DOI:
10.1039/C7SC04749E
(Edge Article)
Chem. Sci., 2018,
9, 1014-1021
Designing interactions by control of protein–ligand complex conformation: tuning arginine–arene interaction geometry for enhanced electrostatic protein–ligand interactions†
Received
2nd November 2017
, Accepted 1st December 2017
First published on 4th December 2017
Abstract
We investigated galectin-3 binding to 3-benzamido-2-O-sulfo-galactoside and -thiodigalactoside ligands using a combination of site-specific mutagenesis, X-ray crystallography, computational approaches, and binding thermodynamics measurements. The results reveal a conformational variability in a surface-exposed arginine (R144) side chain in response to different aromatic C3-substituents of bound galactoside-based ligands. Fluorinated C3-benzamido substituents induced a shift in the side-chain conformation of R144 to allow for an entropically favored electrostatic interaction between its guanidine group and the 2-O-sulfate of the ligand. By contrast, binding of ligands with non-fluorinated substituents did not trigger a conformational change of R144. Hence, a sulfate–arginine electrostatic interaction can be tuned by the choice of ligand C3-benzamido structures to favor specific interaction modes and geometries. These results have important general implications for ligand design, as the proper choice of arginine–aromatic interacting partners opens up for ligand-controlled protein conformation that in turn may be systematically exploited in ligand design.
Introduction
Structure-based ligand design relies on careful study and analysis of a protein (or other target molecule) structure, often in complex with a ligand, obtained by e.g. X-ray diffraction, NMR spectroscopy, and/or homology modeling. Analysis of the physicochemical properties of the protein binding site suggests ideas for molecular design that introduce novel and potentially affinity-enhancing interactions. Hence, such analyses guide the medicinal chemist as to where productive interactions, e.g. hydrogen bonds, ionic interactions, π-interactions etc., may be formed and thus in the design of novel, improved ligands.1 This strategy thus focuses on how to alter the ligand to better match the protein site and not on influencing the protein site to better match the ligand properties. Ligand design is usually based on a static view of protein structure, but significant advances towards handling protein conformational flexibility have been made.2 Protein conformational variability is often observed among a given series of ligand–protein complexes and protein conformational adaptation leading to affinity-enhancing interaction with ligands is frequently observed when analysing protein–ligand structure complexes, for example as in the complex between Oseltamivir and influenza neuraminidase.3 However, the concept of using designed ligand-induced control of protein conformation as a means to create novel interaction sites has less often been a core strategy in ligand discovery. Following our earlier discoveries on conformational variability of aryl–arginine interactions in ligand binding to galectin-3,4,5 here we demonstrate how the choice of phenyl substituents influences the geometry of face-to-face stacking interactions with an arginine guanidino group and how this in turn opens up for formation of a favoured electrostatic interaction between the same arginine and a ligand anionic functionality.
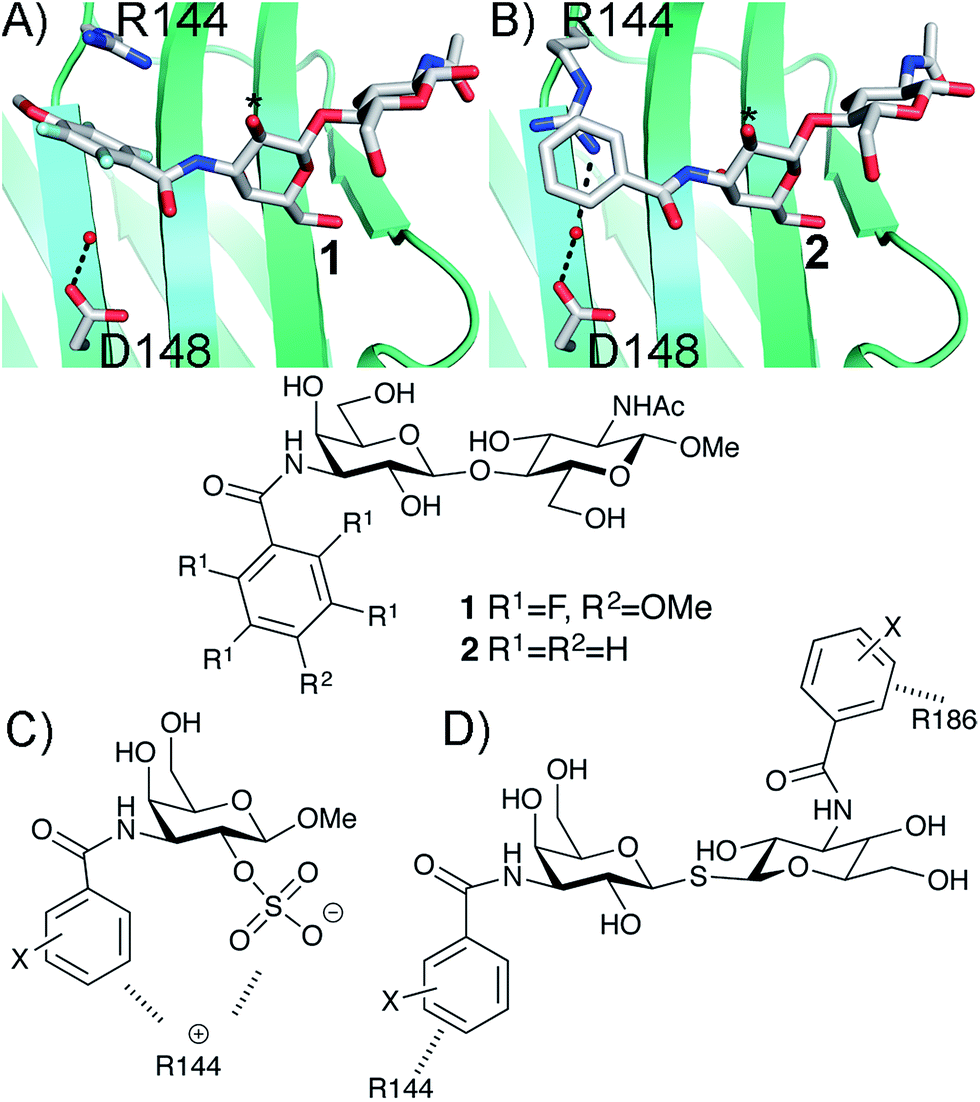
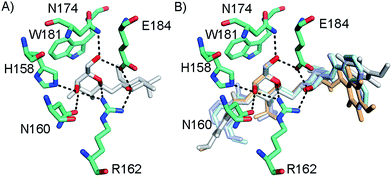
The β-D-galactopyranoside-binding protein galectin-3 belongs to a family of lectins that participates in a number of biologically important processes, of which several are pathological under certain circumstances.6–10 An advantageous feature of galectin-3 is the availability of ample structural and thermodynamic data for galectin-3-ligand complexes, which makes galectin-3 a suitable model protein for investigations of design strategies based on protein–ligand conformational adaptability. Structural analysis of galectin-3 in complex with a LacNAc derivative carrying a 4-methoxy-2,3,5,6-tetrafluoro benzamide at C3 revealed that this ligand benzamido moiety induces a large conformational change in the galectin-3C residue R144, moving it away from a water-mediated salt bridge with N148 on the surface of galectin-3 and positioning the benzamide between the protein backbone and R144 (ref. 5) to form an arginine–arene interaction (Fig. 1A). Later, a structural analysis of galectin-3 complex with a LacNAc derivative carrying an unsubstituted benzamide at C3 revealed that R144 maintains its interaction with N148, while the benzamide moiety is positioned on top of R144,4 again forming an arginine–arene interaction but with an alternative complex geometry (Fig. 1B). Furthermore, galectin-3 conformational dynamics and entropies were affected differently by 4-methoxy-2,3,5,6-tetrafluorobenzamide and unsubstituted benzamide, respectively.4 Hence, the arene properties of the ligand determine the interaction mode and the protein conformation and dynamics. The conformational variability of galectin-3 R144 subsequently led to the hypothesis that compounds with electron-rich groups at the galactose 2-O position could form beneficial interactions with the R144 side-chain conformation properly controlled by the ligand C3 aromatic amide moiety, as shown in Fig. 1A. Indeed, electron-rich substituents at galactose 2-O were evidenced to be affinity-enhancing and this was suggested to be due to interactions with R144 (ref. 11 and 12) (Fig. 1C).
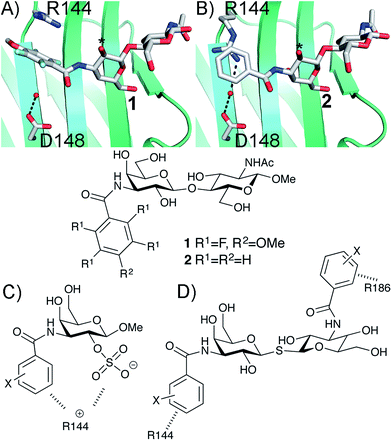 |
| | Fig. 1 X-ray structures4,5 of galectin-3 C-terminal domain in complex with ligands derived from LacNAc carrying (A) a 4-methoxy-2,3,5,6-tetrafluoro-benzamide 2 or (B) a benzamide 1 at C3′,5 revealing the conformational variability in R144. Galactose O2 is indicated with *. (C) Galectin-3 ligands with a sulfate at the 2-O-position were beneficial for the affinity. This was hypothesized to be due to interactions with the positively charged amino acids.11,12 (D) 3,3-Dibenzamido-thiodigalactosides high-affinity ligands of galectin-3 proposed to be due to benzamide interactions with both R144 and R186.13,14 | |
3,3′-Dibenzamido-thiodigalactosides have been shown to be galectin-3 ligands with much higher affinity than the simpler C3-benzamido LacNAc derivatives,13,14 which was explained by two benzamide–arginine interactions involving not only R144 but also R186 (Fig. 1D). The high affinity of the 3,3-dibenzamido-thiodigalactosides for galectin-3 provided an impetus to investigate electron-rich sulfate substituents at one of the galactose 2-O positions of these structures. Herein, we report synthesis, structural, and thermodynamic analysis of 2-sulfo-galactoside and -thiodigalactoside derivatives carrying C3-amides that either induce or do not induce a conformational change in R144 on the surface of galectin-3, as well as the exploitation of such structures to further control R144 conformation and interactions.
Results and discussion
Synthesis and galectin-3 affinity determination of 3-amido-galactosides
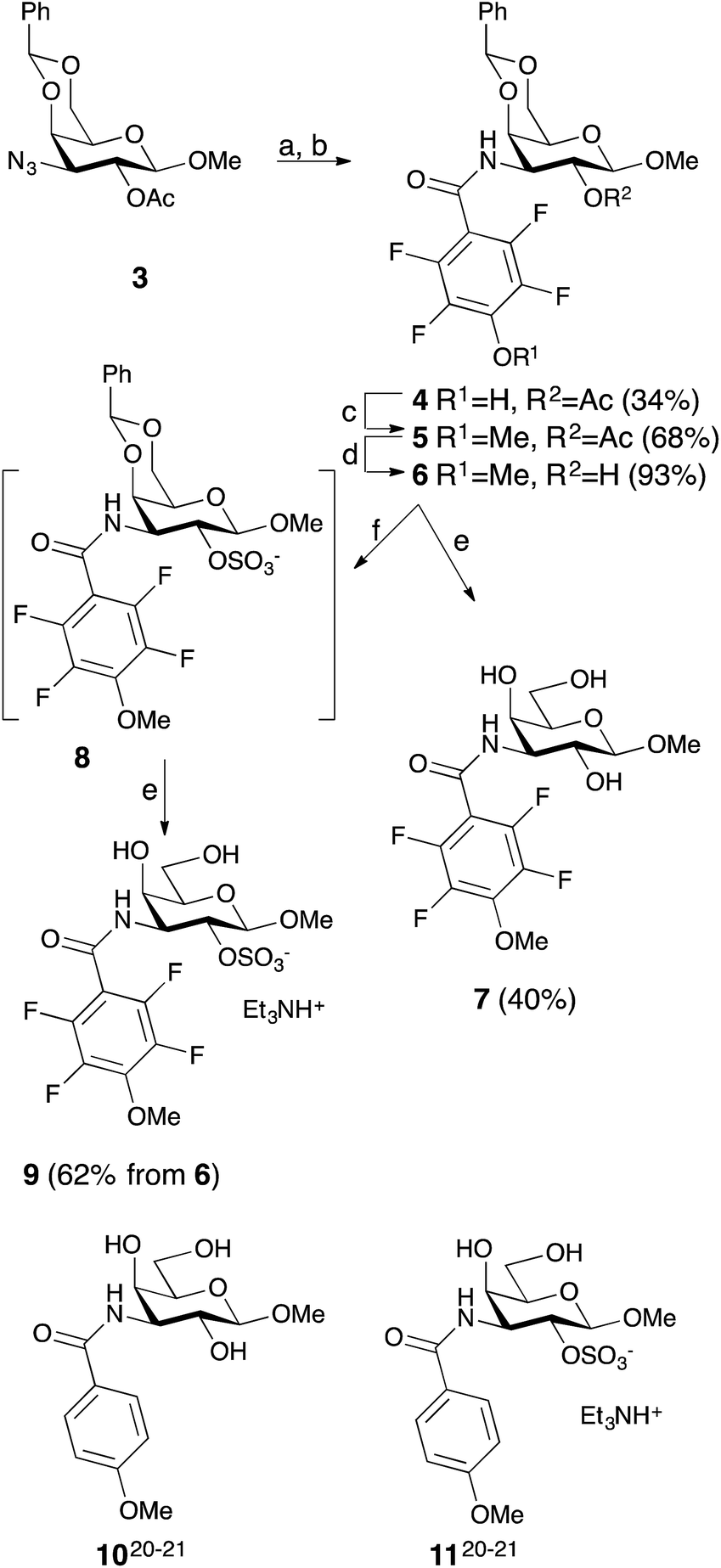
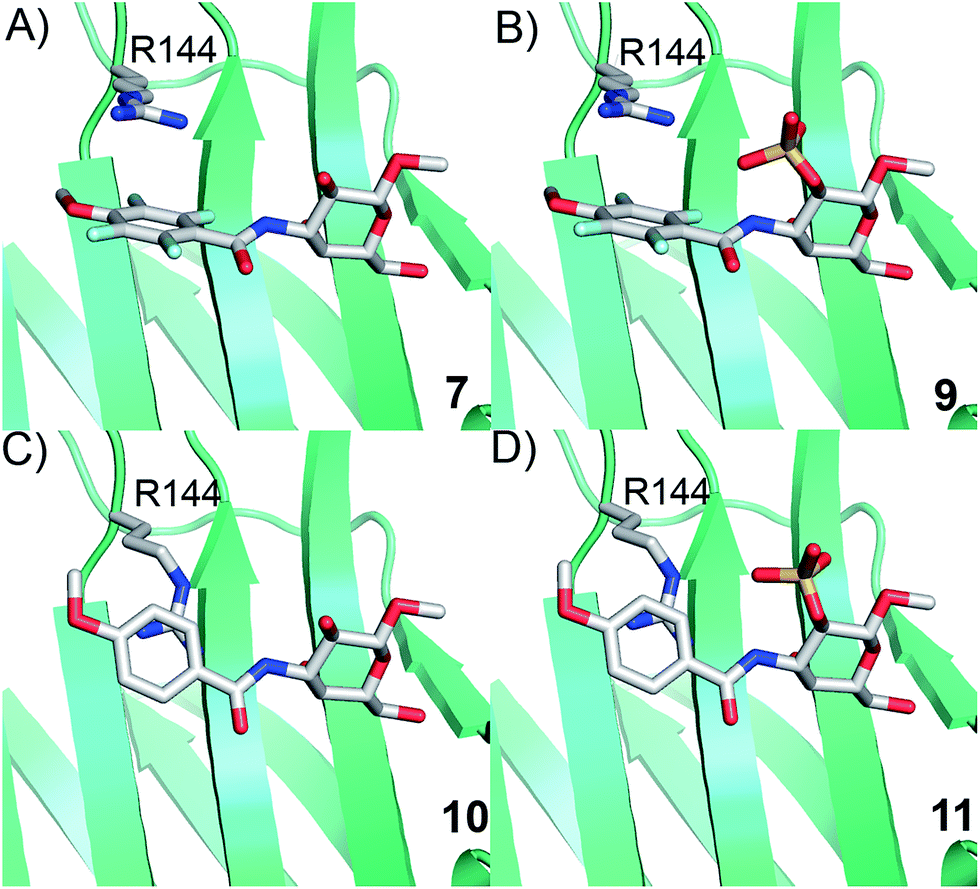
Simplified model compounds, C3 4-methoxy-2,3,5,6-tetrafluorobenzamide-derivatized monogalactosides without (7) and with (9) a 2-O-sulfate, were first synthesized, essentially following routes reported earlier,11,12 and evaluated for galectin-3 binding properties (Scheme 1). The affinities of the monosaccharides 7, 9, 10, and 11 for galectin-3 were determined in a competitive fluorescence polarization assay15,16 and differences in the free energy of binding (ΔΔG) between the fluorinated (7 and 9) and non-fluorinated (10 and 11) monosaccharides were considerably greater when the benzamide is fluorinated (Table 1), supporting a hypothesis that the tetrafluorinated benzamide of 9 induced an R144 conformation more favourable for interacting with the 2-O-sulfate of 9. Although the affinity for the non-sulfated fluorinated 7 was lower by a factor of 2 compared to the non-sulfated and non-fluorinated 10, the addition of a 2-O-sulfate improved the affinity by a factor of 20 for the fluorinated amides (7 → 9) resulting in 9 showing the highest overall affinity. Adding a 2-O-sulfate to the non-fluorinated 10 to obtain the sulfated non-fluorinated 11 only improved the affinity by a factor of 2, similar to earlier observations with a series of p-substituted non-fluorinated analogs.11,12 Taken together, these observations suggest that the fluorinated benzamide in itself is less affinity-enhancing when positioned at a monogalactoside C3 (e.g.7), but in combination with a 2-O-sulfate a strong affinity-enhancing interaction due to a conformational change in R144 and a strengthened interaction between the 2-O-sulfo group and R144 in 9. The non-fluorinated 11 presumably does not reorient R144, thereby precluding the formation of a close interaction between R144 and the 2-O-sulfate of 11, leading to a more modest increase in affinity upon introduction of the sulfate group. Modeled binding modes illustrating this interpretation are shown in Fig. 2. However, the conclusion has to be drawn with some caution as it is based on modeling and an assumption that the fluorinated benzamides of 7 and 9 induce movement of R144 as observed for the corresponding LacNAc derivative.5 Hence, we next investigated the concept in the framework of high-affinity thiodigalactoside ligands,13,14 and included detailed X-ray structural and ITC thermodynamic analyses.
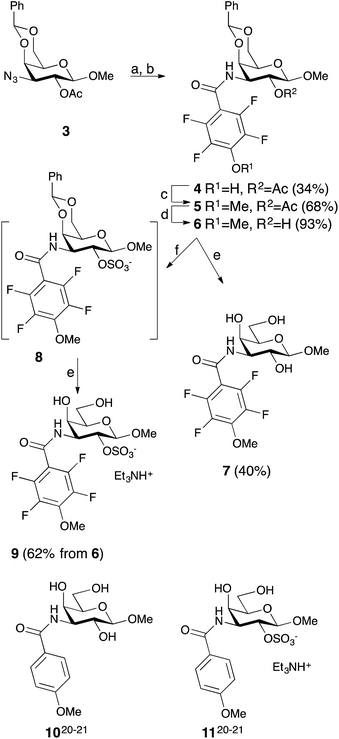 |
| | Scheme 1 Synthesis of the 3-(4-methoxy-2,3,5,6-tetrafluorobenzamido)-β-D-galactosides 7 and 9 and structures of compounds 10 and 11. (a) H2, Pd/C, MeOH, HOAc. (b) 2,3,5,6-Tetrafluoro-4-methoxybenzoyl chloride, pyridine. (c) Me2SO4, K2CO3, acetone. (d) NaOMe, MeOH. (e) HOAc, aq., 67%. (f) SO3–Me3N, DMF. | |
Table 1 Affinities (Kd in μM) of 7, 9, 10, and 11 towards galectin-3 determined in a competitive fluorescence polarization assay.15,16 ΔΔG indicates calculated differences in ΔG (kJ mol−1) between the two fluorinated compounds 7 and 9, and between the non-fluorinated ones 10 and 11
|
|
2-O- |
K
d
|
ΔΔG |
|
7
|
–H |
1300 ± 390 |
|
|
9
|
–SO3– |
65 ± 16 |
−7.4 |
|
10
|
–H |
620 ± 160 |
|
|
11
|
–SO3– |
370 ± 90 |
−1.3 |
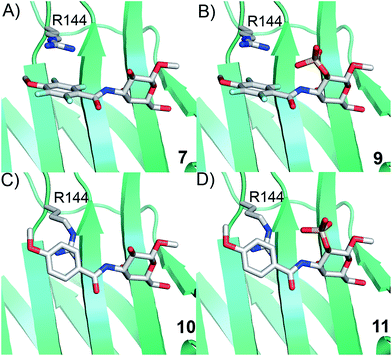 |
| | Fig. 2 Proposed R144 interaction modes for compounds 7, 9, 10, and 11 in complex with galectin-3 (molecular modelling was performed with the MMFFs force field with water implemented in MacroModel version 9.1, Schrödinger, LLC, New York). The galectin-3 structure and ligand and R144 starting conformations were taken from PDB id 1KJL for 10 and 11 and 1KJR for 7 and 9. | |
Synthesis and galectin-3 affinity determination of 3,3′-bis-benzamido-2-O-sulfo-thiodigalactosides – wt, R144K, R144S, R186K, and R186S mutant studies
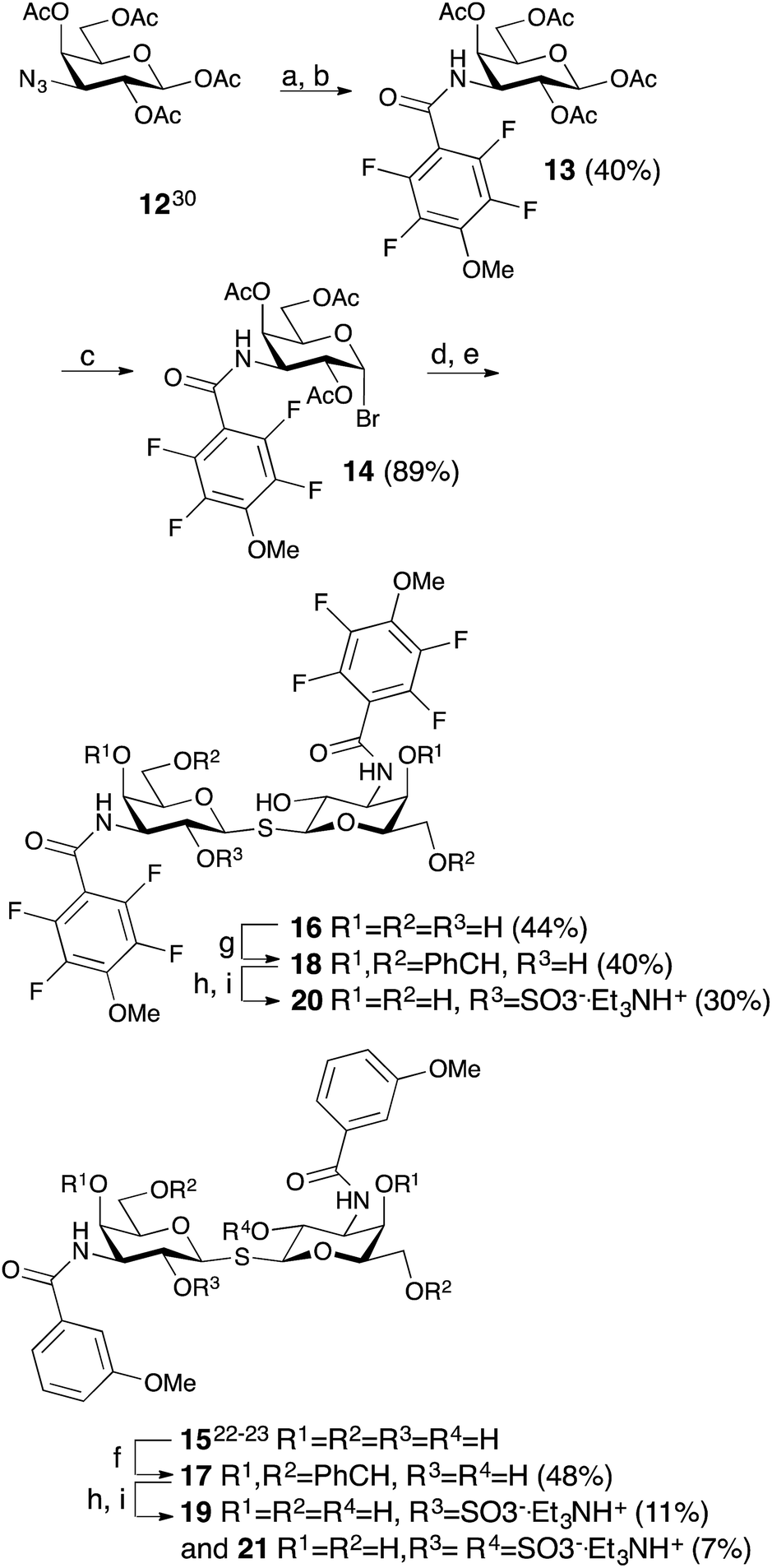
The non-fluorinated bis-3,3′-(3-methoxy-benzamido)-thiodigalactoside derivative 15 is known13,14 and the tetrafluorinated analog 16 was prepared from the known azide 12,17 following the published procedures for 15.11,12 Benzylidene-protection of 15 and 16 to give 17 and 18 followed by sulfation and de-benzylidenation provided the 2-O-sulfates 19 and 20, respectively, together with 2,2′-di-O-sulfate 21 (Scheme 2). Monosulfation of the fluorinated 18 proved more efficient and no disulfate could be isolated.
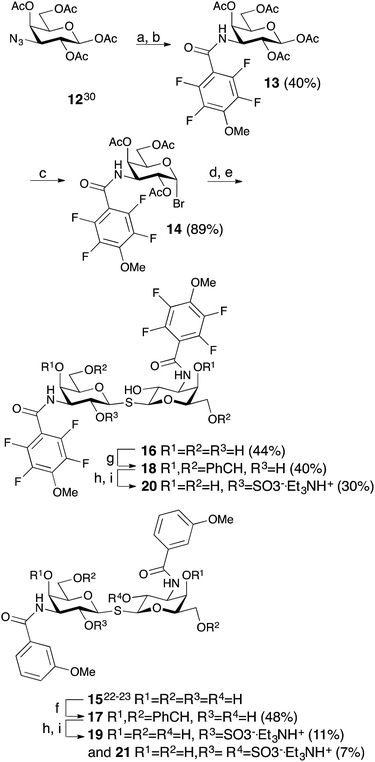 |
| | Scheme 2 Synthesis of thiodigalactosides 15, 16, 19, 20, and 21. (a) H2, Pd/C, MeOH, HOAc. (b) 2,3,5,6-Tetrafluoro-4-methoxybenzoyl chloride, pyridine. (c) HBr, Ac2O, HOAc, CH2Cl2. (d) Na2S, MeCN. (e) NaOMe, MeOH. (f) α,α-dimethoxytoluene, PTSA, MeCN. (g) Benzaldehyde, VO(OTf)2, MeCN. (h) SO3–Me3N, DMF. (i) HOAc, aq., 67%. | |
The introduction of one 2-O-sulfate group on the bis-3,3′-(3-methoxy-benzamido)-thiodigalactoside to give compound 19 did not result in a large affinity improvement over 15 (Table 2). The introduction of a 2-O-sulfate on the bis-3,3′-(4-methoxy-2,3,5,6-tetrafluoro-benzamido)-thiodigalactoside scaffold 20 resulted in a modest affinity improvement over the non-sulfated analog 16, which was only slightly greater in relative terms than that of the bis-3,3′-(3-methoxy-benzamido)-thiodigalactosides 15 and 19 (Table 2). Furthermore, the bis-3,3′-(3-methoxy-benzamido)-thiodigalactosides (15 and 19) displayed a higher affinity for galectin-3 than the fluorinated ones (16 and 20), for both the sulfated and the non-sulfated analogs. Thus, the possible ion pair formed between the 2-O-sulfated bis-3,3′-(4-methoxy-2,3,5,6-tetrafluoro-benzamido)-thiodigalactoside 20 and R144 unexpectedly contributes to only a marginal gain in affinity as compared to the corresponding monosaccharide 9. Furthermore, a higher galectin-3 affinity was observed for the 4-methoxy-2,3,5,6-tetrafluoro-benzamido-LacNAc derivative 2 (Fig. 1, Kd 0.88 μM) than the corresponding 3-methoxy-benzamido-LacNAc derivative (Kd 2.5 μM), which suggests that the added second benzamido moiety in 16 and 20, that is proposed to interact with R186, may negatively influence the affinity for galectin-3. Furthermore, mutant studies support the importance of R144 interacting with both types of benzamide substituents, as a clear decrease in affinity of compounds 15 and 16 is seen for the R144S18 and R144K mutants (Table 2). The sulfated compounds 19 and 20 show a relatively larger loss in affinity for the R144S mutant, but less so for the R144K mutant. This result can be explained by the similar length and charge of lysine and arginine side chains, which both allow for attractive interactions with the sulfate groups of 19 and 20 that cannot occur in the R144S mutant. In case of the R186S18 and R186K mutants, all compounds 15, 16, 19, and 20 have dramatically lower affinity (Table 2), which can be explained by the loss of the favourable interaction between the R186 guanidine group and the benzamide moiety of the ligands.19 By contrast, the limited reduction in affinity observed for the corresponding mutations of R144 (R144S and R144K) might suggest that guanidine–aryl interactions with this residue contributes to free energy of binding to a lesser extent than guanidine–aryl interactions of R186 do. In support of this hypothesis, we have previously observed that R144 undergoes exchange between alternative conformations in all ligand-bound states studied by NMR spectroscopy, whereas R186 is well ordered.4 Altogether, the mutant studies suggest an R144-benzamide interaction in 15, 16, 19, and 20, and the presence of a 2-O-sulfate does affect mutant affinities similarly for both compound pairs (comparing 15 and 19, or 16 and 20).
Table 2 Affinities (Kd in nM) of 15, 16, 19, and 20 towards galectin-3 wt, R144K, R144S, R186K, and R186S, determined in a competitive fluorescence polarization assay.15,16
|
|
wt |
R144S |
R144K |
R186S |
R186K |
|
15
|
110 ± 20 |
215 ± 28 |
220 ± 73 |
42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 7800 000 ± 7800 |
26000 ± 7900 |
|
19
|
57 ± 8 |
550 ± 76 |
76 ± 1 |
33000 ± 5300 |
15000 ± 2200 |
|
16
|
500 ± 51 |
1800 ± 280 |
1300 ± 210 |
79000 ± 1500 |
53000 ± 1300 |
|
20
|
210 ± 40 |
2900 ± 320 |
490 ± 46 |
29000 ± 2900 |
15000 ± 2100 |
Structural analysis of 15, 16, 19, and 20 in complex with galectin-3 C-terminal domain
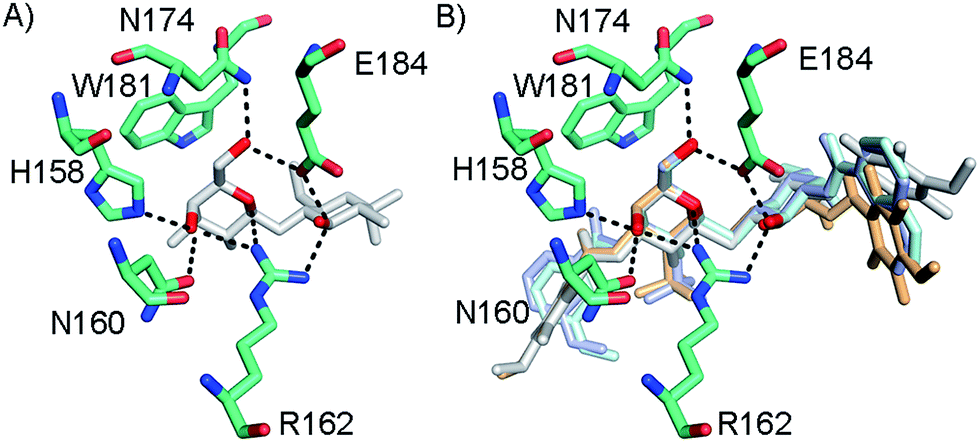
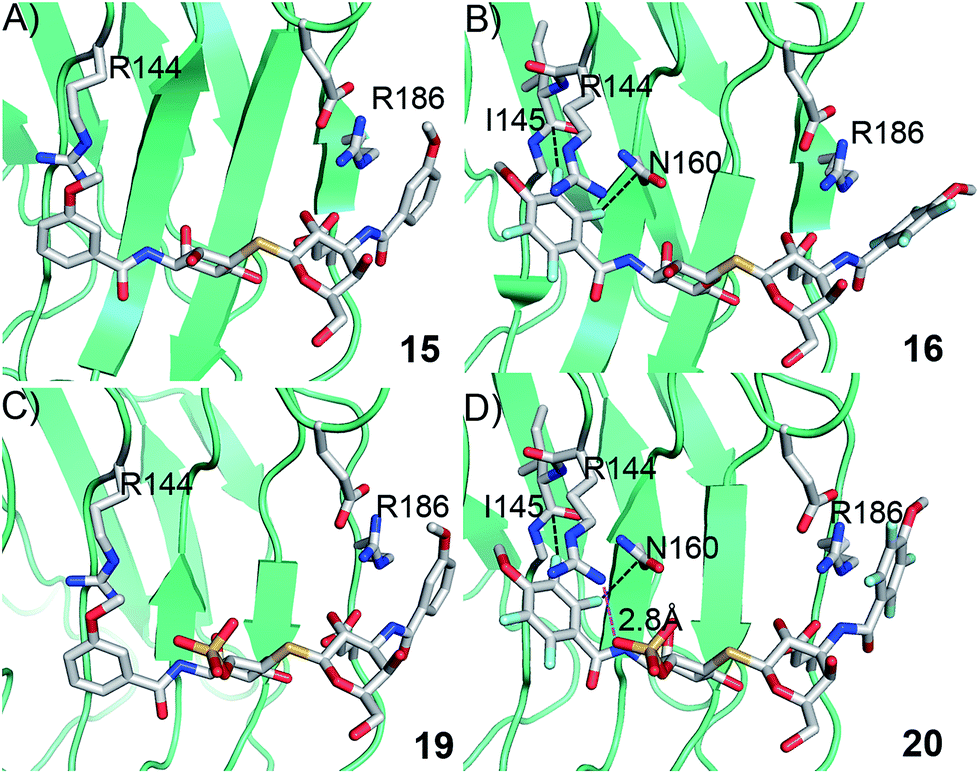
In order to further investigate the hypothesis that the nature of the C3-amide controls the R144 side chain conformation and thus interactions with a 2-O-sulfo group, compounds 15, 16, 19, and 20 were analysed by X-ray crystallography in complex with the galectin-3 C-terminal domain.‡ First, and importantly, the hydrogen bonding patterns to the core thiogalactoside parts of 15, 16, 19, and 20 were virtually identical to those of lactose interacting with galectin-3 (ref. 20) (Fig. 3A–B). The electron densities of the complexes with 15 and 19 clearly revealed an R144 conformation lying along the protein surface and beneath the benzamido moieties, conserving a water-mediated salt bridge to N148, as seen in the lactose complex.20 Hence, the R144 conformation and interaction with one of the benzamides of 15 or 19 are virtually identical to what is observed in complexes with unsubstituted 3′-benzamido-LacNAc.4 Thus, it can be assumed that R144 primarily populates a position that does not allow for a close interaction with the 2-O-sulfo-group of 19 (Fig. 4A and C); the shortest N–O distance between the guanidine of R144 and the sulfate group of 19 is 5.4 Å. In contrast, the complexes with either 16 or 20 revealed an R144 conformation atop one of the 4-methoxy-2,3,5,6-tetrafluoro-benzamide groups, in close proximity to 2-OH and 2-OSO3− of 16 and 20, respectively (Fig. 4B and D). The altered conformational populations of the R144 side-chain in the complexes with 16 and 20, as compared to 15 and 19, may be due to combinations of a desolvation of the fluorinated aromatic ring,21 π–π (ref. 22–24) interactions between the R144 guanidine group and the ligand phenyl group, and the formation of two orthogonal dipolar fluorine-amide bonds25,26 with the backbone amide of I145 (4.0 Å) and side-chain amide of N160 (3.4 Å). Hence, in the crystal structures of complexes with the fluorinated compounds 16 and 20, R144 predominantly populates a conformation that is close to the 2-OSO3− of 20 with a distance of 2.8 Å between the R144 N and sulfate O atoms.
 |
| | Fig. 3 (A) X-ray crystal structures of the galectin-3 C-terminal domain in complex with lactose,20 (B) overlaid X-ray crystal structures of the complexes with 15 (pale cyan), 16 (grey), 19 (light blue), and 20 (light orange) depicting hydrogen bond patterns identical to that of bound lactose. Ligand hydrogen bonding oxygens/hydroxyls are red. | |
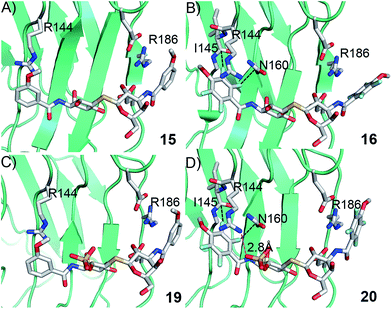 |
| | Fig. 4 X-ray crystal structures of galectin-3 C-terminal domain in complex with compounds (A) 15, (B) 16, (C) 19 and (D) 20, showing the 4-methoxy-2,3,5,6-tetrafluorobenzamide-induced move of the R144 side chain and an ionic interaction at 2.8 Å O–N distance (purple dashed line in panel D) between the 2-O-sulfo-group of 20 and R144; the corresponding distance between 19 and R144 in panel c is 5.4 Å. Two fluorine-amide orthogonal dipolar interactions of 16 and 20 are depicted with black dashed lines for 16 and 20 in (B) and (D). | |
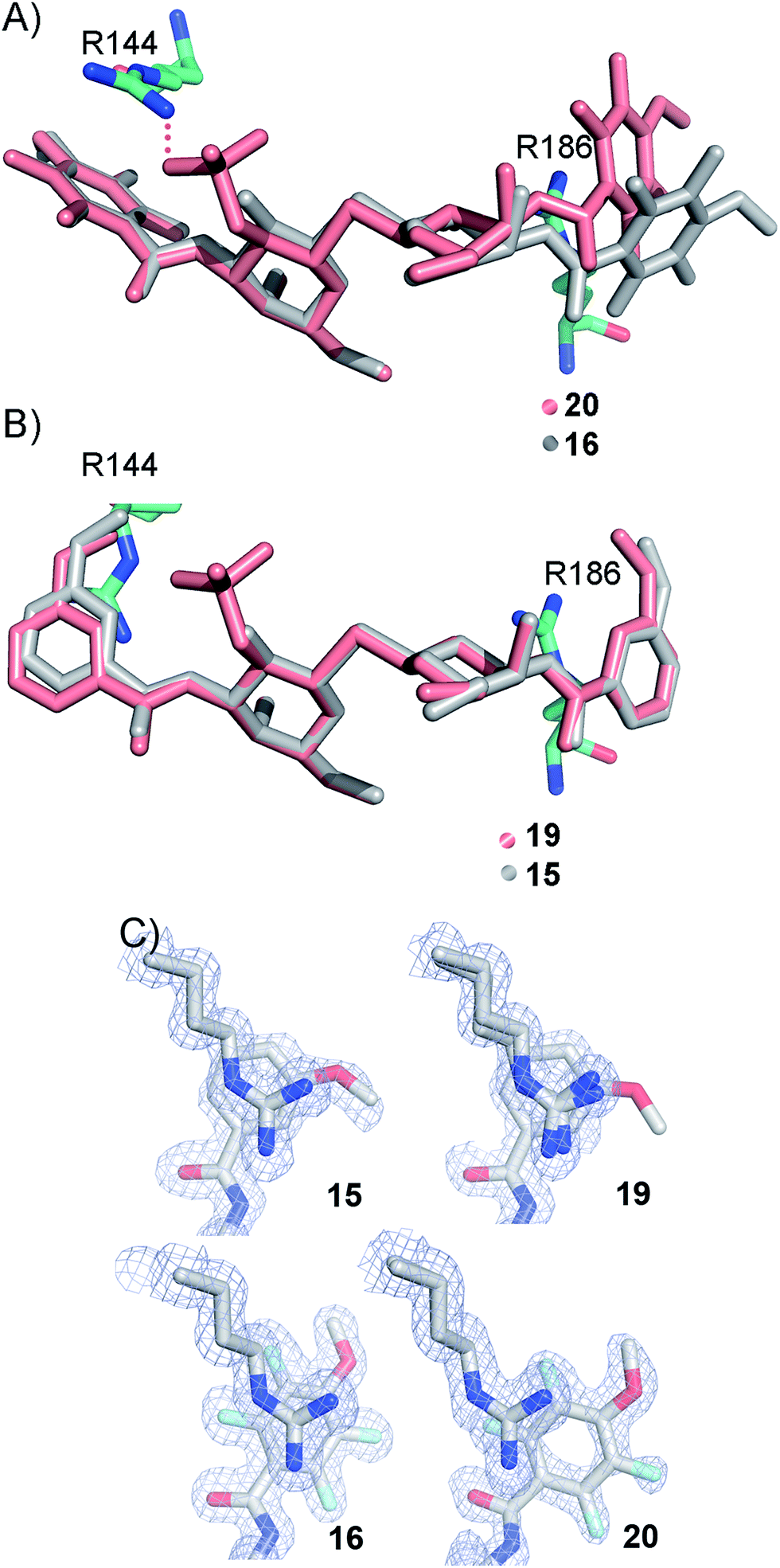
The R186 side chain forms in-plane ion-pairs with E165 and E184, and does not change conformation upon binding of 15, 16, 19, or 20. Face-to-face stacking of one of the benzamides of 15 or 19 onto the R186 side chain is identical for both compounds and strongly supports the hypothesis that such face-to-face stacking of R186 against a ligand phenyl ring is beneficial and contributes to the large affinity gain for bis-amido-thiodigalactosides (e.g.15) over 3′-benzamido-LacNAc derivatives.13,14,19 Alignment of the four complexes of 15, 16, 19, and 20 with galectin-3 reveals that fluorination does not only affect the orientation of R144, but also alters the stacking modes of the benzamido moieties of 16 and 20 onto R186 (Fig. 5A), as compared to the corresponding ones in the complexes of the non-fluorinated 15 and 19 (Fig. 5B). The fluorinated 16 and 20 appears have less optimal π-system overlap with R186 as 16 has the benzamide tilted out of the plane above R186 (Fig. 5B) and 20 is shifted to the edge of the R186 plane (Fig. 5C), which suggests possible explanations for the unexpected relative lower affinity of 16 and 20 for galectin-3 compared to 15 and 19, respectively.
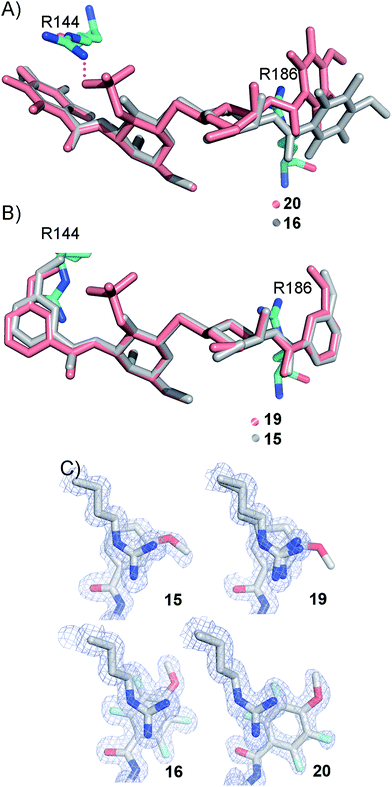 |
| | Fig. 5 (A) Overlaid X-ray crystal structures of galectin-3 complexes with 16 and 20 illustrating differences in benzamide interactions with R186. (B) Overlaid X-ray crystal structures of galectin-3 complexes with 15 and 19 revealing almost identical complex conformations. (C) Experimental electron densities of 15, 19, 16 and 20 over the R186 side chain. | |
Thermodynamic analysis of 3,3′-bis-benzamido-thiodigalactosides 15, 16, 19 and 20
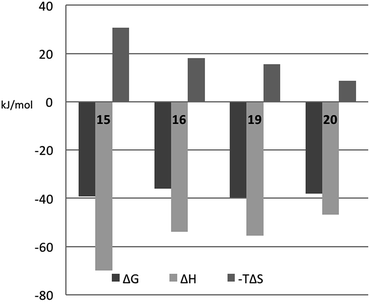
In order to better understand the thermodynamic consequences of the R144 shift in the galectin-3 complexes with 16 and 20 and the induced salt-bridge between R144 and the ligand 2-O-sulfate in 20, thermodynamic parameters of galectin-3 binding to 15, 16, 19, and 20 were determined using isothermal titration microcalorimetry (ITC) (Table 3 and Fig. 6). Importantly, the affinities determined by ITC correspond well with those obtained from the fluorescence polarization assay, thereby validating the use of competitive fluorescence polarization measurements for providing reliable dissociation constants.
Table 3 Isothermal titration microcalorimetry (ITC) and competitive fluorescence polarization (FP) data for compounds 15, 16, 19, and 20 binding to galectin-3 C-terminal domaina
|
|
ΔG (kJ mol−1) |
ΔH (kJ mol−1) |
−TΔS (kJ mol−1) |
n
|
K
d (nM) |
|
ITC data were analyzed by global optimization of multiple data sets. Each compound was fitted using two approaches: the first row shows the results of fitting all parameters to the ITC data, while the second row shows the results obtained when keeping Kd fixed at the value obtained from the competitive FP assay (Table 2).
The relative error in n is less than 3% in all cases.
|
|
15
|
−39.40 ± 0.05 |
−70.0 ± 0.2 |
30.7 ± 0.2 |
0.80 |
126 ± 2 |
|
15
|
−39.7 ± 0.2 |
−69.3 ± 0.5 |
29.6 ± 0.5 |
0.80 |
|
|
19
|
−39.9 ± 0.1 |
−55.4 ± 0.3 |
15.6 ± 0.4 |
1.01 |
103 ± 56 |
|
19
|
−41.3 ± 0.3 |
−53.2 ± 0.4 |
11.9 ± 0.5 |
1.01 |
|
|
16
|
−35.8 ± 0.5 |
−54.0 ± 3.0 |
18.0 ± 3.0 |
0.89 |
604 ± 129 |
|
16
|
−36.3 ± 0.3 |
−52.0 ± 1.0 |
16.0 ± 1.0 |
0.89 |
|
|
20
|
−37.9 ± 0.2 |
−46.8 ± 0.6 |
8.8 ± 0.6 |
1.06 |
261 ± 19 |
|
20
|
−38.5 ± 0.5 |
−46.0 ± 1.0 |
7.2 ± 1.2 |
1.06 |
|
 |
| | Fig. 6 Thermodynamic parameters of binding 15, 16, 19, and 20 to galectin-3, as determined from ITC by fitting all parameters. | |
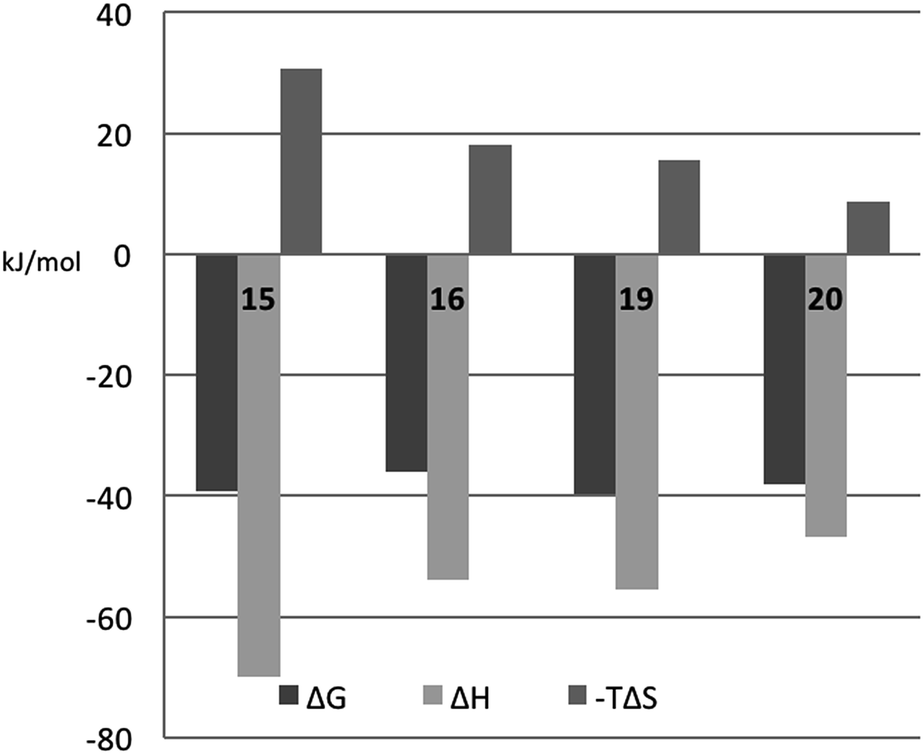
The fluorinated compounds (16 and 20) bind less strongly and with less favourable enthalpy, but also less entropic penalty, than their non-fluorinated counterparts (15 and 19): ΔΔH = 16 kJ mol−1 (16vs.15) and 8 kJ mol−1 (20vs.19) and −TΔΔS = −13 kJ mol−1 (15 and 19) and −7 kJ mol−1 (20vs.19). These results can be rationalized in part by the decreased π-electron density of phenyl rings upon fluorine substitution, which decreases the strength of the cation–π interactions between the guanidine groups of R144 and R186 and the phenyl rings, and by the poor solvation of fluorinated aromatic rings that make binding to the protein relatively more favourable in terms of entropy compared to the non-fluorinated analogues. The former explanation is in line with the structural differences shown in Fig. 5C.
The somewhat higher affinities of the 2-O-sulfated 19 and 20, as compared to the non-sulfated 15 and 16, were also confirmed by ITC, with the effect being slightly greater for the fluorinated ligand, as expected. The affinity increase upon sulfation (15vs.19 and 16vs.20, respectively) is driven by entropy alone, whereas the change in binding enthalpy is unfavourable. These changes in binding enthalpy and entropy induced by the sulfate group, viz. ΔΔH > 0 and −TΔΔS < 0, are expected because the attractive Coulomb interaction between opposite charges (i.e., the guanidine and sulfate groups) is entirely due to favourable entropy and involves unfavourable enthalpy.27,28 We note that the sulfate group of 19 had a much greater influence on both enthalpy and entropy than did the sulfate group of 20. However, analyzing the change in binding free energy (ΔΔG) upon introduction of a 2-O-sulfate to give structures 16 and 20, it is clear that the 2-O-sulfate group of 20 achieves a higher gain in free energy than the 2-O-sulfate group of 19. A useful point of reference is the ratio ΔΔS/ΔΔH,27,28 which should be close to 1.35ΔΔG/(−0.35ΔΔG) = −3.85 in the hypothetical case that ΔΔG is caused entirely by electrostatic interactions. We find that ΔΔS/ΔΔH = −1.3 for the comparison of 20vs.16, while ΔΔS/ΔΔH = −1.0 for the comparison of 19vs.15, thus suggesting that electrostatic effects play a greater role in 20 than in 19, as expected from the crystal structures. However, it should be kept in mind that this simplified analysis neglects all other contributions to binding, such as differential desolvation of the ligands, different interactions in the region surrounding R186, and conformational entropy, all of which surely contribute to ΔΔG.
Conclusions
This investigation demonstrates that it is possible to control a ligand–protein complex structure by choice of aromatic properties of the ligand and thereby favor specific interaction modes and geometries. In the mono-galactoside model system, combining a 2-O-sulfate group with a 4-methoxy-2,3,5,6-tetrafluorobenzamido moiety at C3 leads to a large affinity enhancement as a consequence of the 4-methoxy-2,3,5,6-tetrafluorobenzamido moiety inducing a move of R144 to position its side-chain guanidine moiety ideally for close-range electrostatic interactions with the ligand 2-O-sulfate group. Implementing the combination of a 2-O-sulfate group with a 4-methoxy-2,3,5,6-tetrafluorobenzamido moiety on a thiodigalactoside scaffold did also result in an analogous affinity enhancement, albeit smaller than that observed for the corresponding monosaccharide derivatives. Structural analysis of galectin-3 in complex with a thiodigalactoside carrying fluorinated and non-fluorinated benzamido both with and without 2-O-sulfate groups confirmed that a fluorinated benzamide indeed induces an R144 side-chain move to allow for a close electrostatic interaction with the 2-O-sulfate. This interpretation was supported by thermodynamic data from ITC experiments suggesting that the entropic influence of electrostatic effects is more pronounced in the interaction with the fluorinated benzamide ligand that moves the galectin-3 R144 to a position closer to the ligand sulfate group .
Taken together, this study demonstrates that proper choice of aromatic arginine side-chain interacting partners opens up for ligand-controlled arginine conformations that in turn may be systematically exploited in design of synthetic ligands that induce arginine side chain conformations that form beneficial interactions with electron-rich (anionic) ligand functionalities.
Conflicts of interest
UJN and HL are shareholders in Galecto Biotech AB, a company developing galectin inhibitors.
Acknowledgements
This work was supported by Lund University, the Swedish Research Council (Grants No. 621-2009-5326, 621-2012-2978), the program “Chemistry for Life Sciences” sponsored by the Swedish Strategic Research Foundation, the foundation “Olle Engkvist Byggmästare”, the Royal Physiographic Society, Lund, the European Community's Seventh Framework Program (FP7-2007-2013) under grant agreement no. HEALTH-F2-2011-256986-project acronym PANACREAS, and a project grant awarded by the Knut and Alice Wallenberg Foundation (KAW 2013.0022). We thank the MAX IV laboratory and staff of the I911 beamlines for beamtime and assistance in data collection. We thank Barbro Kahl-Knutsson for assistance with fluorescence anisotropy experiments.
Notes and references
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS PubMed.
- R. Buonfiglio, M. Recanatini and M. Masetti, ChemMedChem, 2015, 10, 1141–1148 CrossRef CAS PubMed.
- J. N. Varghese, P. W. Smith, S. L. Sollis, T. J. Blick, A. Sahasrabudhe, J. L. McKimm-Breschkin and P. M. Colman, Structure, 1998, 6, 735–746 CrossRef CAS PubMed.
- C. Diehl, O. Engström, T. Delaine, M. Håkansson, S. Genheden, K. Modig, H. Leffler, U. Ryde, U. J. Nilsson and M. Akke, J. Am. Chem. Soc., 2010, 132, 14577–14589 CrossRef CAS PubMed.
- P. Sörme, P. Arnoux, B. Kahl-Knutsson, H. Leffler, J. M. Rini and U. J. Nilsson, J. Am. Chem. Soc., 2005, 127, 1737–1743 CrossRef PubMed.
- S. Di Lella, V. Sundblad, J. Cerliani, C. M. Guardia, D. A. Estrin, G. R. Vasta and G. A. Rabinovich, Biochemistry, 2011, 50, 7842–7857 CrossRef CAS PubMed.
- D. Houzelstein, I. R. Goncalves, A. J. Fadden, S. S. Sidhu, D. N. Cooper, K. Drickamer, H. Leffler and F. Poirier, Mol. Biol. Evol., 2004, 21, 1177–1187 CrossRef CAS PubMed.
- H. Leffler, S. Carlsson, M. Hedlund, Y. Qian and F. Poirier, Glycoconjugate J., 2004, 19, 433–440 CrossRef PubMed.
- G. A. Rabinovich and D. O. Croci, Immunity, 2012, 36, 322–335 CrossRef CAS PubMed.
- R. L. Schnaar, J. Leukocyte Biol., 2016, 99, 825–838 CrossRef CAS PubMed.
- C. T. Öberg, H. Leffler and U. J. Nilsson, J. Med. Chem., 2008, 51, 2297–2301 CrossRef PubMed.
- C. T. Öberg, A.-L. Noresson, H. Leffler and U. J. Nilsson, Chem.–Eur. J., 2011, 17, 8139–8144 CrossRef PubMed.
- I. Cumpstey, E. Salomonsson, A. Sundin, H. Leffler and U. J. Nilsson, Chem.–Eur. J., 2008, 14, 4233–4245 CrossRef CAS PubMed.
- I. Cumpstey, A. Sundin, H. Leffler and U. J. Nilsson, Angew. Chem., Int. Ed., 2005, 44, 5110–5112 CrossRef CAS PubMed.
- P. Sörme, B. Kahl-Knutsson, M. Huflejt, U. J. Nilsson and H. Leffler, Anal. Biochem., 2004, 334, 36–47 CrossRef PubMed.
- P. Sörme, B. Kahl-Knutsson, U. Wellmar, U. J. Nilsson and H. Leffler, Methods Enzymol., 2003, 362, 504–512 Search PubMed.
- T. L. Lowary and O. Hindsgaul, Carbohydr. Res., 1994, 251, 33–67 CrossRef CAS PubMed.
- E. Salomonsson, M. Carlsson, V. Osla, R. Hendus-Altenburger, B. Kahl Knutson, C. Oberg, A. Sundin, R. Nilsson, E. Nordberg-Karlsson, U. Nilsson, A. Karlsson, J. M. Rini and H. Leffler, J. Biol. Chem., 2010, 285, 35079–35091 CrossRef CAS PubMed.
- I. Cumpstey, E. Salomonsson, A. Sundin, H. Leffler and U. J. Nilsson, ChemBioChem, 2007, 8, 1389–1398 CrossRef CAS PubMed.
- K. Saraboji, M. Håkansson, S. Genheden, C. Diehl, J. Qvist, U. Weininger, U. Nilsson, H. Leffler, U. Ryde, M. Akke and D. Logan, Biochemistry, 2011, 51, 296–306 CrossRef PubMed.
- J. C. Biffinger, H. W. Kim and S. G. DiMagno, ChemBioChem, 2004, 5, 622–627 CrossRef CAS PubMed.
- D. L. Beene, G. S. Brandt, W. Zhong, N. M. Zacharias, H. A. Lester and D. A. Dougherty, Biochemistry, 2002, 41, 10262–10269 CrossRef CAS PubMed.
- W. Zhong, J. P. Gallivan, Y. Zhang, L. Li, H. A. Lester and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12088–12093 CrossRef CAS.
- M. Giroud, M. Harder, B. Kuhn, W. Haap, N. Trapp, W. B. Schweizer, T. Schirmeister and F. Diederich, ChemMedChem, 2016, 1–6, DOI:10.1002/cmdc.201600132.
- R. Paulini, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 1788–1805 CrossRef CAS PubMed.
- M. Zürcher and F. Diederich, J. Org. Chem., 2008, 73, 4345–4361 CrossRef PubMed.
- V. Petrauskas, E. Maximowitsch and D. Matulis, J. Phys. Chem. B, 2015, 119, 12164–12171 CrossRef CAS PubMed.
- K. A. Sharp, Biopolymers, 1995, 36, 227–243 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis experimental procedures and physical data for compounds. Data processing and refinement statistics for the X-ray crystal structures, 2Fo–Fc electron density maps for 15, 16, 19, and 20 when in complex with galectin-3C, electron densities for the alternate positions of 20 compared to 16, 2Fo–Fc electron density maps for galectin-3 in complex and R144 and R186 interactions with 15, 16, 19, and 20, and copies of 1H NMR spectra for compounds 4, 5, 7, 9, 10, 11, 13, 14, 16, 17, 19–22. See DOI: 10.1039/c7sc04749e |
| ‡ The structures have been deposited in the Protein Data Bank with accession numbers 4BLJ (15), 4BLI (19), 5IUQ (16), and 4BM8 (20). |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d,
M.
Akke
d,
D. T.
Logan
d,
M.
Akke
d,
D. T.
Logan