS PubMed.
- T. Joo, Y. Jia, J.-Y. Yu, D. M. Jonas and G. R. Fleming, J. Phys. Chem., 1996, 100, 2399–2409 CrossRef CAS.
- J. Herek, N. Fraser, T. Pullerits, P. Martinsson, T. Polivka, H. Scheer, R. Cogdell and V. Sundström, Biophys. J., 2000, 78, 2590–2596 CrossRef CAS PubMed.
- S. Jang and R. J. Silbey, J. Phys. Chem., 2003, 118, 9324–9336 CrossRef CAS.
- V. Nagarajan, R. Alden, J. Williams and W. Parson, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13774–13779 CrossRef CAS.
- H.-M. Wu, M. Ratsep, I.-J. Lee, R. Cogdell and G. Small, J. Phys. Chem. B, 1997, 101, 7654–7663 CrossRef CAS.
- C. Hofmann, T. J. Aartsma and J. Köhler, Chem. Phys. Lett., 2004, 395, 373–378 CrossRef CAS.
- E. Harel and G. S. Engel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 706–711 CrossRef CAS PubMed.
- V. Sundström, T. Pullerits and R. van Grondelle, Photosynthetic light-harvesting: reconciling dynamics and structure of purple bacterial LH2 reveals function of photosynthetic unit, J. Phys. Chem. B, 1999, 103(13), 2327–2346 CrossRef.
- S. Hess, M. Chachisvilis, K. Timpmann, M. Jones, G. Fowler, C. Hunter and V. Sundström, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 12333–12337 CrossRef CAS.
- J. T. Kennis, A. M. Streltsov, S. I. Vulto, T. J. Aartsma, T. Nozawa and J. Amesz, J. Phys. Chem. B, 1997, 101, 7827–7834 CrossRef CAS.
- R. Van Grondelle, H. Bergström, V. Sundström and T. Gillbro, Biochim. Biophys. Acta, Bioenerg., 1987, 894, 313–326 CrossRef CAS.
- S. Hess, F. Feldchtein, A. Babin, I. Nurgaleev, T. Pullerits, A. Sergeev and V. Sundström, Chem. Phys. Lett., 1993, 216, 247–257 CrossRef CAS.
- T. J. Pflock, S. Oellerich, J. Southall, R. J. Cogdell, G. M. Ullmann and J. Köhler, J. Phys. Chem. B, 2011, 115, 8813–8820 CrossRef CAS PubMed.
- K. Timpmann, N. W. Woodbury and A. Freiberg, J. Phys. Chem. B, 2000, 104, 9769–9771 CrossRef CAS.
- A. Shreve, J. Trautman, H. A. Frank, T. Owens and A. Albrecht, Biochim. Biophys. Acta, Bioenerg., 1991, 1058, 280–288 CrossRef CAS.
- K. Mukai, S. Abe and H. Sumi, J. Phys. Chem. B, 1999, 103, 6096–6102 CrossRef CAS.
- A. M. Seddon, P. Curnow and P. J. Booth, Biochim. Biophys. Acta, Biomembr., 2004, 1666, 105–117 CrossRef CAS PubMed.
- C. Tanford and J. A. Reynolds, Biochim. Biophys. Acta, Rev. Biomembr., 1976, 457, 133–170 CrossRef CAS.
- N. J. Fraser, P. J. Dominy, B. Ücker, I. Simonin, H. Scheer and R. J. Cogdell, Biochemistry, 1999, 38, 9684–9692 CrossRef CAS PubMed.
- A. Freiberg, M. Rätsep and K. Timpmann, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1471–1482 CrossRef CAS PubMed.
- E. London and H. G. Khorana, J. Biol. Chem., 1982, 257, 7003–7011 CAS.
- L. T. Mimms, G. Zampighi, Y. Nozaki, C. Tanford and J. A. Reynolds, Biochemistry, 1981, 20, 833–840 CrossRef CAS PubMed.
- J.-L. Rigaud, Braz. J. Med. Biol. Res., 2002, 35, 753–766 CrossRef CAS PubMed.
- T. Pflock, M. Dezi, G. Venturoli, R. J. Cogdell, J. Köhler and S. Oellerich, Photosynth. Res., 2008, 95, 291–298 CrossRef CAS PubMed.
- I. Moya, M. Silvestri, O. Vallon, G. Cinque and R. Bassi, Biochemistry, 2001, 40, 12552–12561 CrossRef CAS PubMed.
- J. D. Olsen, J. D. Tucker, J. A. Timney, P. Qian, C. Vassilev and C. N. Hunter, J. Biol. Chem., 2008, 283, 30772–30779 CrossRef CAS PubMed.
- C. N. Hunter, H. Bergstroem, R. Van Grondelle and V. Sundstroem, Biochemistry, 1990, 29, 3203–3207 CrossRef CAS PubMed.
- T. H. Bayburt and S. G. Sligar, FEBS Lett., 2010, 584, 1721–1727 CrossRef CAS PubMed.
- I. Denisov, Y. Grinkova, A. Lazarides and S. Sligar, J. Am. Chem. Soc., 2004, 126, 3477–3487 CrossRef CAS PubMed.
- A. Nath, W. M. Atkins and S. G. Sligar, Biochemistry, 2007, 46, 2059–2069 CrossRef CAS PubMed.
- T. Boldog, S. Grimme, M. Li, S. G. Sligar and G. L. Hazelbauer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11509–11514 CrossRef CAS PubMed.
- I. G. Denisov and S. G. Sligar, Chem. Rev., 2017, 117, 4669–4713 CrossRef CAS PubMed.
- T. H. Bayburt, Y. V. Grinkova and S. G. Sligar, Nano Lett., 2002, 2, 853–856 CrossRef CAS.
- T. Ritchie, Y. Grinkova, T. Bayburt, I. Denisov, J. Zolnerciks, W. Atkins and S. Sligar, Methods Enzymol., 2009, 464, 211–231 CAS.
- R. Zhang, I. D. Sahu, L. Liu, A. Osatuke, R. G. Comer, C. Dabney-Smith and G. A. Lorigan, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 329–333 CrossRef CAS PubMed.
- F. Hagn, M. Etzkorn, T. Raschle and G. Wagner, J. Am. Chem. Soc., 2013, 135, 1919–1925 CrossRef CAS PubMed.
- T. H. Bayburt, Y. V. Grinkova and S. G. Sligar, Arch. Biochem. Biophys., 2006, 450, 215–222 CrossRef CAS PubMed.
- Y. Lu, H. Zhang, D. M. Niedzwiedzki, J. Jiang, R. E. Blankenship and M. L. Gross, Anal. Chem., 2016, 88, 8827–8834 CrossRef CAS PubMed.
- M. F. Richter, J. Baier, R. J. Cogdell, J. Köhler and S. Oellerich, Biophys. J., 2007, 93, 183–191 CrossRef CAS PubMed.
- K. Timpmann, A. Ellervee, T. Pullerits, R. Ruus, V. Sundström and A. Freiberg, J. Phys. Chem. B, 2001, 105, 8436–8444 CrossRef CAS.
- L.-N. Liu, T. J. Aartsma and R. N. Frese, FEBS J., 2008, 275, 3157–3166 CrossRef CAS PubMed.
- M. Z. Papiz, S. M. Prince, T. Howard, R. J. Cogdell and N. W. Isaacs, J. Mol. Biol., 2003, 326, 1523–1538 CrossRef CAS PubMed.
- S. Jang, M. D. Newton and R. J. Silbey, Phys. Rev. Lett., 2004, 92, 218301 CrossRef PubMed.
- L. Cleary and J. Cao, New J. Phys., 2013, 15, 125030 CrossRef.
- S. Bahatyrova, R. N. Frese, A. Siebert, J. D. Olsen, K. O. van der Werf, R. van Grondelle, R. A. Niederman, P. A. Bullough, C. Otto and C. N. Hunter, Nature, 2004, 430, 1058–1062 CrossRef CAS PubMed.
- T. Yeates, H. Komiya, D. Rees, J. Allen and G. Feher, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 6438–6442 CrossRef CAS.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, Rev. Biomembr., 2000, 1469, 159–195 CrossRef CAS.
- V. Cherezov, J. Clogston, M. Z. Papiz and M. Caffrey, J. Mol. Biol., 2006, 357, 1605–1618 CrossRef CAS PubMed.
- P. Timmins, M. Leonhard, H. Weltzien, T. Wacker and W. Welte, FEBS Lett., 1988, 238, 361–368 CrossRef CAS.
- M. Roth, A. Lewit-Bentley, H. Michel, J. Deisenhofer, R. Huber and D. Oesterhelt, Nature, 1989, 340, 659–662 CrossRef CAS.
- J. Lundbaek, P. Birn, J. Girshman, A. Hansen and O. Andersen, Biochemistry, 1996, 35, 3825–3830 CrossRef CAS PubMed.
- M. F. Brown, Chem. Phys. Lipids, 1994, 73, 159–180 CrossRef CAS PubMed.
- M. Şener, J. Strümpfer, J. A. Timney, A. Freiberg, C. N. Hunter and K. Schulten, Biophys. J., 2010, 99, 67–75 CrossRef PubMed.
- E. Lindahl and O. Edholm, J. Chem. Phys., 2000, 113, 3882–3893 CrossRef CAS.
- D. Harries and A. Ben-Shaul, J. Chem. Phys., 1997, 106, 1609–1619 CrossRef CAS.
- R. S. Cantor, Biochemistry, 1997, 36, 2339–2344 CrossRef CAS PubMed.
- R. S. Cantor, Biophys. J., 2002, 82, 2520–2525 CrossRef CAS PubMed.
- J. A. Killian and B. de Kruijff, et al. , Biochim. Biophys. Acta, Biomembr., 2004, 1666, 275–288 CrossRef PubMed.
- S. Scarlata, Braz. J. Med. Biol. Res., 2005, 38, 1203–1208 CrossRef CAS PubMed.
- M. R. Wenk, T. Alt, A. Seelig and J. Seelig, Biophys. J., 1997, 72, 1719–1731 CrossRef CAS PubMed.
- P. Akhtar, M. Dorogi, K. Pawlak, L. Kovács, A. Bóta, T. Kiss, G. Garab and P. H. Lambrev, J. Biol. Chem., 2015, 290, 4877–4886 CrossRef CAS PubMed.
- X.-H. Chen, L. Zhang, Y.-X. Weng, L.-C. Du, M.-P. Ye, G.-Z. Yang, R. Fujii, F. S. Rondonuwu, Y. Koyama and Y.-S. Wu, et al. , Biophys. J., 2005, 88, 4262–4273 CrossRef CAS PubMed.
- R. J. Cogdell, A. Gall and J. Köhler, Q. Rev. Biophys., 2006, 39, 227–324 CrossRef CAS PubMed.
- A. Pandit, M. Reus, T. Morosinotto, R. Bassi, A. R. Holzwarth and H. J. de Groot, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 738–744 CrossRef CAS PubMed.
- E. Crisafi and A. Pandit, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 40–47 CrossRef CAS PubMed.
- A. Pandit, N. Shirzad-Wasei, L. M. Wlodarczyk, H. van Roon, E. J. Boekema, J. P. Dekker and J. Willem, Biophys. J., 2011, 101, 2507–2515 CrossRef CAS PubMed.
- S. Georgakopoulou, R. N. Frese, E. Johnson, C. Koolhaas, R. J. Cogdell, R. van Grondelle and G. van der Zwan, Biophys. J., 2002, 82, 2184–2197 CrossRef CAS PubMed.
- T. Boldog, M. Li and G. L. Hazelbauer, Methods Enzymol., 2007, 423, 317–335 CAS.
- R. J. Cogdell, I. Durant, J. Valentine, J. G. Lindsay and K. Schmidt, Biochim. Biophys. Acta, Bioenerg., 1983, 722, 427–435 CrossRef CAS.
- Y. Saga and K. Hirota, Anal. Sci., 2016, 32, 801–804 CrossRef CAS PubMed.
- K. Kosma, S. A. Trushin, W. Fuß and W. E. Schmid, J. Mod. Opt., 2008, 55, 2141–2177 CrossRef CAS.
- B. Spokoyny, C. J. Koh and E. Harel, Opt. Lett., 2015, 40, 1014–1017 CrossRef CAS PubMed.
- H. D. Vishwasrao, A. A. Heikal, K. A. Kasischke and W. W. Webb, J. Biol. Chem., 2005, 280, 25119–25126 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04814a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |