
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and pH-responsive dissociation of framboidal ABC triblock copolymer vesicles in aqueous solution†
C. J.
Mable
a,
L. A.
Fielding
 c,
M. J.
Derry
a,
O. O.
Mykhaylyk
a,
P.
Chambon
b and
S. P.
Armes
*a
c,
M. J.
Derry
a,
O. O.
Mykhaylyk
a,
P.
Chambon
b and
S. P.
Armes
*a
aDepartment of Chemistry, Uni. of Sheffield, Dainton Building, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: S.P.Armes@sheffield.ac.uk; Tel: +44 (0)114 222 9342
bDepartment of Chemistry, Uni. of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
cSchool of Materials, Uni. of Manchester, Oxford Rd, Manchester, M13 9PL, UK
First published on 13th December 2017
Abstract
A series of pH-responsive all-methacrylic ABC triblock copolymer vesicles were prepared from precursor diblock copolymer vesicles via RAFT seeded emulsion polymerisation. Microphase separation between the two hydrophobic membrane-forming B and C blocks produced a distinctive framboidal morphology, for which the mean globule size can be tuned by adjusting the triblock copolymer composition. These vesicles remain intact at neutral pH, but undergo irreversible dissociation on addition of acid as a result of protonation of the tertiary amine groups located within the third block. Small-angle X-ray scattering (SAXS) was utilised to characterise the morphologies formed at pH 8 and pH 3. According to time-resolved SAXS studies, the acid-induced dissociation of these pH-responsive framboidal vesicles involves appreciable membrane swelling within 50 ms and is complete.
Introduction
It is well-known that amphiphilic block copolymers undergo spontaneous self-assembly in aqueous solution.1–4 For example, many variations on the vesicular morphology have been reported in the literature, including stomatocyte-like,5 Janus-type6 and framboidal vesicles.7–9 Recent advances in polymerisation-induced self-assembly (PISA)10–12 have enabled a wide range of block copolymer nanoparticles to be prepared directly in concentrated aqueous solution via reversible addition-fragmentation chain transfer (RAFT) aqueous dispersion polymerisation.13,14 Such one-pot syntheses are much more convenient than traditional post-polymerisation processing techniques, which typically involve a solvent or pH switch15–18 or thin film rehydration,19,20 and are invariably restricted to dilute solution. For example, a poly(glycerol monomethacrylate) macromolecular chain transfer agent (PGMA macro-CTA) can be chain-extended using 2-hydroxypropyl methacrylate (HPMA).14,21,22 Self-assembly occurs to form PHPMA-core nanoparticles stabilised by the water-soluble PGMA chains. Depending on the precise formulation, a range of copolymer morphologies can be achieved, including spheres, worms and vesicles.21–23 Recently, Chambon et al. utilised RAFT seeded emulsion polymerisation to grow a third water-insoluble-monomer (benzyl methacrylate) from PGMA–PPMA diblock copolymer vesicles. This leads to microphase separation within the vesicle membrane, resulting in the formation of ABC triblock copolymer vesicles with a distinctive framboidal (i.e. raspberry-like) morphology.7 Such model colloids were used to study the effect of varying the surface roughness on Pickering emulsifier performance.9There are many PISA syntheses of thermoresponsive nano-objects in the literature.11,13,24–32 However, there are rather fewer reports of pH-responsive nanoparticles prepared via PISA.8,27,33–37 Moreover, as far as we are aware, there is only one other literature report of pH-responsive framboidal nanoparticles: such phenylboronic acid-functionalised nano-objects were prepared by Hasegawa et al. via aqueous dispersion polymerisation using conventional free radical chemistry.8
Herein we report the chain extension of PGMA–PHPMA diblock copolymer vesicles using varying amounts of 2-(diisopropylamino)ethyl methacrylate (DPA) to produce novel framboidal PGMA–PHPMA–PDPA triblock copolymer vesicles, see Fig. 1. For brevity, such copolymers are hereafter denoted as G58H300Dz, where the subscripts refer to the mean degree of polymerisation, DP, of each block and z is a variable.
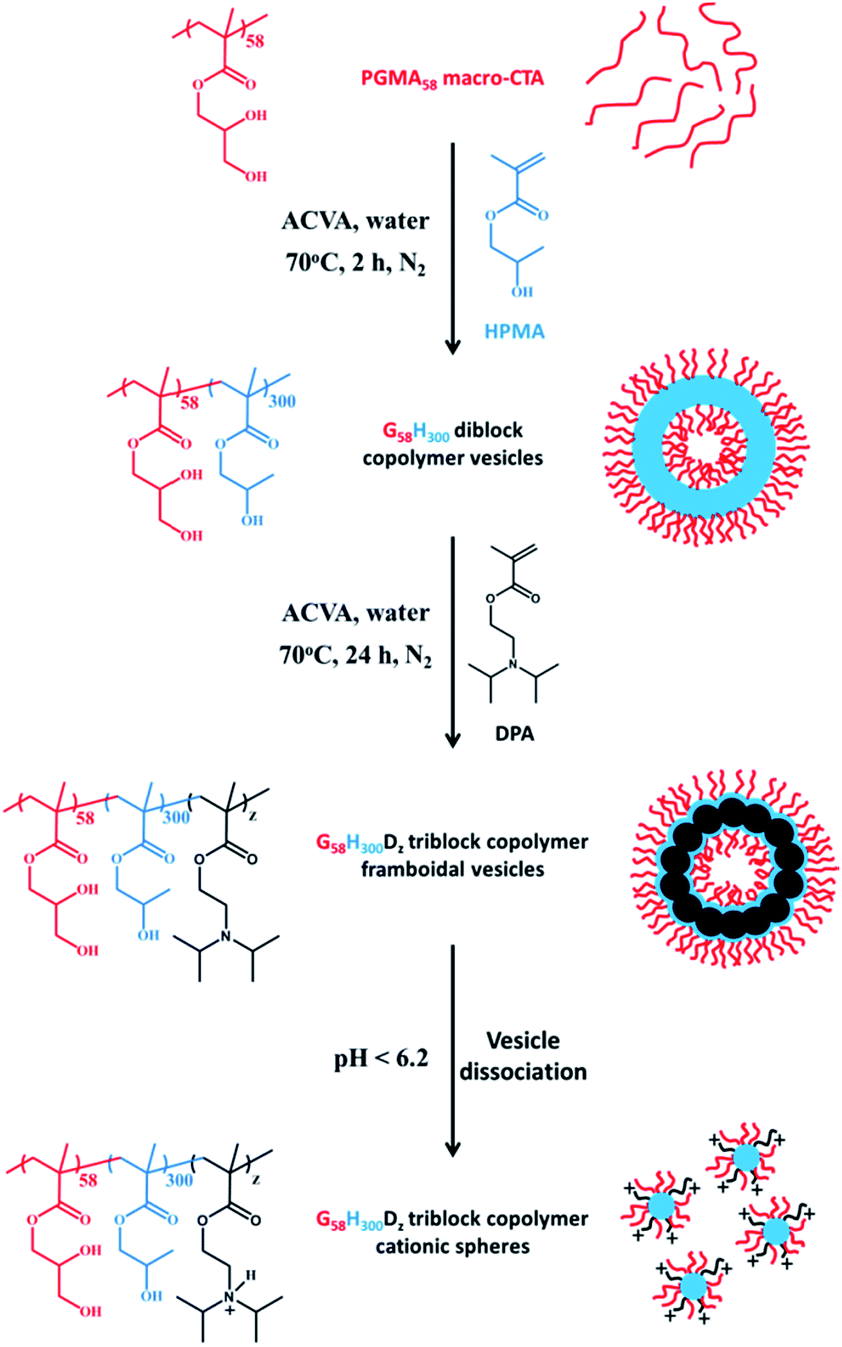 | ||
Fig. 1 Synthetic route to  framboidal triblock copolymer vesicles via polymerisation-induced self-assembly (PISA) in aqueous solution at pH 7–8. Addition of acid causes protonation of the tertiary amine residues, which renders the PDPA block hydrophilic and induces vesicle dissociation. Inset: schematic cartoon of precursor vesicles, framboidal vesicles and cationic spheres. framboidal triblock copolymer vesicles via polymerisation-induced self-assembly (PISA) in aqueous solution at pH 7–8. Addition of acid causes protonation of the tertiary amine residues, which renders the PDPA block hydrophilic and induces vesicle dissociation. Inset: schematic cartoon of precursor vesicles, framboidal vesicles and cationic spheres. | ||
Results and discussion
Initially, a PGMA58 macro-CTA was chain-extended via RAFT aqueous dispersion polymerisation of HPMA at 15% w/w solids to prepare a 100 gram batch of G58H300 diblock copolymer vesicles, see Fig. 1. These G58H300 precursor vesicles were then diluted to 10% w/w and subsequently chain-extended using varying amounts of DPA via RAFT seeded emulsion polymerisation to prepare a series of G58H300Dz triblock copolymers, where z was adjusted from 86 to 460 (allowing for incomplete reaction of the DPA monomer). 1H NMR studies indicated that more than 99% HPMA conversion was achieved within 2 h at 70 °C (as expected based on previous work21), while DPA conversions of 82–92% were achieved within 24 h at 70 °C. 1H NMR studies of the final G58H300Dz triblock copolymers were conducted in CD3OD containing 4% DCl, which is a good solvent for all three blocks (see Fig. S1 in ESI† for assigned spectra).DMF GPC studies conducted using a series of poly(methyl methacrylate) (PMMA) calibration standards indicated that a near-monodisperse G58H300 diblock copolymer precursor (Mw/Mn = 1.12; see Fig. S2 in ESI†) was obtained with a relatively high blocking efficiency. This GPC trace had a weak high molecular weight shoulder, which was attributed to light branching caused by small amounts of dimethacrylate impurity within the HPMA monomer (0.07 mol% as judged by HPLC). In the case of the G58H300Dz triblock copolymers, the PDPA block is DMF-insoluble, while the PGMA block is THF-insoluble, rendering GPC characterisation of this triblock copolymer rather problematic. To render these triblock copolymers THF-soluble for GPC analysis, the PGMA block was derivatised according to a previously reported protocol.38,39 Thus, such G58H300Dz triblock copolymers were dissolved in pyridine, excess benzoic anhydride (four equivalents based on GMA and HPMA residues) was added, and esterification of the hydroxyl groups was allowed to proceed for 24 h at room temperature. The benzoate-protected copolymers obtained by this method were always fully soluble in THF (unlike their precursors). The GPC traces were unimodal and indicated high blocking efficiencies (see Fig. S3 in ESI†). However, dispersities were greater than that expected for RAFT polymerisations (see Table 1). To determine whether this observation was real or merely a GPC artefact, the PGMA58 macro-CTA and G58H300 diblock copolymer precursor were also derivatised so that the DMF GPC and THF GPC protocols could be directly compared (Table 1). Similar number-average molecular weight (Mn) values were obtained for the original homopolymer via DMF GPC and the esterified copolymer via THF GPC, as expected. However, the dispersities are significantly higher for the latter (see Table 1). Thus the dispersities obtained for G58H300Dz triblock copolymers where z is 86, 164 or 249 are likely to be an artefact of the esterification protocol. However, the much higher dispersities (Mw/Mn > 3.0) obtained when z = 356 or 460 are perhaps less likely to be solely owing to such an artefact. For these latter two copolymers, the RAFT polymerisation of the DPA was clearly not well-controlled, giving rise to relatively broad molecular weight distributions. Similar results have been recently reported by Derry et al. for other PISA formulations when targeting relatively high degrees of polymerisation.40
| Copolymer composition | M n (g mol−1) | M w/Mn | D h (PDI)/nm | DLS count rate/kcps | Zeta potential/mV | Globule diameter/nm | Membrane thickness/nm | Vesicle diameterc/nm | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pH 8 | pH 3 | pH 8 | pH 3 | pH 8 | pH 3 | ||||||
| a Data obtained via DMF GPC (against PMMA standards) using a refractive index detector. b Data obtained via THF GPC (against PMMA standards) using a refractive index detector after exhaustive esterification of the hydroxyl groups. c These data are only considered to be approximate, because the SAXS camera length was not long enough to enable accurate calculation of the overall vesicle diameters. | |||||||||||
| G58 macro-CTA | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400a, 14500b 400a, 14500b |
1.13a, 1.36b | |||||||||
| G58H300 | 66700a, 62600b |
1.12a, 1.41b | 396 (0.08) | 388 (0.09) | 55200 |
49000 |
−11 | −2.0 | 16.8 | 383 | |
| G58H300D86 | 71900b |
1.67b | 372 (0.11) | 44 (0.42) | 47900 |
800 | −15 | +34 | 27 | 24.9 | 391 |
| G58H300D164 | 76600b |
1.98b | 379 (0.12) | 67 (0.58) | 46300 |
700 | −19 | +36 | 37 | 36.3 | 402 |
| G58H300D249 | 84800b |
1.98b | 403 (0.07) | 122 (0.49) | 42100 |
1100 | −28 | +37 | 44 | 46.4 | 412 |
| G58H300D356 | 91600 |
3.08b | 409 (0.13) | 293 (0.63) | 28600 |
1600 | −31 | +40 | |||
| G58H300D460 | 119800b |
3.55b | 442 (0.15) | 1413 (0.65) | 19600 |
2600 | −39 | +41 | |||
TEM studies of the G58H300 diblock copolymer revealed a pure vesicular morphology (see Fig. 2). As expected, these precursor vesicles were not pH-responsive: they remained intact at both pH 8 and pH 3. TEM studies of the G58H300Dz triblock copolymers confirmed the formation of framboidal vesicles at pH 8. This distinctive morphology is the result of microphase separation between the PHPMA and PDPA membrane-forming blocks, which becomes more pronounced with increasing z. Unlike the diblock precursor vesicles, these framboidal triblock copolymer vesicles do not remain intact at pH 3: disintegration is observed by TEM (see Fig. 2). Visual inspection of the dispersion is also consistent with loss of the vesicular morphology. At pH 8, the vesicle dispersions are highly turbid as expected, but at pH 3 a relatively clear solution is obtained. When returning to pH 8, a white precipitate is formed, indicating that this pH-responsive behaviour is not reversible, presumably because the framboidal morphology is kinetically-trapped (see Fig. S4 in ESI†).
 | ||
| Fig. 2 Representative TEM images obtained for the G58H300 precursor vesicles and a series of G58H300Dz framboidal vesicles, where z = 86, 164, 249, 356 or 460 (from left to right) at pH 8 (top) and pH 3 (bottom). The scale bars shown in the far left-hand images are correct for all images shown at that particular pH. | ||
Dynamic light scattering (DLS) measurements indicate that vesicle dissolution occurs when the pH is lowered from pH 8 to pH 3 (see Table 1). At pH 8, the G58H300 diblock copolymer vesicles have a hydrodynamic diameter of ∼396 nm. Chain extension with 86 DPA units led initially to a modest reduction in the mean vesicle diameter to 372 nm. Presumably, this initial compaction is the result of a more hydrophobic membrane. For higher z values, the hydrodynamic diameter gradually increases up to 442 nm for G58H300D460. This reflects formation of progressively thicker membranes, with the concomitant evolution of the distinctive framboidal morphology.
At pH 3, the G58H300 precursor vesicles have a comparable hydrodynamic diameter and polydispersity to that observed at pH 8. In contrast, the series of G58H300Dz triblock copolymer vesicles typically exhibit significantly smaller hydrodynamic diameters and higher polydispersities at pH 3 compared to those observed at pH 8. The exception is G58H300D460, for which a relatively large hydrodynamic diameter of 1413 nm is observed. DLS studies were also conducted as a function of pH in the presence of 1 mM KCl (see Fig. 3 and S5 in the ESI†). The G58H300 precursor vesicles maintained a constant hydrodynamic diameter of around 400–500 nm over a wide range of pH, while the scattered light intensity (or count rate) is only reduced by around 20% in acidic solution (see Fig. 3). In contrast, the count rate observed for the G58H300D460 triblock copolymer vesicles is reduced by approximately an order of magnitude (from ∼105 kcps at pH 9 to ∼104 kcps at pH 4), with a concomitant increase in the apparent hydrodynamic diameter from 500 nm up to 1250 nm.
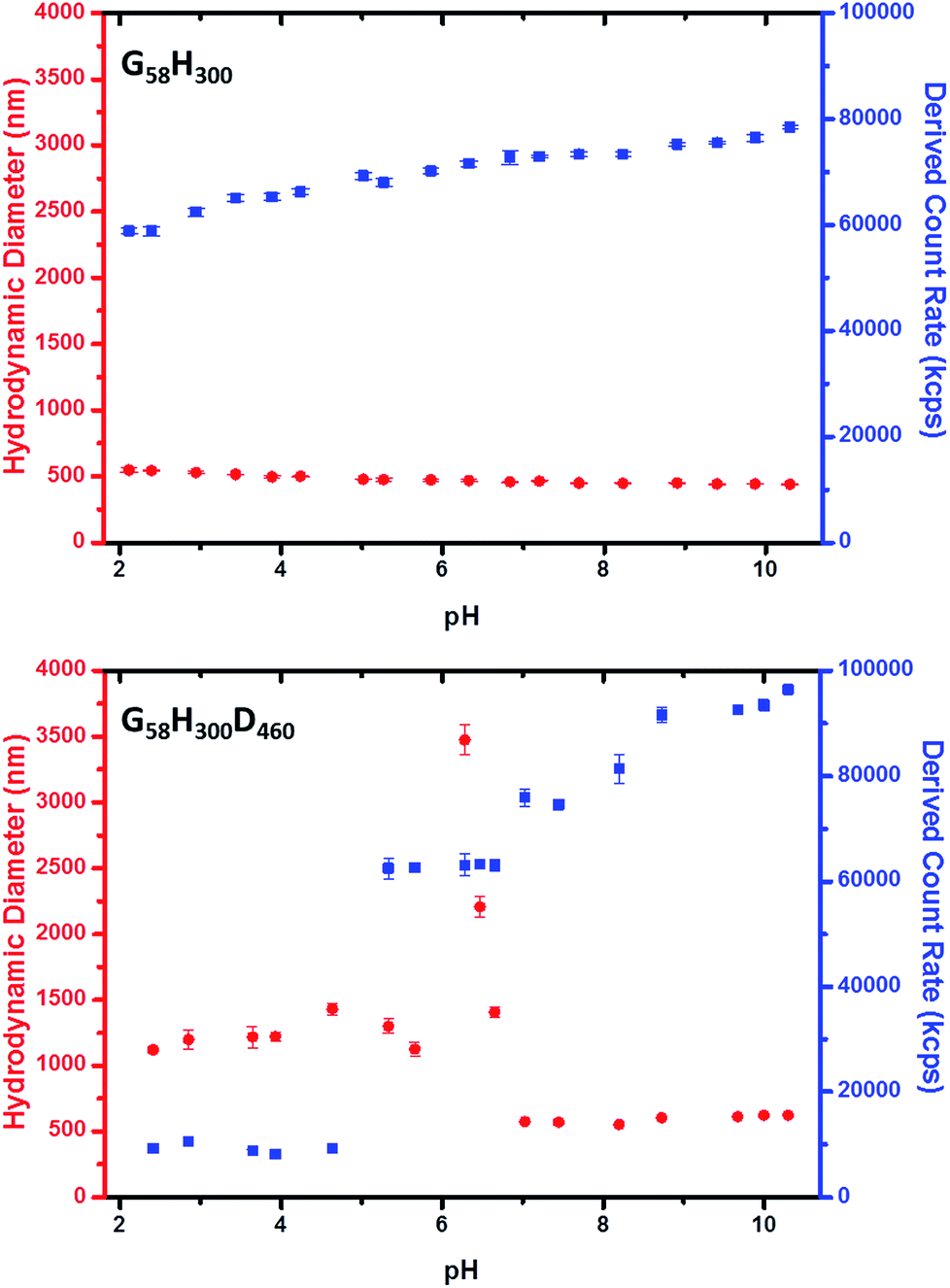 | ||
Fig. 3 Variation in hydrodynamic diameter ( ) and count rate ( ) and count rate ( ) with solution pH for (top) G58H300 diblock copolymer precursor vesicles and (bottom) G58H300D460 triblock copolymer framboidal vesicles. ) with solution pH for (top) G58H300 diblock copolymer precursor vesicles and (bottom) G58H300D460 triblock copolymer framboidal vesicles. | ||
Close inspection of the DLS data suggests that disruption of the framboidal vesicle morphology occurs when the pH is lowered below the pKa of the PDPA block, which is approximately 6.2 (see Fig. 3). At around this pKa value, the hydrophobic PDPA chains within the vesicle membrane gradually become protonated and therefore acquire cationic character (see Fig. 1). The partially protonated vesicles pass through their isoelectric point (IEP) at around pH 6, which induces flocculation. Below the IEP, partial vesicle disintegration occurs and relatively loose, weakly-interacting colloidal aggregates of rather ill-defined morphology are formed, as judged by TEM and DLS studies (see Fig. 2 and 3). It is emphasised that molecularly-dissolved triblock copolymer chains are not produced at low pH, because the relatively long central PHPMA block retains its weakly hydrophobic character under these conditions.
Aqueous electrophoresis studies were conducted on the G58H300 precursor vesicles and both the G58H300D86 and G58H300D460 framboidal vesicles as a function of pH (see Fig. 4). The latter two samples exhibited isoelectric points at around pH 6, with the positive zeta potentials of +34 to +42 mV observed at low pH being attributed to protonation of the tertiary amine groups located within in the PDPA block. This is consistent with the observation of a dramatic reduction in aggregate size in each case, as judged by TEM (see Fig. 2) and DLS (see Fig. 3 and S4 in ESI†).
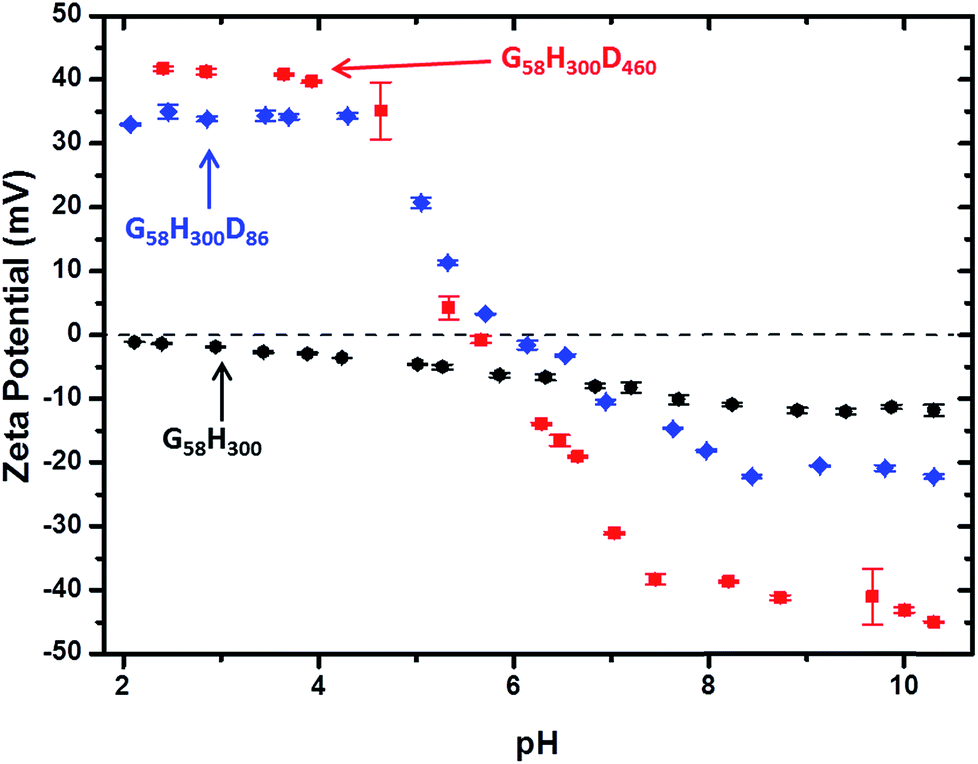 | ||
Fig. 4 Zeta potential vs. pH curves obtained for: ( ) G58H300 diblock copolymer precursor vesicles, ( ) G58H300 diblock copolymer precursor vesicles, ( ) G58H300D86 triblock copolymer vesicles and ( ) G58H300D86 triblock copolymer vesicles and ( ) G58H300D460 triblock copolymer vesicles. ) G58H300D460 triblock copolymer vesicles. | ||
In contrast, the diblock copolymer precursor vesicles did not exhibit any IEP, as expected. Above around pH 6, all three types of vesicles remained intact and exhibited anionic character, possibly owing to selective adsorption of hydroxide ions onto the vesicle surface.41 Aqueous electrophoresis studies were also conducted on the other copolymers (see Fig. S6 in ESI†). Considering the TEM, DLS and zeta potential data together, three distinct physical states for these G58H300Dz triblock copolymer particles can be identified. Small cationic copolymer aggregates are formed below pH 5, weakly cationic copolymer vesicles are obtained at pH 5–6, and anionic framboidal vesicles are produced above pH 6.2.
SAXS analyses (see Fig. 5) were conducted to obtain reliable framboidal vesicle globule dimensions and also to provide further insight with regard to the copolymer morphology formed at pH 3. TEM images indicate that the G58H300 precursor vesicles have smooth membranes. In contrast, the highly distinctive framboidal morphology possessed by the G58H300Dz triblock copolymer vesicles dried at pH 8 is comparable to that of the polymer core-particulate silica shell particles reported by Balmer and co-workers.42–45 In this earlier work, Monte Carlo simulations were utilised to demonstrate42 that the SAXS patterns obtained for such nanocomposite particles could be described by a two-population model. This model is represented by a superposition of two scattering signals originating from a core–shell structure comprising a spherical latex core surrounded by a shell composed of small spherical silica nanoparticles (population 1) and the many silica nanoparticles that form this shell (population 2).
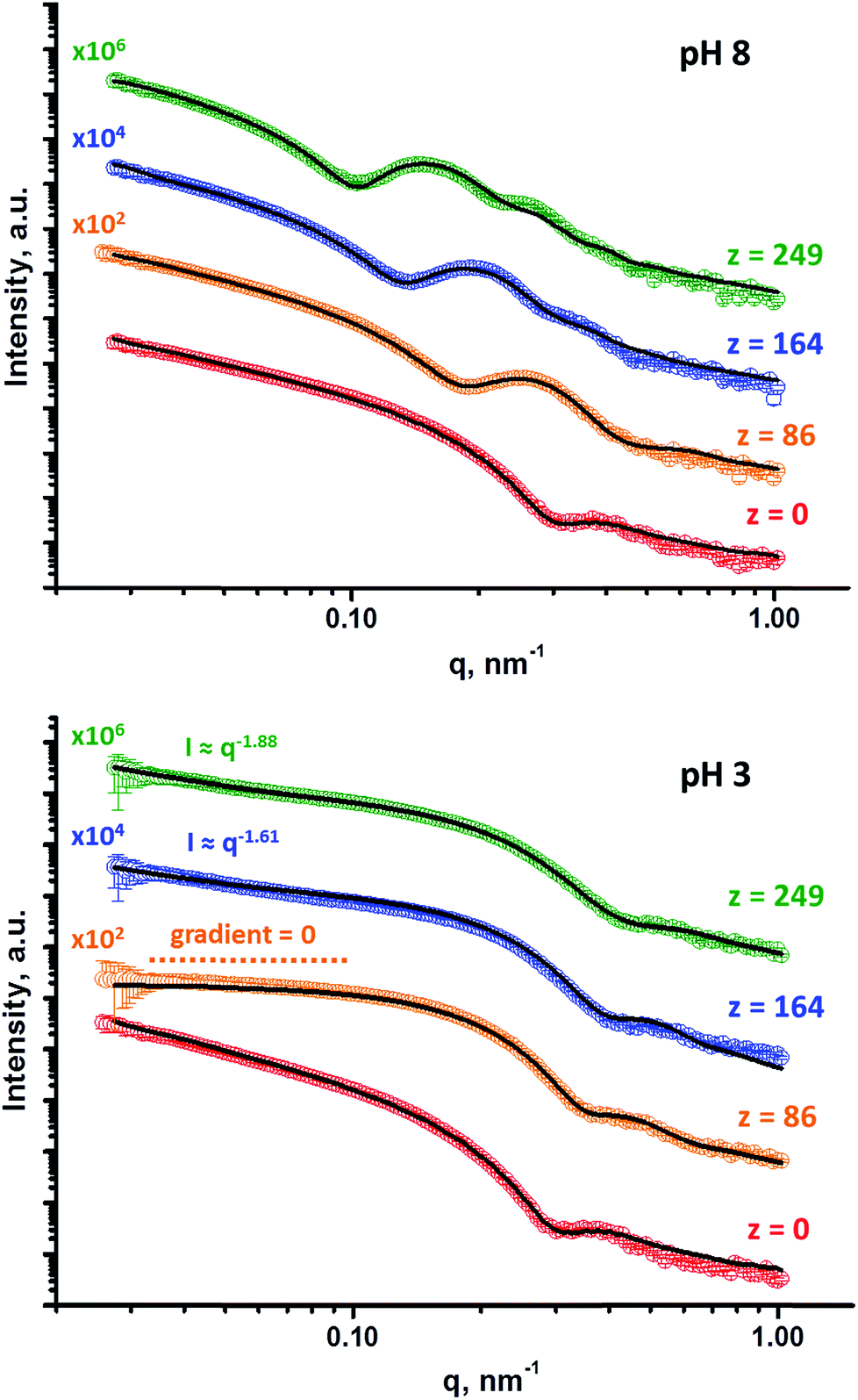 | ||
| Fig. 5 SAXS patterns obtained at pH 8 and pH 3 for 1.0% w/w aqueous dispersions of G58H300 diblock copolymer precursor vesicles (where z = 0) and a series of framboidal G58H300Dz triblock copolymer vesicles, where z = 86, 164, 125, 249. Solid lines represent fitting curves. At pH 8, a single vesicle model was sufficient for z = 0, whereas the superposition of a vesicle model and a spherical micelle model were required for higher z values. At pH 3, a single vesicle model was sufficient for z = 0 and a single spherical micelle model was sufficient for z = 86, whereas superposition of a spherical micelle and a mass fractal model was required for higher z values. For clarity, these SAXS patterns are shifted upward by an arbitrary factor, as indicated in the figure. | ||
A similar approach was recently adopted by Mable et al. for the SAXS analysis of closely-related pH-invariant framboidal triblock copolymer vesicles.9 In this more recent study, population 1 represented the vesicles and population 2 described the micelle-like spherical globules formed within the vesicle membrane. Herein, we use the same two-population SAXS model for fitting the G58H300Dz patterns recorded at pH 8.
Population 1 corresponds to smooth vesicles and is thus also appropriate for SAXS analysis of the G58H300 diblock copolymer precursor. This approach46 produced a reasonably good fit to the SAXS pattern over six orders of magnitude of X-ray scattering intensity (Fig. 5, pH 8, red data). The volume-average vesicle diameter was estimated to be 383 nm (unfortunately, the camera length used to collect these SAXS data was not long enough to provide a more reliable value for this parameter). Nevertheless, this is consistent with both TEM observations (Fig. 2) and DLS data (Table 1). According to TEM analysis, the mean vesicle diameter was about 350 nm, while DLS studies indicated a mean hydrodynamic vesicle diameter (Dh) of 396 nm with a polydispersity index (PDI) of 0.08. The radius of gyration (Rg) of the G58 corona block was determined to be 2.4 nm from model fitting of the G58H300 SAXS pattern. This experimental value is comparable to a theoretical estimate: the projected contour length of a single GMA monomer is 0.255 nm (two carbon bonds in all-trans conformation), the total contour length of a G58 block, LPGMA = 58 × 0.255 nm = 14.79 nm and the Kuhn length of 1.53 nm, based on the literature value for poly(methyl methacrylate),47 result in an estimated Rg of (14.79 × 1.53/6)1/2, or 1.94 nm. The water volume fraction, xsol, within the membrane is approximately 0.50 according to the SAXS data fit. This is relatively high and reflects the weakly hydrophobic nature of the PHPMA block.24 As expected, when acid is added to the aqueous dispersion of G58H300 diblock copolymer vesicles to lower its solution pH, these vesicles remain unchanged because they are not pH-responsive (see Fig. 5, pH 3, red data).
Superposition of scattering signals from populations 1 and 2 (corresponding to vesicles and spherical micelles, respectively),9,48 was essential to produce satisfactory fits to the SAXS patterns obtained for the G58H300Dz framboidal vesicles, where z is 86, 164 or 249 (Fig. 5, pH 8). Previously, we assumed that both the Rg of the PGMA block and the water content within the hydrophobic part of the vesicle membrane remained constant, regardless of whether closely-related triblock copolymer vesicles were smooth or framboidal.9 A similar assumption was made in the current study, and the Rg and xsol values determined for the G58H300 precursor vesicles were fixed when fitting the SAXS patterns recorded for the G58H300Dz framboidal vesicles. This self-consistent analytical approach indicated that the thickness of the hydrophobic component of the vesicle membrane (Tmc) increased when targeting higher DPs for the PDPA block (see Table S1 in the ESI†). In addition, both TEM observations (Fig. 2) and DLS studies (Table 1) suggest that the vesicle diameter remained virtually constant over all copolymer compositions. Taken together, these data suggest that the vesicle growth mechanism involves a gradual reduction in the vesicle lumen volume, as reported by Warren and co-workers where non-framboidal G55Hy vesicles, for y = 200 to 2000.49 Similar observations were reported by Mable et al. when chain-extending G63H350 precursor vesicles with benzyl methacrylate (B) to obtain G63H350Bz framboidal vesicles, where z ranged from 25 to 400.9 The nanoscale phase separation that occurs within the vesicle membrane described by the spherical micelle model (population 2) can be verified by SAXS analysis. Both the spherical micelle radius (Rs) and the relative concentration of the second population (c2/c1) increase when targeting higher PDPA block volume fractions, VPDPA, (see Table S1 in the ESI†). SAXS analysis indicates that the mean micelle/globule diameter (Ds = 2Rs + 4Rg) for the G58H300Dz framboidal vesicles increases from 27 nm to 44 nm as z is varied from 86 to 249.
Nevertheless, some deviations between the fitting pattern and the experimental pattern are discernible for G58H300D249. The pronounced feature observed in the experimental SAXS pattern at q ∼ 0.25 nm−1 cannot be fully reproduced by the model fit. In principle, growth of the PDPA block within the hydrophobic membrane may drive its strong segregation from the PHPMA block, producing two regions of differing electron density within the spherical globule cores. If this explanation is correct, the inner core is likely to be the highly hydrophobic PDPA block while the outer core should contain the weakly hydrophobic PHPMA block. To examine this hypothesis, the two-population model was further refined. For population 1, the hydrophobic component of the vesicle membrane was assumed to comprise an inner PDPA layer surrounded by two outer PHPMA layers. Similarly, the hydrophobic spherical micelle core associated with population 2 was assumed to have a core–shell structure, whereby the core contained the PDPA blocks and the PHPMA blocks were located within the shell. However, this more sophisticated model did not produce an improved data fit compared to the original two-population model. This suggests that the deviation observed between the experimental and fitting patterns is not related to strong segregation between these two hydrophobic blocks. It is perhaps worth emphasising here that the two-population model describes the vesicles and spherical micelles independently: it does not include cross terms between these two structural features. In the literature, attempts have been made to account for such cross terms for other complex multicomponent particles.50 However, various additional parameters such as vesicle and spherical micelle polydispersities are required for such models. This significantly complicates the analysis and is considered to be beyond the scope of the current study.
It was also difficult to obtain satisfactory fits to the SAXS patterns obtained for G58H300D356 and G58H300D460 framboidal vesicles using the basic two-population model. As recently reported in the literature, the vesicle growth mechanism during PISA leads to a gradual reduction in the volume of the vesicle lumen.40 Because these two vesicles possess relatively long PDPA blocks, the vesicle membrane is rather thick, resulting in a substantially reduced vesicle lumen volume. Moreover, these framboidal vesicles can no longer be described as spherical globules located on a vesicle surface. Instead, the micelle-like globules become so large that these nanoparticles are essentially a cluster (aggregate) of pseudo-spherical globules. For example, the c2/c1 ratio increases from 0.273 to 0.997 as z is varied from 86 to 249. Thus, the relative concentration of micelle-like globules is essentially the same as that of the vesicles when z = 249. For higher PDPA block DPs (e.g. z = 356 or 460), the c2/c1 ratio increases further, indicating a significant reduction in the relative vesicle concentration. Similar findings were reported recently by Mable et al. when analysing G63H350Bz framboidal vesicles. The two-population model did not provide a satisfactory fit to the SAXS pattern recorded for vesicles prepared when targeting the longest PBzMA block DP (z = 400), most likely for the same reason.9 Instead, an alternative ‘aggregated sphere’ model should be used to obtain satisfactory SAXS data fits. Given this literature precedent, the two G58H300Dz triblock copolymers with the longest PDPA blocks (i.e. z = 356 and 460) are not considered to be genuine framboidal vesicles.
TEM, DLS and zeta potential data suggest that relatively small, rather ill-defined cationic aggregates are formed below pH 5 by this series of G58H300Dz triblock copolymers. In order to provide further morphological insights, SAXS patterns were recorded at pH 3. The low q gradient of the SAXS pattern obtained for the G58H350D86 triblock copolymer at this pH tends to zero (see Fig. 5, pH 3, orange data), which is characteristic of spherical micelles.51 The spherical micelle model48,52 provided a satisfactory fit to this SAXS pattern over three orders of magnitude of X-ray scattering intensity. The SAXS-derived mean sphere diameter Ds (where Ds = 2Rs + 4Rg) was calculated to be 33.6 ± 3.2 nm, which is consistent with that reported by DLS (44 nm, see Table 1) given that these two techniques report different moments of the size distribution. However, the SAXS patterns recorded for G58H300D164 and G58H300D249 at pH 3 show a significant upturn in X-ray scattering intensity at low q compared to the pattern obtained for G58H300D86 at pH 3. This suggests that scattering objects larger than the spherical micelles have been formed, hence the spherical micelle model alone is not appropriate for data analysis of these two copolymer compositions. TEM analysis (Fig. 2, pH 3) indicates the formation of mass fractals comprising aggregates of weakly-interacting spherical micelles.53 Thus, these two SAXS patterns were fitted using a superposition of the spherical micelle model and a truncated power law function representing a mass fractal structure [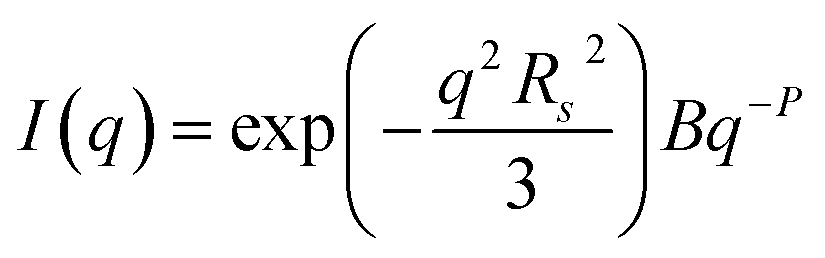 , where I(q) is the scattered X-ray intensity, B is a constant and P is an exponent corresponding to the mass fractal dimension; the exponent term associated with Rs provides a high q cut-off for the power component54]. This approach gave satisfactory fits to the data over nearly four orders of magnitude of X-ray scattering intensity (see Fig. 5, pH 3, blue and green data). Smaller micelles were formed at pH 3 when increasing the DP of the PDPA block. For example, Ds was determined to be 30.8 ± 2.8 nm and 26.9 ± 2.6 nm for z = 164 and 249, respectively (Table S1†). This is in good agreement with the well-known principles of block copolymer self-assembly: longer stabiliser blocks favour the formation of micelles with lower aggregation numbers.55–57 Moreover, the Percus–Yevick effective volume fraction (FPY) increased from 0.08 to 0.12 on increasing z from 164 to 249, indicating that the smaller spherical micelles become more aggregated under these conditions. This is consistent with the observed increase in the mass fractal dimensions (P) from 1.61 to 1.88, suggesting the formation of denser fractals when z = 249. In summary, the precise copolymer morphology obtained at pH 3 is strongly dependent on the DP of the PDPA block. However, we do not have a satisfactory explanation for the formation of these mass fractals (as opposed to non-interacting spherical micelles) at the present time.
, where I(q) is the scattered X-ray intensity, B is a constant and P is an exponent corresponding to the mass fractal dimension; the exponent term associated with Rs provides a high q cut-off for the power component54]. This approach gave satisfactory fits to the data over nearly four orders of magnitude of X-ray scattering intensity (see Fig. 5, pH 3, blue and green data). Smaller micelles were formed at pH 3 when increasing the DP of the PDPA block. For example, Ds was determined to be 30.8 ± 2.8 nm and 26.9 ± 2.6 nm for z = 164 and 249, respectively (Table S1†). This is in good agreement with the well-known principles of block copolymer self-assembly: longer stabiliser blocks favour the formation of micelles with lower aggregation numbers.55–57 Moreover, the Percus–Yevick effective volume fraction (FPY) increased from 0.08 to 0.12 on increasing z from 164 to 249, indicating that the smaller spherical micelles become more aggregated under these conditions. This is consistent with the observed increase in the mass fractal dimensions (P) from 1.61 to 1.88, suggesting the formation of denser fractals when z = 249. In summary, the precise copolymer morphology obtained at pH 3 is strongly dependent on the DP of the PDPA block. However, we do not have a satisfactory explanation for the formation of these mass fractals (as opposed to non-interacting spherical micelles) at the present time.
A final SAXS experiment utilising a stopped-flow set-up was conducted to examine the precise time scale on which such framboidal vesicles dissociate after addition of acid. This experiment was conducted using a similar framboidal triblock copolymer to those reported in this work. More specifically, a framboidal G58H250D184 triblock copolymer was employed. 1H NMR studies indicated that 92% DPA conversion was achieved within 24 h at 70 °C for this particular copolymer synthesis (see Fig. S7a in ESI†). TEM studies confirmed that framboidal vesicles were present at pH 8 (see Fig. S7b in ESI†) and indicated the formation of mass fractals at pH 3 (see Fig. S7c in ESI†). These observations are fully consistent with those discussed above for similar G58H250Dz framboidal vesicles.
The initial SAXS pattern obtained for these G58H250D184 vesicles at pH 8 (see the uppermost pattern shown in Fig. 6) resembles that recorded for the G58H300D164 vesicles (see Fig. 5a). To analyse the kinetics of the acid-induced disintegration of these G58H250D184 framboidal vesicles using an HCl/DPA molar ratio of 1.50, a stopped-flow cell was mounted on the synchrotron beamline and SAXS patterns were recorded every 10 ms for 1.0 s. However, the first pattern was excluded, because the dead time for the stopped-flow cell set-up was determined to be 16 ms. Selected SAXS patterns are displayed in Fig. 6.
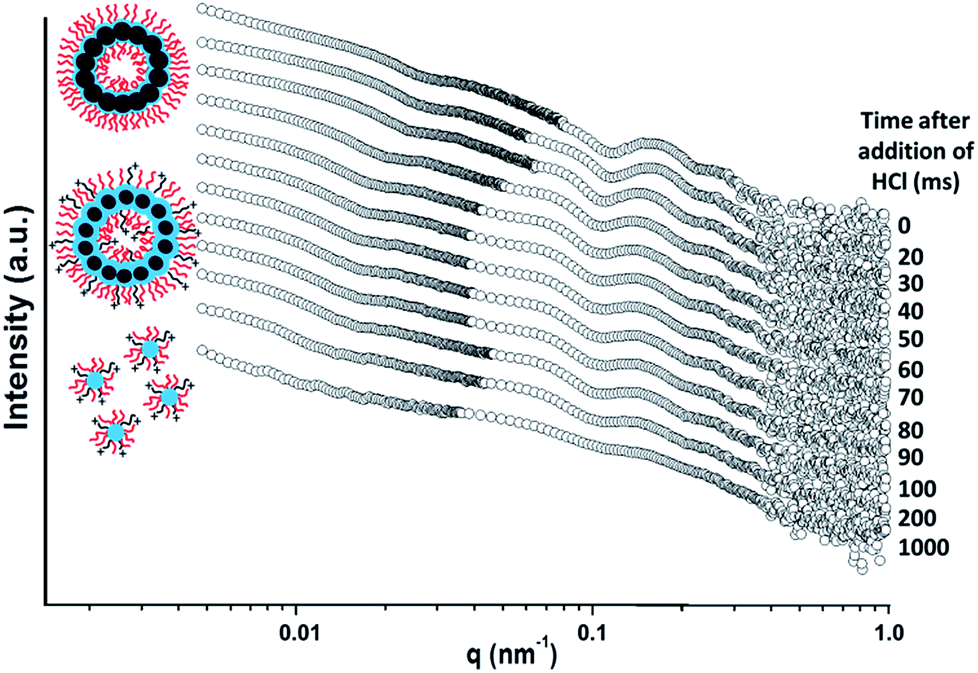 | ||
| Fig. 6 Time-resolved SAXS patterns obtained for a 0.27% w/w aqueous dispersion of G58H250D184 triblock copolymer vesicles after addition of acid using an HCl/DPA molar ratio of 1.50. A scattering pattern was obtained for the G58H250D184 vesicles prior to addition of acid. Scattering patterns were then recorded every 10 ms for 1.0 s. Selected SAXS patterns are shown after various reaction times (for clarity, these patterns are shifted by an arbitrary scaling factor). The framboidal vesicle diameter increases and the vesicle membrane swells as the tertiary amine groups within the PDPA chains become protonated until vesicle disintegration occurs to form mass fractals comprising cationic spherical micelles. [N.B. PGMA = red, PHPMA = blue, PDPA = black]. | ||
Detailed analysis of these SAXS patterns has not been attempted, because the complexity of the evolving multi-component system (framboidal vesicles, swollen vesicles and weakly interacting cationic spheres) precludes data fitting to existing scattering models. Nevertheless, the kinetics of vesicle dissociation can be assessed from these curves. The inflection in the first (upper) SAXS pattern at q ∼ 0.03 nm−1 represents the overall vesicle diameter. This feature shifts to lower q over time, indicating the formation of larger vesicles after addition of HCl. Similarly, the local minimum in the first SAXS pattern at q ∼ 0.1 nm−1 corresponding to the mean membrane thickness (Tm) shifts to lower q. This is consistent with substantial membrane swelling due to water ingress as the tertiary amine groups within the PDPA block become protonated and hence hydrophilic. Just 1.0 s after HCl addition, there are no longer any vesicles present within the aqueous dispersion. Instead, rapid vesicle dissociation produces mass fractal aggregates of ill-defined spherical micelles, as indicated by the static measurements discussed above.
Experimental
Materials
All reagents were used as received unless otherwise stated. 2-(Diisopropylamino)ethyl methacrylate (DPA), deuterium chloride (DCl; 20% w/w in D2O) and 4,4′-azobis-4-cyanopentanoic acid (ACVA) were purchased from Sigma-Aldrich (UK). DPA inhibitor was removed by passing this monomer through an inhibitor removal column. Ethanol, dichloromethane, dimethyl sulfoxide (DMSO) and dimethyl formamide (DMF) were purchased from Fisher Scientific (UK). 2-Cyano-2-propyl dithiobenzoate (CPDB) was purchased from Strem Chemicals (Cambridge, UK). Glycerol monomethacrylate (GMA) was kindly donated by GEO Specialty Chemicals (Hythe, UK) and used without further purification. 2-Hydroxypropyl methacrylate (HPMA) was purchased from Alfa Aesar (UK) and contained 0.07% dimethacrylate impurity, as judged by high performance liquid chromatography (HPLC). Deuterated methanol (CD3OD) was purchased from Goss Scientific (UK). Deionised water was obtained using an Elga Elgastat Option 3A water purifier; its pH was approximately 6.2 and its surface tension was around 72.0 mN m−1 at 20 °C.400 g mol−1, Mw/Mn = 1.13) was purified by precipitation into excess dichloromethane. A mean DP of 58 was calculated for this macro-CTA using 1H NMR.
Characterisation
000 g mol−1). Chromatograms were analysed using Varian Cirrus GPC software (version 3.3) provided by the instrument manufacturer (Agilent). The THF GPC set-up comprised two 5 μm (30 cm) Mixed C columns and a WellChrom K-2301 refractive index detector operating at 950 ± 30 nm. The mobile phase contained 2.0% v/v triethylamine and 0.05% w/v butylhydroxytoluene (BHT) and the flow rate was 1.0 mL min−1. A series of ten near-monodisperse poly(methyl methacrylate) standards (Mp values ranging from 645 to 2186000 g mol−1) were used for calibration.
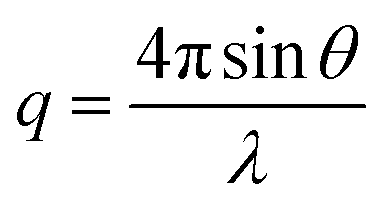 is the modulus of the scattering vector and θ is one-half of the scattering angle. Either polycarbonate or glass capillaries with a diameter of 2.0 mm were used as sample holders. Time-resolved SAXS measurements were performed at ID02 station using a Biologic stopped-flow apparatus. In such experiments, a glass cuvette of 1.4 mm path length was used as the sample holder. Static and time-resolved SAXS measurements were conducted on copolymer dispersions diluted to 1.0% w/w and 0.27% w/w, respectively (with the latter value being the final concentration once aqueous HCl had been added to the aqueous copolymer dispersion in the chamber). Two camera lengths were used to collect the stopped-flow time-resolved SAXS data. The resulting two data sets were subsequently merged at q ∼ 0.015 nm−1. SAXS patterns recorded at these two camera lengths overlapped well (see Fig. 6 and S8 in the ESI†), indicating excellent reproducibility. X-ray scattering data were reduced (integrated, normalised, background-subtracted) using either standard routines from the ID02 beamline or Nika SAS macros for Igor Pro, and were further analysed by Irena SAS macros for Igor Pro.58
is the modulus of the scattering vector and θ is one-half of the scattering angle. Either polycarbonate or glass capillaries with a diameter of 2.0 mm were used as sample holders. Time-resolved SAXS measurements were performed at ID02 station using a Biologic stopped-flow apparatus. In such experiments, a glass cuvette of 1.4 mm path length was used as the sample holder. Static and time-resolved SAXS measurements were conducted on copolymer dispersions diluted to 1.0% w/w and 0.27% w/w, respectively (with the latter value being the final concentration once aqueous HCl had been added to the aqueous copolymer dispersion in the chamber). Two camera lengths were used to collect the stopped-flow time-resolved SAXS data. The resulting two data sets were subsequently merged at q ∼ 0.015 nm−1. SAXS patterns recorded at these two camera lengths overlapped well (see Fig. 6 and S8 in the ESI†), indicating excellent reproducibility. X-ray scattering data were reduced (integrated, normalised, background-subtracted) using either standard routines from the ID02 beamline or Nika SAS macros for Igor Pro, and were further analysed by Irena SAS macros for Igor Pro.58
Conclusions
Polymerisation-induced self-assembly has been used to prepare pH-responsive ABC triblock copolymer vesicles via RAFT seeded emulsion polymerisation of DPA within PGMA–PHPMA precursor vesicles. Only relatively poor control over the copolymer molecular weight distribution was achieved during growth of the third block. Nevertheless, TEM and SAXS studies indicate that the final PGMA–PHPMA–PDPA vesicles exhibit a distinctive framboidal morphology at around neutral pH, with the globule size correlating with the target DP of the PDPA block. On addition of acid, the tertiary amine groups within this block become protonated and hence hydrophilic. This drives vesicular disintegration: only ill-defined aggregated spheres are observed by post mortem TEM analysis and this dramatic change in copolymer morphology is irreversible. SAXS studies confirm the presence of a fractal-like morphology at pH 3, whose features depend on the DP of the PDPA block. Furthermore, time-resolved SAXS studies performed after addition of acid reveal a substantial increase in vesicle diameter and a significantly thicker vesicle membrane after just 50 ms, with vesicle disintegration being complete within one second.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank Christopher Hill and Svetomir Tzokov at the University of Sheffield Biomedical Science Electron Microscopy Suite for their TEM assistance. The authors are grateful to ESRF (France) for providing SAXS beam time, specifically stations BM26 and ID02. SPA thanks the European Research Council for an ERC Advanced Investigator grant (PISA 320372) to support CJM and also EPSRC for a Platform grant (EP/J007846/1) to support MJD.References
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. K. Brannan and F. S. Bates, Macromolecules, 2004, 37, 8816–8819 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- K. T. Kim, J. Zhu, S. A. Meeuwissen, J. J. L. M. Cornelissen, D. J. Pochan, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2010, 132, 12522–12524 CrossRef CAS PubMed.
- D. A. Christian, A. Tian, W. G. Ellenbroek, I. Levental, K. Rajagopal, P. A. Janmey, A. J. Liu, T. Baumgart and D. E. Discher, Nat. Mater., 2009, 8, 843 CrossRef CAS PubMed.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- U. Hasegawa, T. Nishida and A. J. van der Vlies, Macromolecules, 2015, 48, 4388–4393 CrossRef CAS.
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- G. Delaittre, M. Save and B. Charleux, Macromol. Rapid Commun., 2007, 28, 1528–1533 CrossRef CAS.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS PubMed.
- Y. T. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- J. Du, Y. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982–17983 CrossRef CAS PubMed.
- A. Blanazs, M. Massignani, G. Battaglia, S. P. Armes and A. J. Ryan, Adv. Funct. Mater., 2009, 19, 2906–2914 CrossRef CAS.
- K. E. B. Doncom, C. F. Hansell, P. Theato and R. K. O'Reilly, Polym. Chem., 2012, 3, 3007–3015 RSC.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef CAS.
- G. Battaglia and A. J. Ryan, J. Am. Chem. Soc., 2005, 127, 8757–8764 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 12450–12453 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2013, 136, 1023–1033 CrossRef PubMed.
- G. Liu, Q. Qiu and Z. An, Polym. Chem., 2012, 3, 504–513 RSC.
- G. Delaittre, M. Save, M. Gaborieau, P. Castignolles, J. Rieger and B. Charleux, Polym. Chem., 2012, 3, 1526–1538 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- V. J. Cunningham, L. P. D. Ratcliffe, A. Blanazs, N. J. Warren, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Polym. Chem., 2014, 5, 6307–6317 RSC.
- M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Angew. Chem., Int. Ed., 2017, 56, 1746–1750 CrossRef CAS PubMed.
- Y. W. Pei, N. C. Dharsana, J. A. Van Hensbergen, R. P. Burford, P. J. Roth and A. B. Lowe, Soft Matter, 2014, 10, 5787–5796 RSC.
- Y. W. Pei, K. Jarrett, M. Saunders, P. J. Roth, C. E. Buckley and A. B. Lowe, Polym. Chem., 2016, 7, 2740–2750 RSC.
- J. R. Lovett, N. J. Warren, S. P. Armes, M. J. Smallridge and R. B. Cracknell, Macromolecules, 2016, 49, 1016–1025 CrossRef CAS PubMed.
- N. J. W. Penfold, J. R. Lovett, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2017, 8, 272–282 RSC.
- N. J. W. Penfold, J. R. Lovett, N. J. Warren, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2016, 7, 79–88 RSC.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2015, 54, 1279–1283 CrossRef CAS PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 2nd edn, 1991 Search PubMed.
- H. Zhang and E. Ruckenstein, Macromolecules, 2000, 33, 4738–4744 CrossRef CAS.
- M. Save, J. V. M. Weaver, S. P. Armes and P. McKenna, Macromolecules, 2002, 35, 1152–1159 CrossRef CAS.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- K. G. Marinova, R. G. Alargova, N. D. Denkov, O. D. Velev, D. N. Petsev, I. B. Ivanov and R. P. Borwankar, Langmuir, 1996, 12, 2045–2051 CrossRef CAS.
- J. A. Balmer, O. O. Mykhaylyk, A. Schmid, S. P. Armes, J. P. A. Fairclough and A. J. Ryan, Langmuir, 2011, 27, 8075–8089 CrossRef CAS PubMed.
- J. A. Balmer, O. O. Mykhaylyk, J. P. A. Fairclough, A. J. Ryan, S. P. Armes, M. W. Murray, K. A. Murray and N. S. J. Williams, J. Am. Chem. Soc., 2010, 132, 2166–2168 CrossRef CAS PubMed.
- L. A. Fielding, O. O. Mykhaylyk, S. P. Armes, P. W. Fowler, V. Mittal and S. Fitzpatrick, Langmuir, 2012, 28, 2536–2544 CrossRef CAS PubMed.
- L. A. Fielding, O. O. Mykhaylyk, A. Schmid, D. Pontoni, S. P. Armes and P. W. Fowler, Chem. Mater., 2014, 26, 1270–1277 CrossRef CAS.
- J. Bang, S. Jain, Z. Li, T. P. Lodge, J. S. Pedersen, E. Kesselman and Y. Talmon, Macromolecules, 2006, 39, 1199–1208 CrossRef CAS.
- L. J. Fetters, D. J. Lohse and R. H. Colby, in Physical Properties of Polymers Handbook, ed. J. Mark, Springer New York, 2007, ch. 25, pp. 447–454, DOI:10.1007/978-0-387-69002-5_25.
- J. S. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929–1937 CrossRef CAS PubMed.
- K. Larson-Smith, A. Jackson and D. C. Pozzo, J. Colloid Interface Sci., 2010, 343, 36–41 CrossRef CAS PubMed.
- O. Glatter and O. Kratky, Small-Angle X-ray Scattering, Academic Press, London, 1982 Search PubMed.
- V. J. Cunningham, L. P. D. Ratcliffe, A. Blanazs, N. J. Warren, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Polym. Chem., 2014, 5, 6307–6317 RSC.
- D. J. Growney, O. O. Mykhaylyk, T. Derouineau, L. A. Fielding, A. J. Smith, N. Aragrag, G. D. Lamb and S. P. Armes, Macromolecules, 2015, 48, 3691–3704 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1995, 28, 717–728 CrossRef CAS.
- Z. S. Gao and A. Eisenberg, Macromolecules, 1993, 26, 7353–7360 CrossRef CAS.
- Z. Tuzar and P. Kratochvil, Adv. Colloid Interface Sci., 1976, 6, 201–232 CrossRef CAS.
- S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195–217 CrossRef.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details for the synthesis and characterisation of the diblock and triblock copolymer vesicles, assigned 1H NMR spectra, GPC traces, digital images of dispersions, additional plots of pH vs. hydrodynamic diameter, count rate and zeta potential, a table of SAXS parameters, extra data for G58H250D184 and un-merged SAXS patterns are provided. See DOI: 10.1039/c7sc04788f |
| This journal is © The Royal Society of Chemistry 2018 |