
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric total syntheses of myrtucommuacetalone, myrtucommuacetalone B, and callistrilones A, C, D and E†
Min-Jing
Cheng
ab,
Jia-Qing
Cao
a,
Xin-Yi
Yang
c,
Li-Ping
Zhong
b,
Li-Jun
Hu
b,
Xi
Lu
c,
Bao-Long
Hou
b,
Ya-Jian
Hu
b,
Ying
Wang
a,
Xue-Fu
You
c,
Lei
Wang
*a,
Wen-Cai
Ye
 *a and
Chuang-Chuang
Li
*b
*a and
Chuang-Chuang
Li
*b
aCollege of Pharmacy, Jinan University, Guangzhou 510632, China. E-mail: cpuwanglei@126.com; chyewc@gmail.com
bDepartment of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: ccli@sustc.edu.cn
cInstitute of Medicinal Biotechnology, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China
First published on 27th November 2017
Abstract
Herein, we describe a concise catalytic approach to the first asymmetric total syntheses of myrtucommuacetalone, myrtucommuacetalone B, and callistrilones A, C, D and E. The syntheses proceed in only 5–7 steps from the readily available compound 11, without the need for protecting groups. Key features of the syntheses include a unique organocatalytic asymmetric Friedel–Crafts-type Michael addition with high enantioselectivity and a broad substrate scope, a novel Michael-ketalization-annulation cascade reaction, and an oxidative [3 + 2] cycloaddition. Furthermore, the new compound 7 exhibited potent antibacterial activities against several multidrug-resistant strains (MRSA, VISA and VRE), and showed greater potency than vancomycin.
Introduction
Polycyclic polymethylated phloroglucinols (PPPs) are a class of natural product isolated mainly from the plants of the Myrtaceae and Guttiferae families. A diverse range of more than 70 PPPs have been isolated. These natural products have become attractive targets for chemists because of their complex structural features, and some PPPs have been reported to exhibit biological activities.1,2 For example, myrtucommulone A (1), which was first isolated in 1974, is highly active against Gram-positive bacteria and cancer cells.1a,b Myrtucommuacetalone (2) exhibits inhibitory activity towards the production of nitric oxide (NO˙), as well as pronounced antiproliferative activity against T-cells.3 Structurally, the naturally occurring PPPs 1–7 are based on a diverse range of complex scaffolds (Fig. 1). Myrtucommuacetalone (2) and myrtucommuacetalone B (3) consist of a synthetically challenging and unprecedented bridged furochromene moiety with a fascinating polycyclic ketal skeleton and a 2-oxabicyclo[3.3.1]nonane scaffold. Callistrilones (4–7) are based on a previously unknown carbon skeleton consisting of a unique [1]benzofuro[2,3-a]xanthene ring system.4 Furthermore, compounds 4 and 5 consist of a sterically compact 6/6/6/5/6/3-fused hexacyclic skeleton, containing six stereocenters with one tetrasubstituted center. Based on their structural complexity, the construction of compounds belonging to this family represents a synthetic challenge.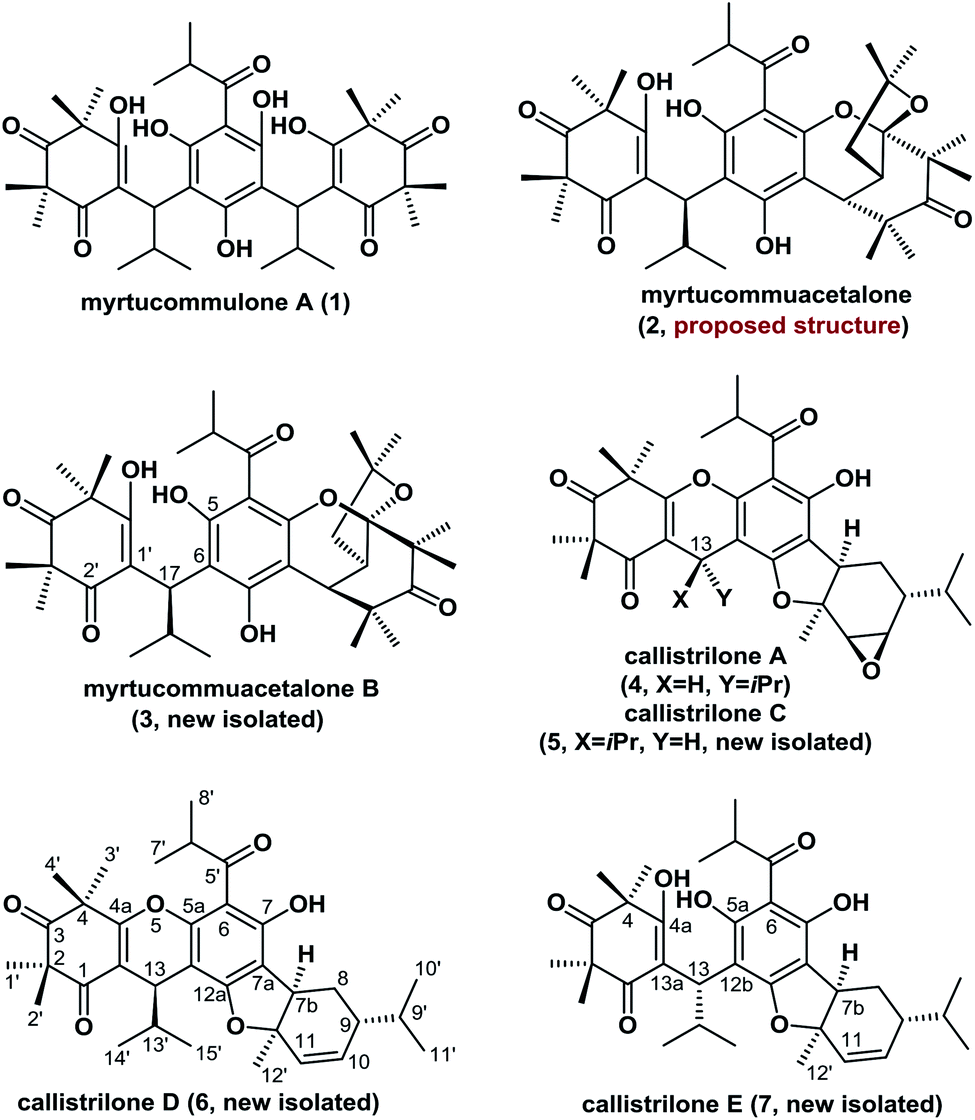 | ||
| Fig. 1 Representative polycyclic polymethylated phloroglucinols. | ||
The synthetically challenging structural motifs of these PPPs together with their promising pharmacological properties have attracted considerable interest from the synthetic community.5,6 In 2010, Jauch et al. reported the first total synthesis of myrtucommulone A (1).5a However, the asymmetric total syntheses of compounds 2–7 are yet to be reported and the catalytic asymmetric and divergent syntheses of PPPs have not been achieved. In our continuing efforts towards the synthesis of biologically active natural products,7 herein we describe the isolation and identification of four novel PPPs (3 and 5–7) from the plants Callistemon rigidus and Myrtus communis. Furthermore, we report the first catalytic asymmetric total syntheses of myrtucommuacetalone, myrtucommuacetalone B and callistrilones A, C, D and E in only 5–7 steps. Notably, the new compound 7 was found to exhibit antibacterial activity against multidrug-resistant strains.
Results and discussion
Isolation and structural elucidation of compounds 3 and 5–7
The new compounds 3 and 5–7 were isolated from the plants C. rigidus and M. communis (Myrtaceae) by column chromatography and preparative high-performance liquid chromatography [see ESI†].The molecular formula of compound 3 was established to be C38H52O9 based on its HRESIMS data (m/z 653.3688 [M + H]+, calcd for C38H53O9: 653.3684). The IR spectrum suggested the presence of aromatic (1592 and 1470 cm−1), hydroxyl (3202 cm−1), and carbonyl groups (1707 cm−1). Comparison of the 1H and 13C NMR data of 3 with those of myrtucommuacetalone (2) revealed that their chemical shifts were similar,3 except for differences in the C-17, C-5 and C-2′ signals, indicating that 3 was a C-17 epimer of 2. This conclusion was further confirmed by X-ray diffraction analysis (see ESI†) and by our total synthesis.
The HRESIMS of 7 showed a quasimolecular ion peak at m/z 567.3328 [M + H]+ (calcd for C34H47O7: 567.3316), consistent with the molecular formula C34H46O7. The 1H and 13C NMR spectra of 7 displayed two sets of signals in a ratio of approximately 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, which suggested that this compound exists as a pair of rotamers owing to the intramolecular hydrogen-bonding. Comprehensive analysis of the NMR data of 7 indicated that it shared the same framework as callistrilone A (4),4 however, the signals for the epoxy carbons in 4 were replaced by signals for olefinic carbons and two additional hydroxyl signals were present in 7. The HMBCs between OH-4a and C-4/C-13a, between OH-5a and C-6/C-12b, and between H-12′ and C-7b/C-11 confirmed its planar structure (Fig. S1–S7†). The unambiguous structural assignments and stereochemistry of 7 could be elucidated by X-ray diffraction (see ESI†) and the successful total synthesis.
:4, which suggested that this compound exists as a pair of rotamers owing to the intramolecular hydrogen-bonding. Comprehensive analysis of the NMR data of 7 indicated that it shared the same framework as callistrilone A (4),4 however, the signals for the epoxy carbons in 4 were replaced by signals for olefinic carbons and two additional hydroxyl signals were present in 7. The HMBCs between OH-4a and C-4/C-13a, between OH-5a and C-6/C-12b, and between H-12′ and C-7b/C-11 confirmed its planar structure (Fig. S1–S7†). The unambiguous structural assignments and stereochemistry of 7 could be elucidated by X-ray diffraction (see ESI†) and the successful total synthesis.
Comparison of the NMR data of 5 and 6 with those of the known compound callistrilone A (4) suggested that they possessed a similar framework.4 A comprehensive analysis of their 1H–1H COSY, HSQC, HMBC, and NOESY spectra led us to conclude that 5 is the C-13 epimer of 4, and the epoxy group in 5 is replaced by olefinic carbons in 6 (see ESI†), as confirmed by our asymmetric total synthesis.
Retrosynthetic analysis of PPPs 2–7
Retrosynthetically (Fig. 2), the bridged polycyclic ketal skeletons in 2 and 3 could be synthesized from 8, by an unreported Michael-ketalization-annulation cascade reaction8 with compound 9 (see the proposed pathway in Scheme 1). Callistrilone A (4) could be synthesized from ent-8 by a biomimetic oxidative [3 + 2] cycloaddition9 with commercially available (−)-α-phellandrene (10), followed by cyclization and epoxidation. In addition, several other natural PPPs isolated from Myrtaceae plants (such as 5–7) could be constructed in a similar manner through a few simple functional-group transformations.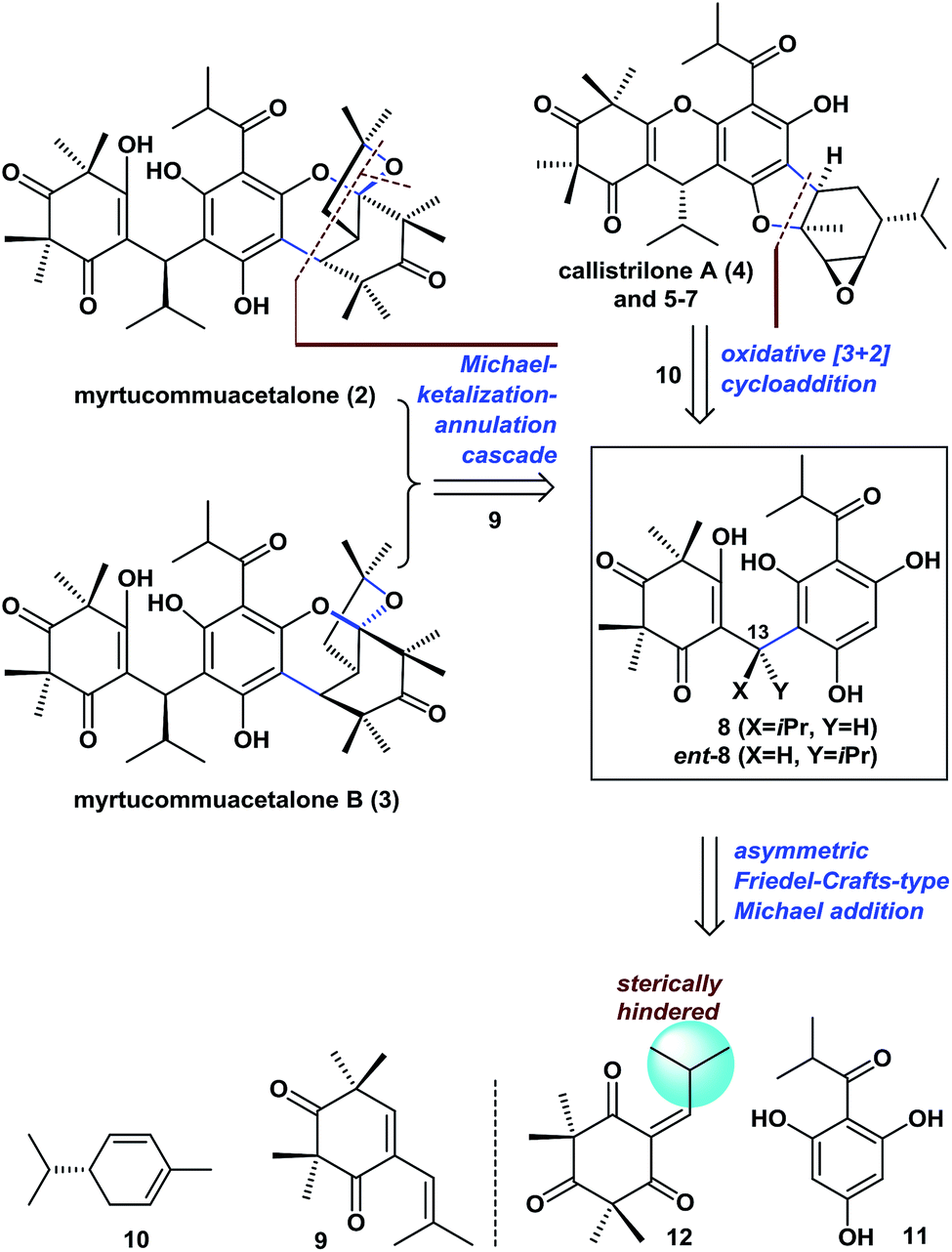 | ||
| Fig. 2 Retrosynthetic analysis of PPPs 2–7. | ||
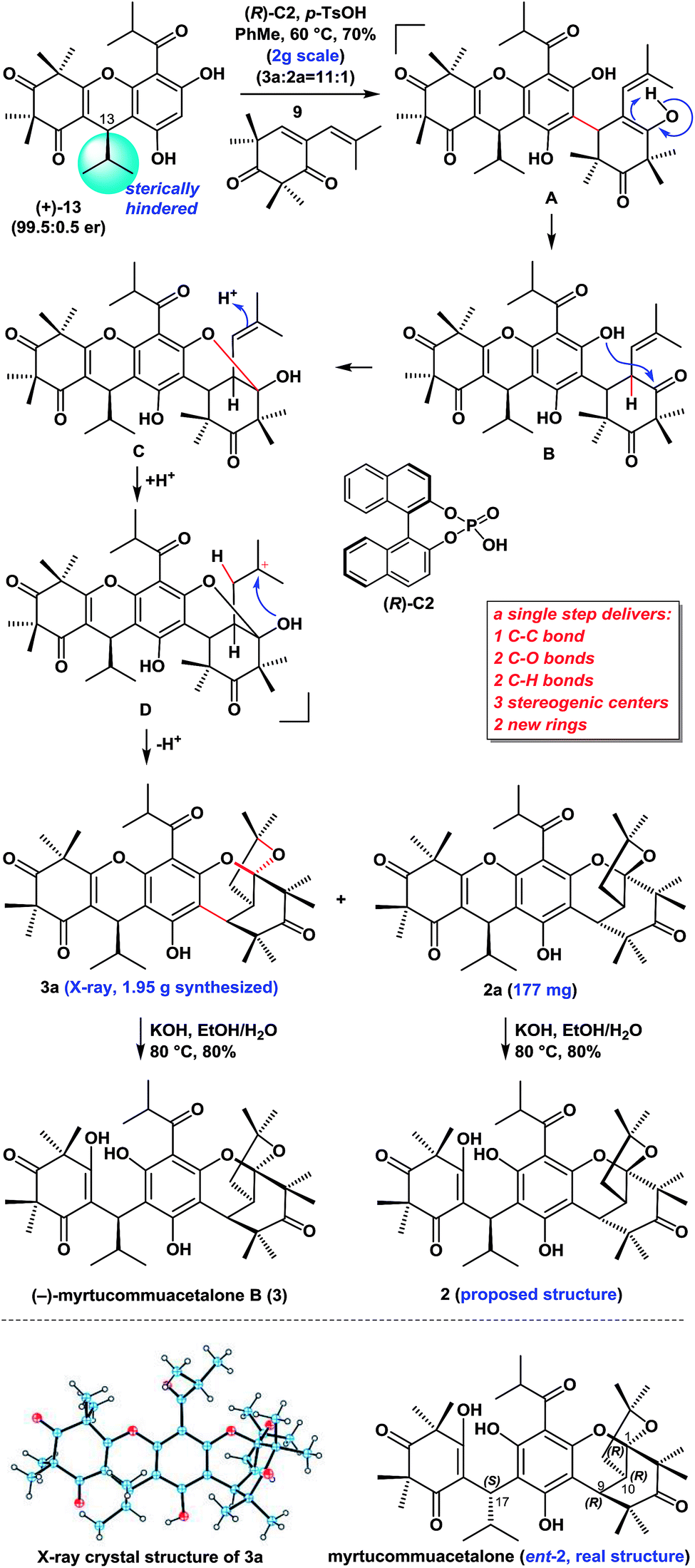 | ||
| Scheme 1 Asymmetric syntheses of 2 and 3. | ||
To achieve the enantioselective synthesis of 2–7, it is essential to prepare 8 or ent-8 with high enantioselectivity from compounds 11 and 12, by Friedel–Crafts-type Michael (FCM) additions (Fig. 2). In recent years, enantioselective FCM additions with active aromatics have been intensively investigated.10 However, current approaches10g–n give only poor yields and/or poor enantioselectivities if the FCM acceptor is sterically hindered or alkyl substituted, as is the case for 12. Moreover, there have been few literature reports concerning phloroglucinol derivatives as FCM donors. In particular, the three unprotected hydroxyl groups in 11, which can undergo a competing oxa-Michael addition to give unexpected products, make this FCM addition more difficult. Recently, Jauch et al. reported the enantioselective synthesis of 8 with an 81:19 enantiomeric ratio (er), through the use of an excess (3 equiv.) of a chiral Al–Li–BINOL (1,1′-bi-2-naphthol) complex. These conditions resulted in an inseparable mixture of chiral (+)-1 with an 85:15 er and meso-1 in a ratio of 59:41.5b Thus, the catalytic and highly enantioselective synthesis of 8 and 2–7 remains a challenge to be addressed and is in demand.
Organocatalytic enantioselective FCM additions
With compounds 11 and 1211 in hand, we proceeded to investigate the enantioselective FCM addition. Inspired by previous elegant work by Luo and co-workers,12 we envisioned that chiral phosphoric acids (CPAs),13 which have been used as powerful organocatalysts for numerous reactions over the past 12 years, might be able to facilitate this transformation enantioselectively. We initially conducted the FCM addition reaction of 11 and 12 in PhMe at −40 °C in the presence of 10 mol% of phosphoric acid (S)-C1 (entry 1, Table 1). Encouragingly, despite the high steric hindrance of 12, the reaction proceeded smoothly to give 8, followed by p-TsOH-mediated cyclization to give (+)-myrtucommulone B (13)1m in 35% yield with an 82.5:17.5 er (see ESI† for details). This proof of principle outcome showed that control of the C13 chirality of 8 (or 13) was possible through the use of a chiral phosphoric acid-catalyzed asymmetric FCM addition. Next, we turned our attention to the effects of the substituents and axial chiral backbone of the catalysts to improve the enantioselectivity. As shown in Table 1, the electron-donating/withdrawing properties and steric bulk of the aromatic-ring substituents, as well as the nature of the backbone, had a considerable influence on the enantioselectivity. We further optimized the reaction conditions by changing the solvents and adding Lewis acids.14 After extensive experimentation, we identified the following protocol to be optimal (entry 31): when 11 was treated with 12 in the presence of catalytic (S)-C15 (10 mol%) and AlF3 (100 mol%) with 3 Å MS in PhMe at −70 °C for 6 days, followed by TsOH-mediated cyclization, (+)-(13R)-131m was obtained in a 75% isolated yield with a 95:5 er (2.0 g scale). After the recrystallization of 13, its er value was improved to 99.5:0.5. Moreover, the catalyst (S)-C15 was easily recoverable and could be reused several times without a considerable loss of activity. We also achieved the highly enantioselective synthesis of (−)-(13S)-ent-13, using (R)-C16 as the catalyst, according to the same procedure as above (entry 32). Notably, the route described above allowed for the facile synthesis of 10 g of both (+)-13 and (−)-ent-13 (see ESI† for details), thereby highlighting the robust nature of this chemistry.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Additive | T (°C) | Solvent | Time | Yieldb (%) | er |
|
a Unless otherwise stated, the reactions of entries 1–30 were performed with 11 (0.1 mmol), 12 (0.2 mmol) and CPA (0.01 mmol) in toluene (2 mL) at −76 °C to 26 °C.
b Isolated yield.
c Determined by chiral HPLC analysis with a ChiralCel OD-H column (i-PrOH/n-hexane = 5:95, 0.8 mL min−1).
d 4 Å MS (35 mg) was added.
e 3 Å MS (35 mg) was added.
f 5 Å MS (35 mg) was added.
g The reactions of entries 31 and 32 were performed with 11 (2.0 g, 10.2 mmol), 12 (20.4 mmol) and (S)-C15 or (R)-C16 (1.02 mmol) in toluene (204 mL) at −70 °C.
h The er could be easily improved by recrystallization.
|
|||||||
| 1 | (S)-C1 | −40 | Toluene | 24 h | 35 | 82.5:17.5 |
|
| 2 | (S)-C2 | −40–26 | Toluene | 24 h | 73 | 50:50 |
|
| 3 | (S)-C3 | −40 | Toluene | 24 h | Trace | — | |
| 4 | (S)-C4 | −40 | Toluene | 24 h | Trace | — | |
| 5 | (S)-C5 | −40 | Toluene | 24 h | 18 | 52.5:47.5 |
|
| 6 | (S)-C6 | −40 | Toluene | 24 h | 24 | 55:45 |
|
| 7 | (R)-C7 | −40 | Toluene | 24 h | 30 | 40:60 |
|
| 8 | (S)-C8 | −40 | Toluene | 24 h | 36 | 60:40 |
|
| 9 | (S)-C9 | −40 | Toluene | 24 h | Trace | — | |
| 10 | (S)-C10 | −40 | Toluene | 24 h | 23 | 62:38 |
|
| 11 | (S)-C11 | −40 | Toluene | 24 h | 21 | 0 | |
| 12 | (S)-C12 | −40 | Toluene | 24 h | Trace | — | |
| 13 | (S)-C13 | −40 | Toluene | 24 h | 25 | 64:36 |
|
| 14 | (S)-C14 | −40 | Toluene | 24 h | 18 | 50:50 |
|
| 15 | (S)-C15 | −40 | Toluene | 24 h | 73 | 88:12 |
|
| 16 | (S)-C15 | −60 | Toluene | 108 h | 76 | 91:09 |
|
| 17 | (S)-C15 | −60 | DCM | 108 h | 70 | 89.5:10.5 |
|
| 18 | (S)-C15 | −60 | CHCl3 | 108 h | 30 | 91:09 |
|
| 19 | (S)-C15d | −60 | Toluene | 108 h | 74 | 92:08 |
|
| 20 | (S)-C15d | MgF2 (10%) | −60 | Toluene | 108 h | 76 | 93:07 |
| 22 | (S)-C15d | Fe(OTf)3 (10%) | −60 | Toluene | 108 h | 74 | 91:09 |
| 23 | (S)-C15d | Zn(OTf)2 (10%) | −60 | Toluene | 108 h | 72 | 92.5:7.5 |
| 24 | (S)-C15d | AIF3 (10%) | −60 | Toluene | 72 h | 71 | 93.5:6.5 |
| 25 | (S)-C15d | CaF2 (10%) | −60 | Toluene | 72 h | 74 | 92.5:7.5 |
| 26 | (S)-C15d | AlF3 (10%) | −76 | Toluene | 7 d | 55 | 95.5:4.5 |
| 27 | (S)-C15e | AlF3 (10%) | −76 | Toluene | 6 d | 25 | 96.5:3.5 |
| 28 | (S)-C15f | AlF3 (10%) | −76 | Toluene | 6 d | 28 | 96:04 |
| 29 | (S)-C15e | AIF3 (100%) | −70 | Toluene | 6 d | 77 | 95:05 |
| 30 | (R)-C16e | AIF3 (100%) | −70 | Toluene | 6 d | 76 | 5:95 |
| 31 | (S)-C15e,g | AlF3 (100%) | −70 | Toluene | 6 d | 75 | 95:5 (99.5:0.5)h |
| 32 | (R)-C16e,g | AlF3 (100%) | −70 | Toluene | 6 d | 75 | 5:95 (0.5:99.5)h |
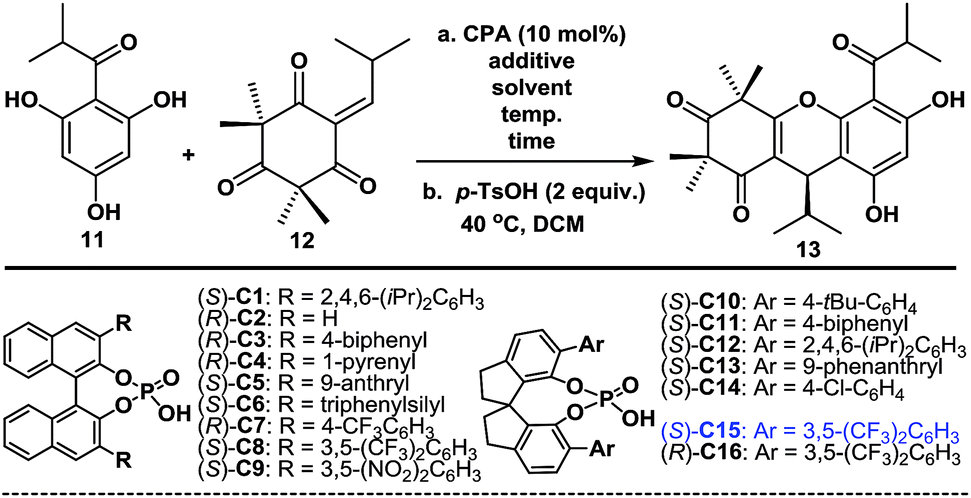
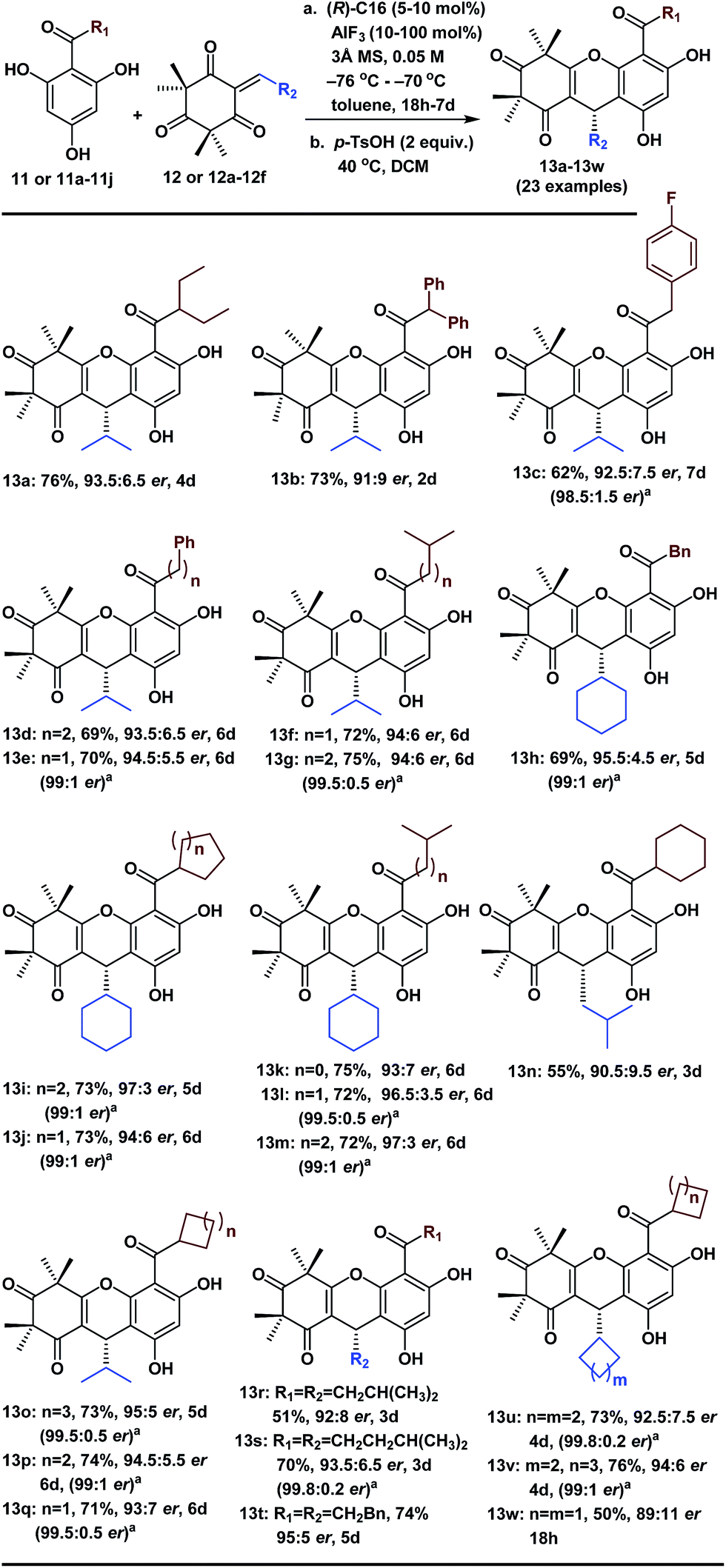
With the optimized conditions in hand, we next examined the substrate scope of various substituted acylphloroglucinols. As shown in Table 2, different substituents of the substrates (11 or 11a–11j, see ESI† for details) were tolerated in the FCM addition with the Michael reaction acceptor 12, to give the corresponding products 13a–13g and 13o–13q in good yields with er values of 91:9 to 95:5. Hence, our method has broad generality for the synthesis of polycyclic polymethylated phloroglucinol derivatives. Notably, when the R1 group was CHX2 (X = Et or Ph), the reactions were accelerated (13a and 13b); however, the er values were slightly lower. Interestingly, when the R1 group was a 5-, 6- or 7-membered ring, there were no notable effects on the enantioselectivity of the FCM additions (13o, 13p and 13q).
| a The er could be easily improved by recrystallization. |
|---|
|
Moreover, to validate the generality of this transformation, we evaluated the use of various sterically hindered Michael reaction acceptors (12 or 12a–12f, see ESI† for details) with the various Michael reaction donors (11 and 11a–11j). Most of the reactions reached completion within 6 days and gave the desired products 13h–13m and 13s–13v in good yields with good or excellent enantioselectivities. Notably, the products (13n, 13r and 13w) were isolated in lower yields because the byproducts of the double-FCM additions increased as the reaction time was extended. When the R2 group was a 6-, 5- or 4-membered ring with a different cyclic tension, there were considerable effects on the reaction rates (13j took 6 days, 13u took 4 days and 13w took 18 h). Furthermore, in many cases, after the recrystallization of the corresponding phloroglucinol products, the er improved to 99:1–99.8:0.2. These polycyclic polymethylated phloroglucinol derivatives will be beneficial for the asymmetric synthesis of other diverse PPPs.1
Asymmetric syntheses of myrtucommuacetalone (2) and myrtucommuacetalone B (3)
With compounds 911 and (+)-13 in hand, we proceeded to investigate our proposed syntheses of the natural myrtucommuacetalone (2) and myrtucommuacetalone B (3) (Scheme 1). We subsequently evaluated the proposed Michael-ketalization-annulation cascade sequence using (+)-13 and 9 as substrates with a variety of catalysts (i.e., TFA, p-TsOH, CSA, AcOH and some chiral phosphoric acids, as shown in Table 1) and solvents (i.e., DCM, DCE, THF, DME, CHCl3 and PhMe). Rewardingly, we found that the treatment of a mixture of 13 and 9 with (R)-C2 and TsOH (1:1.5) in PhMe at 60 °C resulted in the expected Michael-ketalization-annulation cascade reaction to give the desired diastereoisomers 3a and 2a with sterically compact hexacyclic skeletons in a combined yield of 70% (2.0 g) and a ratio of 11:1. However, it is worth mentioning that without the chiral acid (R)-C2, the treatment of 13 and 9 with p-TsOH (1.5 equiv.) in similar conditions resulted in the formation of 3a and 2a in a combined yield of 60% and a ratio of 4:1. These compounds were readily separated by recrystallization. The structure of 3a was unambiguously confirmed by X-ray crystallography.
We envisaged that (+)-13 might undergo a Michael (or FCM) addition to 9 in the presence of (R)-C2 and p-TsOH to generate intermediate B (presumably from A). The subsequent intramolecular attack of the less hindered free phenolic hydroxyl group on the less hindered carbonyl group in B would then yield the hemiacetal C, whose trisubstituted alkene would be protonated to give intermediate D. The hydroxyl group of the hemiacetal in D would undergo the annulation to yield the ketalization products 3a and 2a. We reasoned that the sterically hindered isopropyl group at C13 in (+)-13 was critical for this diastereo- and regioselective outcome. This route therefore provided facile access to a total of 2.13 g of 3a and 2a (see the ESI† for details), thereby highlighting the robust nature of this chemistry. Notably this new cascade reaction constructed five new chemical bonds, two new rings, and three stereogenic centers with high diastereoselectivity6f and regioselectivity in a single step.
The treatment of compounds 3a and 2a with KOH in EtOH/H2O at 80 °C gave (−)-myrtucommuacetalone B (3) and myrtucommuacetalone (2, proposed structure), respectively, in good yields. The 1H and 13C NMR spectra of 2 were identical to those of the natural product, however the sign of its optical rotation was the opposite of that reported in the literature {synthetic: [α]25D = +24 (c = 0.1, CHCl3); natural: [α]30D = −33 (c = 1.0, CHCl3)}.3 Notably, the absolute configuration of naturally occurring 2 has not been reported previously.3,6f The absolute configuration of naturally occurring myrtucommuacetalone was therefore determined to be 1R, 9R, 10R, 17S based on our total synthesis.
Asymmetric syntheses of callistrilones A, C, D and E
Moving forward, we proceeded with our proposed syntheses of the remaining natural PPPs 4–7 (Scheme 2). We initially investigated the intermolecular oxidative [3 + 2] cycloaddition of ent-13 or 13 with 10 using various conditions from the literature that have previously been applied to achieve the construction of several related systems.9,7b Disappointingly, these conditions failed to afford the desired product in this particular case. Eventually however, the treatment of ent-13 or 13 and 10 with Ag2CO315 in refluxing MeCN afforded the angular product 6a or callistrilone D (6) diastereo- and regioselectively as a single product in a 45% yield (1.0 g scale). Furthermore, compounds 6a and 6 were treated with NaI/oxone for iodohydroxylation of the disubstituted double bond, followed by the diastereoselective cyclization with NaH as a base, to give both the desired callistrilone A (4) and callistrilone C (5) in a 60% overall yield. Notably, the expected direct epoxidation of compound 6a or 6 with meta-chloroperoxybenzoic acid (mCPBA) or dimethyldioxirane (DMDO) failed to afford the desired product 4 or 5. Pleasingly, the treatment of 6a with KOH in EtOH/H2O gave callistrilone E (7). The structures of synthetic 6 and 4 were also confirmed by X-ray crystallography.
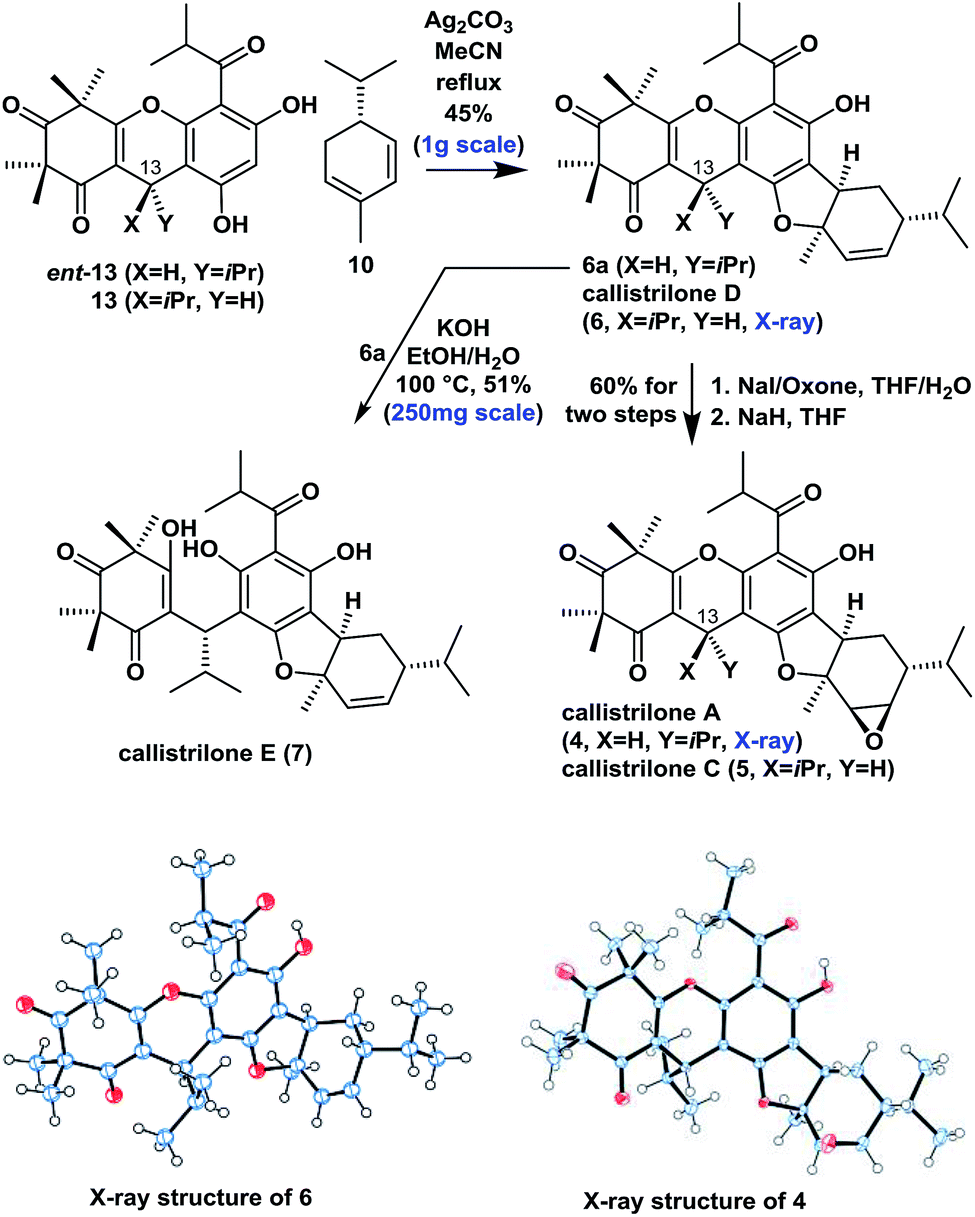 | ||
| Scheme 2 Asymmetric synthesis of PPPs 4–7. | ||
Antibacterial activity assay
The emergence of multidrug-resistant bacteria has become a major threat to public health. It is therefore important to develop a better understanding of chemicals that show activity against drug-resistant bacterial strains such as methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-intermediate S. aureus (VISA), and vancomycin-resistant Enterococcus faecium (VRE).16 A major focus in current antibiotic development is based on the screening of natural products.17 Therefore, the antibacterial activities of the compounds prepared in the current study were evaluated against six Gram-positive and five Gram-negative bacteria (Table 3). Among the compounds, 7 exhibited pronounced antibacterial activities against all Gram-positive bacteria, including three multidrug-resistant strains, with MIC values in the range of 0.25 to 2 μg mL−1. Notably, compound 7 exhibited greater antibacterial activity against multidrug-resistant strains (MRSA, VISA and VRE) than vancomycin, which is currently considered to be the last resort for treatment of Gram-positive bacterial infections. This compound therefore represents a promising lead compound for the development of antibacterial agents.| Compound | S. aureus ATCC 33591 (MRSA) | S. aureus ATCC 700699 (VISA) | E. faecium ATCC 700221 (VRE) | S. aureus ATCC 29213 (MSSA) | E. faecalis ATCC 29212 | S. epidermidis |
|---|---|---|---|---|---|---|
| 2 | 8 | 8 | 64 | 8 | 64 | 16 |
| 3 | 16 | 16 | >128 | 16 | >128 | >128 |
| 4 | 32 | 32 | 64 | 32 | 64 | >128 |
| 5 | >128 | >128 | >128 | >128 | >128 | >128 |
| 6 | >128 | >128 | >128 | >128 | >128 | >128 |
| 7 | 0.25 | 0.25 | 0.5 | 0.25 | 2 | 2 |
| Vancomycin | 2 | 8 | >128 | 1 | 4 | 2 |
Conclusions
We have developed a new approach for the highly concise, catalytic and first total asymmetric synthesis of myrtucommuacetalone, myrtucommuacetalone B and callistrilones A, C, D and E, in only 5–7 steps. Our route shows good step, redox and atom economy from simple building blocks (9–12) and avoids the need for protecting groups.18 This synthetic strategy was enabled by a unique organocatalytic asymmetric Friedel–Crafts-type Michael addition to synthesize 8 (95:5 er, after recrystallization of 13: 99.5:0.5 er), a versatile biomimetic synthetic precursor for the construction of some other PPPs. Notably, a Michael-ketalization-annulation cascade reaction was established as the key step in the efficient formation of the difficult to construct bridged furochromene moiety, together with the polycyclic ketal skeleton of myrtucommuacetalone B, with high diastereoselectivity and regioselectivity. A diastereo- and regioselective oxidative [3 + 2] cycloaddition allowed for the facile construction of the unusual and sterically compact 6/6/6/5/6-fused pentacyclic skeleton of the callistrilones. Based on our total synthesis, the absolute configuration of myrtucommuacetalone (2) was determined. Notably, the new compound 7 exhibited considerable antibacterial activity against Gram-positive bacteria and showed greater antibacterial activity against multidrug-resistant strains (i.e., MRSA, VISA and VRE) than that of vancomycin. This work will serve as a platform for the catalytic asymmetric synthesis of a diverse range of PPPs1 and for further systematic evaluation of their biological activities. These investigations are underway and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is dedicated to Professor Qi-Lin Zhou on the occasion of his 60th birthday. This work was supported by the Natural Science Foundation of China (Grant no. 21522204, 21672095 and U1401225), Guangdong Science and Technology Department (2016A050503011) and the Shenzhen Science and Technology Innovation Committee (Grant no. JCYJ20170412152454807, JSGG20160301103446375 and KQTD2015071710315717).Notes and references
- (a) Y. Kashman, A. Rotstein and A. Lifshitz, Tetrahedron, 1974, 30, 991 CrossRef CAS; (b) A. Rotstein, A. Lifshitz and Y. Kashman, Antimicrob. Agents Chemother., 1974, 6, 539 CrossRef CAS PubMed; for selected isolations, see: (c) M. Makino and Y. Fujimoto, Phytochemistry, 1999, 50, 273 CrossRef CAS; (d) C. K. Lee, Tetrahedron Lett., 1999, 40, 7255 CrossRef CAS; (e) H. Ito, H. Iwamori, N. Kasajima, M. Kaneda and T. Yoshida, Tetrahedron, 2004, 60, 9971 CrossRef CAS; (f) L. Larsen, M. H. Benn, M. Parvez and N. B. Perry, Org. Biomol. Chem., 2005, 3, 3236 RSC; (g) F. Shaheen, M. Ahmad, S. N. Khan, S. S. Hussain, S. Anjum, B. Tashkhodjaev, K. Turguniv, M. N. SultanChoudhary, M. I. Choudhary and A.-U. Rahman, Eur. J. Org. Chem., 2006, 2371 CrossRef CAS; (h) A. R. Carroll, S. Urban, J. Lamb, R. Moni, G. P. Guymer, P. I. Forster and R. J. Quinn, J. Nat. Prod., 2008, 71, 881 CrossRef CAS PubMed; (i) A. R. Carroll, J. Lamb, R. Moni, G. P. Guymer, P. I. Forster and R. J. Quinn, J. Nat. Prod., 2008, 71, 1564 CrossRef CAS PubMed; (j) A. Hiranrat and W. Mahabusarakam, Tetrahedron, 2008, 64, 11193 CrossRef CAS; (k) F. Cottiglia, L. Casu, M. Leonti, P. Caboni, C. Floris, B. Busonera, P. Farci, A. Ouhtit and G. Sanna, J. Nat. Prod., 2012, 75, 225 CrossRef CAS PubMed; (l) A. Hiranrat, W. Mahabusarakam, A. R. Carroll, S. Duffy and V. M. Avery, J. Org. Chem., 2012, 77, 680 CrossRef CAS PubMed; (m) M. Hans, M. Charpentier, V. Huch, J. Jauch, T. Bruhn, G. Bringmann and D. Quandt, J. Nat. Prod., 2015, 78, 2381 CrossRef CAS PubMed; (n) C. Liu, S. Ang, X.-J. Huang, H.-Y. Tian, Y.-Y. Deng, D.-M. Zhang, Y. Wang, W.-C. Ye and L. Wang, Org. Lett., 2016, 18, 4004 CrossRef CAS PubMed; (o) Y.-L. Zhang, C. Chen, X.-B. Wang, L. Wu, M.-H. Yang, J. Luo, C. Zhang, H.-B. Sun, J.-G. Luo and L.-Y. Kong, Org. Lett., 2016, 18, 4068 CrossRef CAS PubMed; (p) X.-J. Qin, H. Liu, Q. Yu, H. Yan, J.-F. Tang, L.-K. An, A. Khan, Q.-R. Chen, X.-J. Hao and H.-Y. Liu, Tetrahedron, 2017, 73, 1803 CrossRef CAS.
- (a) A. Rosa, M. Deiana, V. Casu, G. Corona, G. Appendino, F. Bianchi, M. Ballero and M. A. Dessi, Free Radical Res., 2003, 37, 1013 CrossRef CAS; (b) C. Feißt, L. Franke, G. Appendino and O. Werz, J. Pharmacol. Exp. Ther., 2005, 315, 389 CrossRef PubMed; (c) I. Tretiakova, D. Blaesius, L. Maxia, S. Wesselborg, K. Schulze-Osthoff, J. Cinatl, M. Michaelis and O. Werz, Apoptosis, 2008, 13, 119 CrossRef CAS PubMed; (d) A. Rossi, R. Di Paola, E. Mazzon, T. Genovese, R. Caminiti, P. Bramanti, C. Pergola, A. Koeberle, O. Werz, L. Sautebin and S. Cuzzocrea, J. Pharmacol. Exp. Ther., 2009, 329, 76 CrossRef CAS PubMed; (e) A. Koeberle, F. Pollastro, H. Northoff and O. Werz, Br. J. Pharmacol., 2009, 156, 952 CrossRef CAS PubMed; (f) K. Izgi, B. Iskender, J. Jauch, S. Sezen, M. Cakir, M. Charpentier, H. Canatan and C. SakalarIzgi, J. Biochem. Mol. Toxicol., 2015, 29, 432 CrossRef CAS PubMed.
- M. I. Choudhary, N. Khan, M. Ahmad, S. Yousuf, H. K. Fun, S. Soomro, M. Asif, M. A. Mesaik and F. Shaheen, Org. Lett., 2013, 15, 1862 CrossRef CAS PubMed.
- J.-Q. Cao, X.-J. Huang, Y.-T. Li, Y. Wang, L. Wang, R.-W. Jiang and W.-C. Ye, Org. Lett., 2016, 18, 120 CrossRef CAS PubMed.
- (a) H. Muller, M. Paul, D. Hartmann, V. Huch, D. Blaesius, A. Koeberle, O. Werz and J. Jauch, Angew. Chem., Int. Ed., 2010, 49, 2045 CrossRef PubMed; (b) M. Charpentier, M. Hans and J. Jauch, Eur. J. Org. Chem., 2013, 4078 CrossRef CAS.
- (a) M. Morkunas, L. Dube, F. Gotz and M. E. Maier, Tetrahedron, 2013, 69, 8559 CrossRef CAS; (b) A. Gervais, K. E. Lazarski and J. A. Porco, J. Org. Chem., 2015, 80, 9584 CrossRef CAS PubMed; (c) H. Tan, H. Liu, X. Chen, Y. Yuan, K. Chen and S. Qiu, Org. Lett., 2015, 17, 4050 CrossRef CAS PubMed; (d) H. C. Lam, J. T. Spence and J. H. George, Angew. Chem., Int. Ed., 2016, 55, 10368 CrossRef CAS PubMed; (e) L. Lv, Y. Li, Y. Zhang and Z. Xie, Tetrahedron, 2017, 73, 3691 CrossRef CAS; when we were preparing this manuscript, Tan and co-workers reported a racemic total synthesis of myrtucommuacetalone, see: (f) H. Liu, L. Huo, B. Yang, Y. Yuan, W. Zhang, Z. Xu, S. Qiu and H. Tan, Org. Lett., 2017, 19, 4786 CrossRef CAS PubMed; for a review, see: (g) I. P. Singh, J. Sidana, S. B. Bharate and W. J. Foley, Nat. Prod. Rep., 2010, 27, 393 RSC.
- (a) B. Chen, X. Liu, Y. Hu, D. Zhang, L. Deng, J. Lu, L. Min, W. Ye and C.-C. Li, Chem. Sci., 2017, 8, 4961 RSC; (b) C. Qiao, W. Zhang, J.-C. Han and C.-C. Li, Org. Lett., 2016, 18, 4932 CrossRef CAS PubMed; (c) J. C. Han, F. Li and C.-C. Li, J. Am. Chem. Soc., 2014, 136, 13610 CrossRef CAS PubMed; (d) G. Liu, G. J. Mei, R. Chen, H. Yuan, Z. Yang and C.-C. Li, Org. Lett., 2014, 16, 4380 CrossRef CAS PubMed; (e) H. Wei, C. Qiao, G. Liu, Z. Yang and C.-C. Li, Angew. Chem., Int. Ed., 2013, 52, 620 CrossRef CAS PubMed.
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS PubMed.
- (a) G. Buchi and P.-S. Chu, J. Org. Chem., 1978, 43, 3717 CrossRef CAS; for reviews, see: (b) V. Nair and A. Deepthi, Chem. Rev., 2007, 107, 1862 CrossRef CAS PubMed; (c) B. B. Snider, Chem. Rev., 1996, 96, 339 CrossRef CAS PubMed; (d) J. Iqbal, B. Bhatia and N. K. Nayyar, Chem. Rev., 1994, 94, 519 CrossRef CAS; for selected examples, see: (e) E. I. Heiba and R. M. Dessau, J. Org. Chem., 1974, 39, 3456 CrossRef CAS; (f) Y. R. Lee, B. S. Kim and D. H. Kim, Tetrahedron, 2000, 56, 8845 CrossRef CAS; (g) V. Nair, P. M. Treesa, D. Maliakal and N. P. Rath, Tetrahedron, 2001, 57, 7705 CrossRef CAS; (h) T. R. Blum, Y. Zhu, S. A. Nordeen and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 11056 CrossRef CAS PubMed.
- For reviews, see: (a) T. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS PubMed; (b) M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS PubMed; (c) D. Almasi, D. Alonso and A. C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS; (d) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed; (e) S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC; (f) V. Terrasson, R. M. Figueiredo and J. M. Campagne, Eur. J. Org. Chem., 2010, 2635 CrossRef CAS. For some representative examples, see: (g) R. P. Herrera, V. Sgarzani, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 6576 CrossRef CAS PubMed; (h) E. M. Fleming, T. McCabe and S. J. Connon, Tetrahedron Lett., 2006, 47, 7037 CrossRef CAS; (i) D.-P. Li, Y.-C. Guo, Y. Ding and W.-J. Xiao, Chem. Commun., 2006, 799 RSC; (j) S. Sasaki, T. Yamauchi and K. Higashiyama, Tetrahedron Lett., 2010, 51, 2326 CrossRef CAS; (k) E. Paradisi, P. Righi, A. Mazzanti, S. Ranieri and G. Bencivenni, Chem. Commun., 2012, 48, 11178 RSC; (l) I. Aillaud, D. M. Barber, A. L. Thompson and D. J. Dixon, Org. Lett., 2013, 15, 2946 CrossRef CAS PubMed; (m) L. Yu, X. Xie, S. Wu, R. Wang, W. He, D. Qin, Q. Liu and L. Jing, Tetrahedron Lett., 2013, 54, 3675 CrossRef CAS; (n) T. Arai, A. Tsuchida, T. Miyazaki and A. Awata, Org. Lett., 2017, 19, 758 CrossRef CAS PubMed.
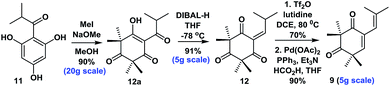
- When we were preparing this manuscript, Xie and co-workers reported an elegant synthesis of 12, see ref. 6e. The readily available starting material 11 was methylated and treated with DIBAL-H in THF, followed by in situ elimination to give 12 in 91% yield. The subsequent reaction of 12 with Tf2O and 2,6-lutidine in refluxing DCE, followed by treatment with a mixture of Pd(OAc)2, PPh3, HCO2H and Et3N, afforded 9 in 90% yield (see ESI for details†)
 .
. - J. Lv, X. Li, L. Zhong, S.-Z. Luo and J.-P. Cheng, Org. Lett., 2010, 12, 1096 CrossRef CAS PubMed.
- For pioneering work on chiral phosphoric acid catalysis, see: (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566 CrossRef CAS PubMed; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356 CrossRef CAS PubMed; for the use of spinol-derived phosphoric acids, see: (c) F. Xu, D. Huang, C. Han, W. Shen, X. Lin and Y. Wang, J. Org. Chem., 2010, 75, 8677 CrossRef CAS PubMed. For typical reviews, see: (d) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed; (e) M. Terada, Chem. Commun., 2008, 4097 RSC; (f) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed.
- L. Hong, W.-S. Sun, D.-X. Yang, G.-F. Li and R. Wang, Chem. Rev., 2016, 116, 4006 CrossRef CAS PubMed.
- B.-C. Hong, I.-C. Shen and J.-H. Liao, Tetrahedron Lett., 2001, 42, 935 CrossRef CAS.
- C. A. Arias and B. E. Murray, N. Engl. J. Med., 2009, 360, 439 CrossRef CAS PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111 CrossRef CAS PubMed.
- (a) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC; (b) I. S. Young and P. S. Baran, Nat. Chem. Biol., 2009, 1, 193 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1526145, 1526146, 1582656, 1582657 and 1582658. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04672c |
| This journal is © The Royal Society of Chemistry 2018 |