
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of DNA nanostructure-based interfacial probes for the electrochemical detection of nucleic acids directly in whole blood†
Chao
Li
a,
Xiaolu
Hu
a,
Jianyang
Lu
a,
Xiaoxia
Mao
c,
Yang
Xiang
*a,
Yongqian
Shu
*b and
Genxi
Li
 *ac
*ac
aState Key Laboratory of Pharmaceutical Biotechnology, Collaborative Innovation Center of Chemistry for Life Sciences, Department of Biochemistry, Nanjing University, Nanjing 210093, P. R. China. E-mail: xiangy@nju.edu.cn; genxili@nju.edu.cn; Tel: +86-25-83593596
bDepartment of Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, P. R. China. E-mail: yongqianshu531@163.com
cLaboratory of Biosensing Technology, School of Life Sciences, Shanghai University, Shanghai, 200444, China
First published on 28th November 2017
Abstract
Here we report a robust and sensitive DNA nanostructure-based electrochemical (E-nanoDNA) sensor that utilizes tetrahedral DNA nanostructures (TDNs) as an interfacial probe to detect biomolecules in a single-step procedure. In this study, we have firstly demonstrated that the use of TDNs can significantly suppress electrochemical background signals compared to traditional linear DNA probes upon introduction of base mismatches in the edges of TDNs. After further optimization of the two functional strands in the TDNs, quantitative, one-step detection of DNA can be achieved in the picomolar range in less than 10 min, and directly in complex media. Moreover, the baseline drift of this biosensor can be greatly decreased even after several hours in flowing whole blood in vitro, which suggests that the sensor holds potential to be employed in live animals. Furthermore, through replacing functional strands with aptamers or other DNA elements, this E-nanoDNA sensor can be easily used to probe various analytes, broadening the application range of the proposed sensor.
Introduction
Electrochemical DNA (E-DNA) sensors have provided new opportunities for simple, sensitive, and portable detection of biomolecules relevant to clinical diagnosis and monitoring the treatment of diseases.1 Usually, the signal generation of these sensors originates from binding-induced changes in the dynamics of the surface immobilized, redox reporter-labeled DNA probe, which affect the flexibility of the probe–target complex, leading to a remarkable change in the redox current.2,3 So, these E-DNA sensors are inherently fast, reagentless, portable and cost-effective. However, a crucial issue in E-DNA sensors also exists, i.e., the decreased accessibility of analytes to the probes immobilized on heterogeneous electrode surfaces compared with analyte–probe recognition in homogeneous solutions,4,5 thus it is easily interfered with by endogenous substances (i.e., nucleases, protein, etc.), leading to compromised analytical performance. Although many innovative probe designs such as hairpin probes,6,7 single or double-stranded probes,8,9 triple-stem probes,10 triblock probes,11 pseudoknot probes,12,13 and antigen-labeled probes14,15 have all proved successful at biomolecule (i.e., DNA, proteins, small molecules, etc.) detection, novel strategies that have the capacity to improve the recognition abilities of such heterogeneous surface probes are still required.16 Moreover, recently, a new challenge has been proposed for E-DNA sensors, which are expected to be used to realize real-time analysis of analytes in living animals,17,18 since conventional probes may suffer poor robustness in complex matrices (i.e., whole blood), impairing the analytical performance of E-DNA sensors. Therefore, the development of novel electrochemical probes for the construction of E-DNA sensors which may meet the increasing requirements of bioanalysis is highly desirable.As a matter of fact, since the orientation and distribution of traditional linear DNA probes on the electrode surface are difficult to control, Fan and co-workers have reported a novel electrochemical sensor based on 3D nanostructure DNA probes.19–21 In their design, a tetrahedral DNA nanostructure (TDN) containing a capture probe at one vertex and three thiol groups at the other vertices was designed and immobilized on the gold electrode surface. Thanks to the rigid and arrayed DNA nanostructures, greatly improved sensitivity was achieved compared to that of conventional E-DNA sensors based on single-stranded DNA (ssDNA) considering the entanglement between the probes and the localized aggregation of the probes. Nevertheless, since the signal generation of this sensor necessarily depends on the formation of a sandwich structure, namely “DNA tetrahedron/target molecule/signal probe/enzyme”, it requires multiple incubation and washing steps and the addition of an exogenous biotin-labeled DNA strand and streptavidin-labeled enzyme, making the whole assay expensive, laborious and time-consuming, which impedes its use in point-of-care testing (POCT).
Herein, we present a novel DNA nanostructure-based electrochemical (E-nanoDNA) sensor that overcomes the drawbacks of the two types of sensor mentioned above. Building upon the pioneering works by the Plaxco and Fan groups, we have added DNA nanostructures to the E-DNA sensor. To do so, we designed a TDN with two additional stands appended to the two vertices (Scheme 1, left). The top one functions as the capture strand (a) and the other one at the bottom acts as the assistant strand (b). This allows us to flexibly add recognition elements that specifically bind the analyte of interest through choosing appropriate DNA elements (i.e., DNA, aptamer, DNAzyme, etc.) and covalently modifying the electrochemical species. The functional part of strand (a) has a relatively long sequence [9 nucleotides (nt)] and the melting temperature of the strand (a):target duplex is 41.2 °C. Strand (b) has a relatively short sequence (6 nt) and the melting temperature of the strand (b):target duplex is less than 1 °C (1 μM DNA in 10 mM sodium phosphate, 0.5 M NaCl and 1.5 mM MgCl2 pH 7.4 buffer). By using three thiolated ssDNAs as anchoring legs, thiol modifications on the three vertices of the “bottom” face of the TDN are designed as anchoring units for immobilization on the electrode surface. In the absence of a target, strand (a) sequesters the methylene blue (MB) molecule from the electrode surface due to the strong steric effect induced by the large and rigid DNA nanostructures, producing a feeble MB redox current. Upon target binding, the observed MB redox current increases significantly. The proposed sensor underlying this signaling functions as follows. The target is firstly captured by strand (a) through DNA hybridization. Then, short strand (b) binds with the strand (a)/target complex to form an intact structure (Tm = 63.3 °C) and provides traction to pull down strand (a), increasing the chance of the MB colliding with the electrode surface and facilitating electron transfer (Scheme 1, right). This E-nanoDNA sensor has combined the advantages of traditional E-DNA sensors and TDN-based electrochemical sensors. First, it can more efficiently capture analytes and suppress background current, indicating better analytical performance. Second, the DNA nanostructure is more stable and less sensitive to biofouling than linear DNA probes in complex matrices, promoting the reproducibility of the sensor. Third, testing can be achieved by only one-step incubation without requiring additional reagents and washing steps, greatly reducing the operating difficulty.
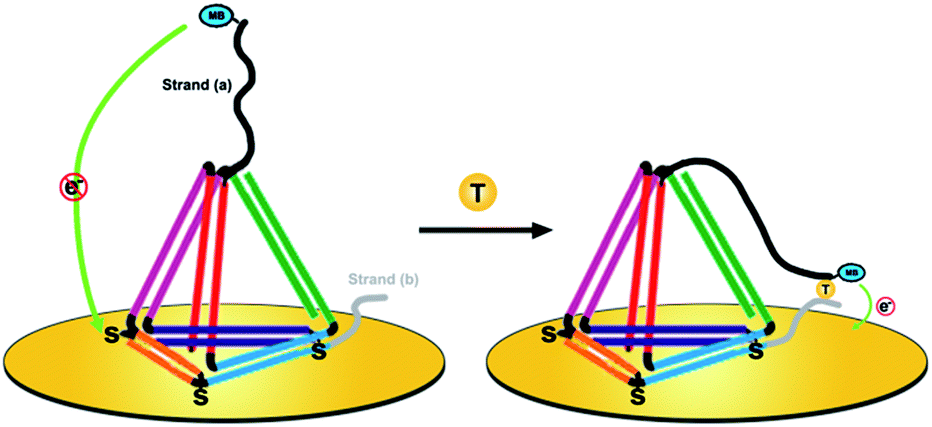 | ||
| Scheme 1 The E-nanoDNA sensor contains an electrode-bound, redox-reporter modified tetrahedral DNA nanostructure, with two functional strands (black and grey), that undergoes a target binding-induced conformational change on the electrode surface. This conformational change pulls the reporter (methylene blue, MB) close to the electrode surface (yellow disk), thereby producing a target-dependent change in current when the sensor is interrogated by square wave voltammetry. | ||
Results and discussion
To improve the analytical performance of the E-nanoDNA sensor, two different strategies can be employed: lowered background signal and amplified detection. In the first case, considering that previous E-DNA sensors have widely employed a floppy or relatively rigid DNA probe (i.e., ssDNA or dsDNA), large electrochemical signals come from dynamic collision between the electrode and the redox moiety without the addition of target, attenuating the signal gain. The hypothesis was that increasing the size of the DNA probe can efficiently reduce the chance of a collision between the electrode and the redox moiety, thereby reducing the background current. Therefore, a series of DNA probes with different sizes – ssDNA25, TDN10, TDN20, and TDN30 (ssDNA has 25 nt and each edge of the TDN contains 10, 20, or 30 base pairs, respectively) – were employed to test the background signal. All of the sequences used were rationally designed with the aid of structural prediction tools (NUPACK software) to avoid undesired secondary structures appearing at the edges. Because each base pair is separated by 0.34 nm in the double helix, the edge lengths of these TDNs were 3.4, 6.8, and 10.2 nm, respectively. Since the persistence length of ssDNA is about 1 nm, we could estimate the height of these probes on the electrode surface to be 2, 3.8, 6.5, and 9.3 nm (a six-carbon spacer forms a layer of ∼1 nm). The TDNs were assembled with four different ssDNAs through a facile annealing process. Gel electrophoresis, atomic force microscopy (AFM), and dynamic light scattering (DLS) results confirmed the successful formation of the DNA tetrahedron (Fig. S1†). Fig. 1A compares the background signals of the sensor modified with ssDNA25, TDN10, TDN20, and TDN30. The surface coverage of these probes, determined by a quartz crystal microbalance (Fig. S2†), was maintained within the range of (2.5 ± 0.4) × 1012 molecules per cm−2 throughout this study, thus the nanospacing between the DNA probes was ∼5.0 nm, which is advantageous for capturing analyte. The electrochemical behavior was investigated by square wave voltammetry (SWV), which allows highly sensitive detection of very low concentrations of a redox probe. Compared to the background peak current of 1887 nA with the ssDNA25, as hypothesized, it was indeed possible to reduce the background signal by 2.1-fold using TDN10. This result demonstrates that the large framework of the tetrahedron indeed prevented direct collision between MB and the electrode surface. However, the background peak currents aren’t obviously attenuated when using TDN20 and TDN30, though they have larger spatial structures. This is probably owing to the fact that MB molecules can directly pass the electron through the DNA backbone, leading to a large background signal.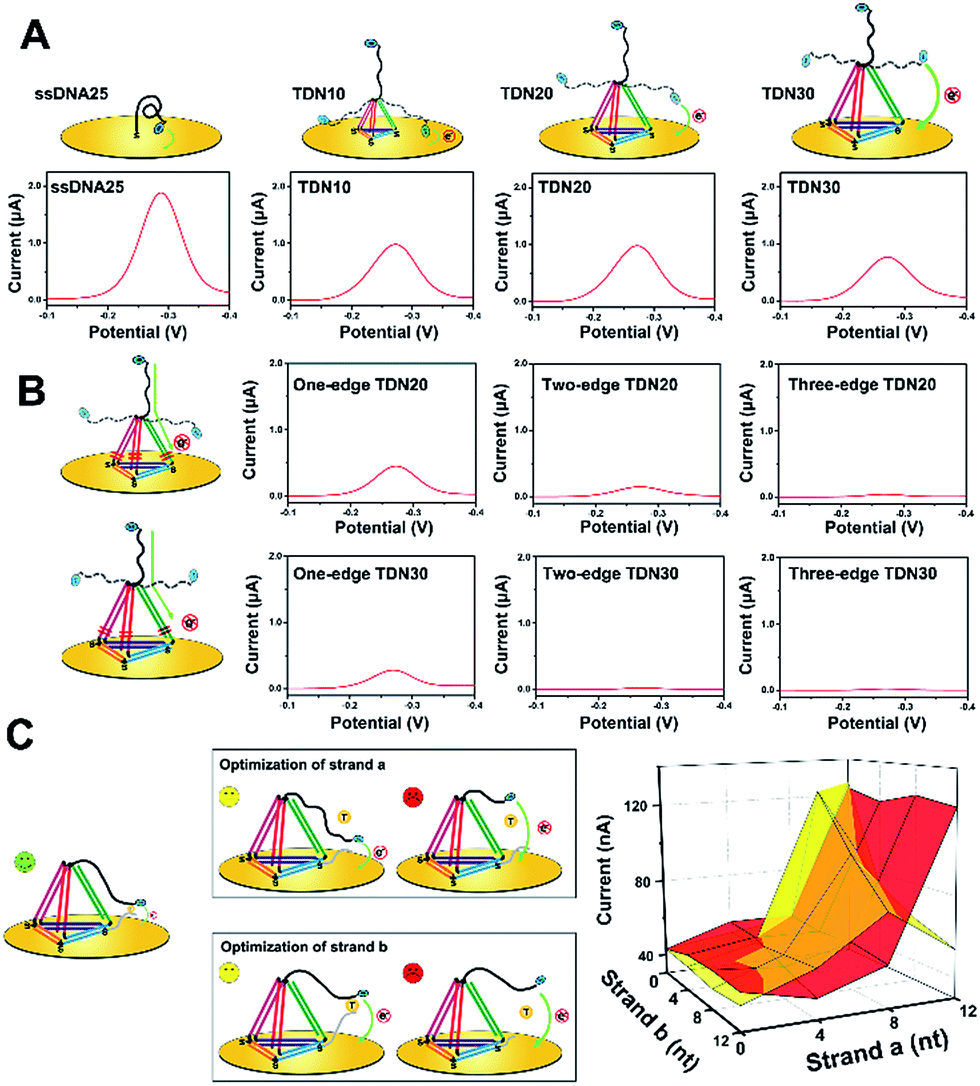 | ||
| Fig. 1 Optimization of the E-nanoDNA sensor through reducing background signals and promoting positive signals in the presence of analyte. (A) Background signals obtained from sensors modified with different probes: ssDNA25, TDN10, TDN20 and TDN30. (B) Background signals obtained from sensors modified with TDNs containing a different number of one-base mismatches. (C) Optimization of the spacer lengths of strand (a) and strand (b) for better analytical performance in the absence (red) or presence (yellow) of target (1 nM). | ||
To find out the real reasons for the above results, our next attempt was to introduce a one-base-pair mismatch in the edge of the TDN20 and TDN30 at the bottom site to block charge passing through the DNA double helices, since MB transporting electrons are very sensitive to perturbation of π-stacking in the duplex (Fig. 1B, left).22,23 As hypothesized, the introduction of a one-base-pair mismatch in the one edge of TDN20 and TDN30 successfully blocked the electron transfer of MB via stacked nucleobases, resulting in the decrease of the background peak current from 764 nA and 695 nA down to 387 nA and 234 nA, respectively (Fig. 1B). Furthermore, when a one-base-pair mismatch is introduced into the two or three edges of TDN20 and TDN30, the background signal is reduced close to the baseline. Considering that the large TDN30 requires the synthesis of a long DNA sequence and that the synthesis of larger TDN (e.g., TDN40) requires a more complex method, a TDN20 containing three one-base-pair mismatches was chosen as the electrochemical probe.
Next, in order to amplify the positive signal, the MB head, in principle, needed to be pulled as close as possible to the underlying electrode, where the electron could be transferred by tunneling between the redox label and the gold electrode. So, the spacer length of strand (a) and strand (b) should be carefully optimized. When the strands are too short, the formation of an intact “strand (a)/strand (b)/target” complex is prohibited, failing to generate any detectable signals. In the case of strand (a) with a long spacer (i.e., 12 nt), a large background signal was observed, which indicates that strand (a) may directly interact with the electrode surface (Fig. 1C, upper).
However, the sensor can also respond to the target molecules and obtain a reduced current, which indicates that the sensor becomes a “signal-off” model. As for the spacer of strand (b), increasing the length of the spacer (>4 bp) also led to a reduced signaling current in the presence of analyte (Fig. 1C, bottom), indicating that the formed complex is pulled away from the electrode surface, decreasing the chance of an interaction of MB with the electrode surface. Therefore, the spacer lengths of strand (a) and strand (b) were selected as 8 nt and 4 nt, respectively.
After determination of the DNA nanostructure, the sensor performance was first evaluated by detection of DNA molecules. We observed that the new E-nanoDNA sensor responded rapidly (∼8 min, Fig. S3†) and robustly (standard deviation across three electrodes <5%) to its target. The signaling current increased significantly as we titrated the sensor with increasing amounts of target DNA (Fig. 2A). The detection limit of the current sensor was 300 fM (>3 SD, standard deviation), and the useful dynamic range spanned four orders of magnitude (Fig. 2A, inset). This sensitivity is superior to that of many developed E-DNA sensors or DNA nanostructure-based electrochemical sensors (Table S2†). In contrast, control experiments revealed that the addition of a one-base mismatched target at a concentration of 50 nM (which would be a saturating target concentration for the perfect match) only produced a signal change of ∼64% (Fig. 2B). The E-nanoDNA sensor was highly resistant to the complex sample (i.e., serum), with a feeble alteration of the background noise and nearly the same hybridization signal for the 1 nM target, in the presence of 100% serum (Fig. S4†).
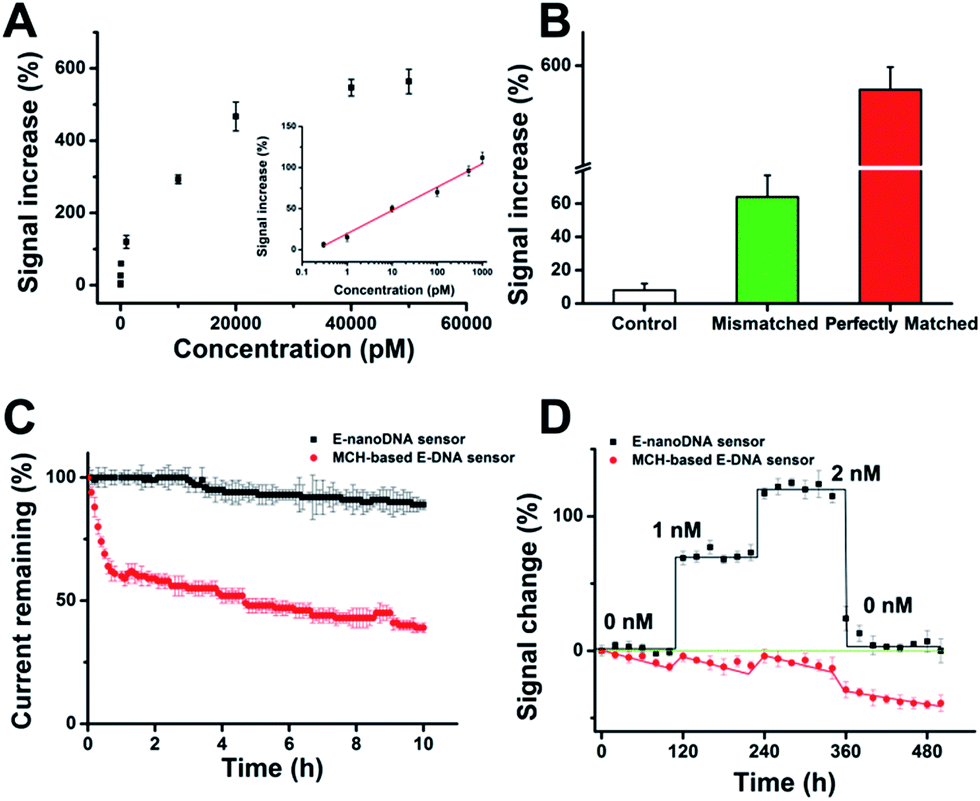 | ||
| Fig. 2 Application of the E-nanoDNA sensor for DNA detection. (A) The dynamic range of the E-nanoDNA sensor covers target concentrations from 300 fM to 50 nM. Inset: detection of DNA in the low concentration range. (B) Comparison of electrochemical signals obtained by the E-nanoDNA sensor with a control strand (random strand, 50 nM), a one-base mismatched strand (50 nM), and a perfectly matched probe (50 nM). (C) When challenged in flowing whole blood (no target), the E-nanoDNA sensors (black) exhibit less than 11% current drift over 10 h, while the original signal of conventional MCH-based E-DNA sensors (red) loses around 61%. (D) The improved drift performance of the E-nanoDNA sensors allows real-time analysis in whole blood. Shown are data recorded in vitro in flowing whole blood samples spiked with target DNA at different time points. The error bars show the standard deviation of at least three independently matched electrodes to record this data set. | ||
More importantly, Plaxco et al. have demonstrated that traditional 6-mercapto-1-hexanol (MCH)-based E-DNA sensors suffer from severe drift when employed in flowing whole blood,24 which greatly limits their application in continuous, real-time, and chip-based measurements. Due to the robustness and antifouling ability of the DNA nanostructure, we were motivated to investigate whether the E-nanoDNA sensor could eliminate the drift seen in the flowing sample without intricate correction algorithms, thereby resulting in stable analytical performance over hours of successive operation. Fig. 2C demonstrates that the E-nanoDNA sensor exhibited excellent baseline stability over the course of 10 h in flowing whole blood, while the signal from the MCH-based sensor was reduced to ∼39%. Under these same conditions, the E-nanoDNA sensor responded quantitatively to the target in flowing whole blood and returned quantitatively to its original baseline when the target was removed (Fig. 2D), achieving nanomolar precision in the measurement of DNA; however, the MCH-based sensor shows evident drift in both the presence and absence of the target.
Subsequently, we believe that this new E-nanoDNA sensor may provide a versatile platform for the analysis of a wide range of biomolecules by taking advantage of aptamers. To assemble an aptamer-based E-nanoDNA (EA-nanoDNA) sensor, we replaced the DNA probe with an anti-adenosine triphosphate (ATP) aptamer sequence that was divided into two different fragments, and assembled the aptasensor on gold surfaces. In the absence of ATP, the split aptamer strands don’t interact with each other. Upon introduction of ATP, the two fragments are induced to form a strong aptamer–target complex, generating a large signaling current. As shown in Fig. 3, this EA-nanoDNA sensor showed excellent sensitivity toward ATP, with a remarkable detection limit of 5 nM (Fig. 3B), which is lower than those of the ssDNA aptamer-based sensors.25,26 The specificity was further tested in the presence of possible interference, such as CTP, GTP, and UTP. The alteration in the electrochemical response induced by the non-specific binding between the ATP analogues above was much lower than that of the response induced by the specific binding of ATP (Fig. 3B, inset), demonstrating the excellent selectivity of the EA-nanoDNA sensor.
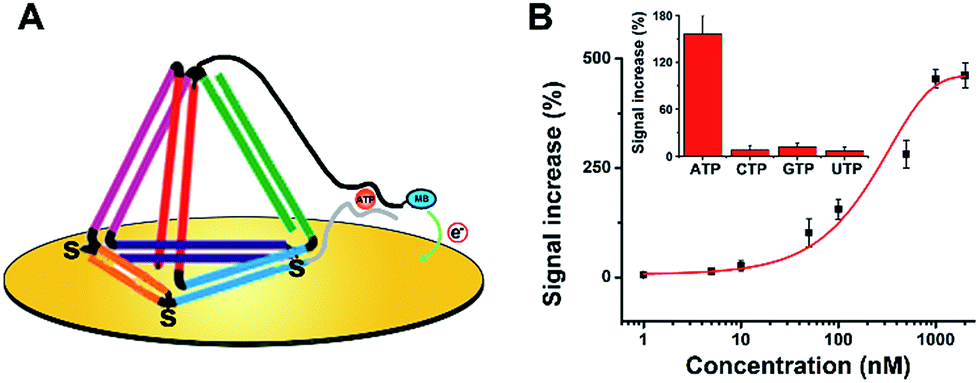 | ||
| Fig. 3 The application of the EA-nanoDNA sensor for ATP detection. (A) The EA-nanoDNA sensor-based ATP detection scheme. (B) Quantitative results for detecting varying concentrations of ATP. Inset: a selectivity study using CTP, GTP, and UTP as control molecules, [ATP, CTP, GTP and UTP] = 100 nM. | ||
Conclusions
In summary, we have constructed a novel DNA nanostructure-based E-DNA sensor for direct detection of biomolecules. In this system, two additional strands extended from the two vertices of the TDN folds around the analyte bringing the MB molecule into close proximity to the electrode surface affording an electrochemical response. Unlike previous reagentless electrochemical E-DNA sensors based on ssDNA or DNA hairpins with weak rigidity, this approach provides a more robust platform with improved analytical performance. Compared with previous DNA tetrahedron-based electrochemical sensors using enzyme-turnover as signal generation, this method requires no expensive, additional reagent and multiple incubation and washing steps, thus greatly simplifying the operation procedures, which is highly desirable for POC diagnostic sensors. However, currently, this type of E-sensor still relies on the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model and one target molecule only generates one signal event, so the sensitivity is inferior to enzyme-assisted ones. Therefore, our next work will focus on optimization of the electrode surface and attempt to employ amplification elements into the proposed sensor. We also believe that other DNA nanostructures, such as DNA boxes, DNA nanosheets, DNA prisms, etc., can also be used as electrochemical probes after appropriate design, which may further improve the performance of the proposed biosensor. All together, these results are the first example of the application of DNA nanostructures as bifunctional probes for the electrochemical detection of biomolecules of interest and can pave the way for the design of other innovative analytical systems.
:1 binding model and one target molecule only generates one signal event, so the sensitivity is inferior to enzyme-assisted ones. Therefore, our next work will focus on optimization of the electrode surface and attempt to employ amplification elements into the proposed sensor. We also believe that other DNA nanostructures, such as DNA boxes, DNA nanosheets, DNA prisms, etc., can also be used as electrochemical probes after appropriate design, which may further improve the performance of the proposed biosensor. All together, these results are the first example of the application of DNA nanostructures as bifunctional probes for the electrochemical detection of biomolecules of interest and can pave the way for the design of other innovative analytical systems.
Experimental
Materials and reagents
DNA oligonucleotides were synthesized and purified by Sangon Biotechnology Inc. (Shanghai, China). 6-Mercaptohexanol (MCH), tris(2-carboxyethyl)phosphine hydrochloride (TCEP), adenosine triphosphate (ATP) and hexaammineruthenium(III) chloride (RuHex) were purchased from Sigma. Foetal bovine serum (FBS) was purchased from Life Technologies (Gibco). Blood was collected from lab volunteers. All of the other chemicals were of analytical grade and were used without further purification. All of the solutions were prepared with Milli-Q water from a Millipore system.The buffers employed were as follows: the DNA tetrahedron preparation and immobilization buffer was 10 mM Tris–HCl and 10 mM MgCl2, pH 7.4. The electrodes were washed in 0.2 M PBS. The hybridization reactions were performed in 10 mM Tris–HCl, 0.5 M NaCl, and 1.5 mM MgCl2, pH 7.4. Electrochemical detection for methylene blue (MB) was performed in 20 mM PBS and 0.5 M NaCl, pH 7.4.
Preparation of DNA tetrahedron
Equal quantities of the four strands for the formation of the tetrahedrons were mixed in preparation buffer at a final concentration of 1 μM and then heated to 95 °C for 10 min and immediately cooled to 4 °C. Characterization of the synthesized DNA tetrahedron was used for atomic force microscopy (AFM) and a dynamic light scattering (DLS) study.Preparation of modified gold electrodes and surface area determination
Gold electrodes (1 mm in diameter) were polished on a microcloth with 0.3 μm and 0.05 μm γ-alumina. The polished electrodes were then sonicated in ethanol and pure water for 2 min each. Lastly, the electrodes were electrochemically cleaned in 0.5 M H2SO4 by scanning the potential between the oxidation and reduction of gold, −0.35 V and 1.5 V, respectively. All of the electrochemical measurements were carried out with a CHI 660D electrochemical workstation (CH Instruments Inc., Austin). A three-electrode configuration was employed in all of the experiments and involved a gold working electrode, a platinum wire counter electrode and a saturated calomel reference electrode (SCE).Electrode modification
A 10 μL volume of 1 μM tetrahedron probes in immobilization buffer (containing 5 mM TCEP) was dropped onto the surface of the cleaned gold electrodes and incubated for 2 h at room temperature. The modification for the single-stranded DNA (ssDNA) was the same as that for the tetrahedrons, and the concentration was also 1 μM. However, for the modification of the ssDNA, the modified electrodes were then exposed to a 1 mM MCH solution at room temperature for 1 hour to replace non-specific interactions and form a self-assembled monolayer (SAM). Between each step, the electrode was rinsed and dried with N2. Then, the modified electrodes were incubated with different concentrations of target probe at 37 °C for 10 min.Target detection
For the DNA and ATP assays, the modified electrodes were incubated in target solutions of various concentrations for 10 min at 37 °C. The electrodes were then extensively rinsed with washing buffer and subjected to electrochemical measurements.Electrochemical measurements
Square wave voltammetry (SWV) was performed using a potential window of −0.1 to −0.4 V (versus SCE), a 1 mV potential step, a 50 mV amplitude and a 20 Hz frequency.Real-time detection of DNA in continuously flowing blood
For real-time DNA detection, samples of blood were prepared in syringes and doped with different concentrations of DNA. The syringe was connected to a closed-loop system equipped with flow-cell screen-printed electrodes (DropSens, Spain). All of the experiments were conducted in a closed-loop system with a continuous flow of whole blood (1 mL s−1) using a circulator pump to mimic the circulation in the vasculature. At each concentration, SWV measurements were performed every 15 s scanning from −0.15 to −0.55 V, and the peak reduction current was recorded using a computer-controlled data acquisition system. After the target response experiments the sensors were regenerated by washing with fresh blood.Live subject statement
All of the blood tests described in the project were performed in the laboratory of Professor Genxi Li, as approved by the Nanjing Drum Tower Hospital of Nanjing University Medical School Ethics Committee.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21235003, 81672570 and J1210026), the Science Foundation of Jiangsu Province, China (Grant No. BM2015023), and Fundamental Research Funds for the Central Universities in China (Grant No. 021414380001).References
- J. Wang, Biosens. Bioelectron., 2006, 21, 1887–1892 CrossRef CAS PubMed.
- A. A. Lubin and K. W. Plaxco, Acc. Chem. Res., 2010, 43, 496–505 CrossRef CAS PubMed.
- K. Hsieh, B. S. Ferguson, M. Eisenstein, K. W. Plaxco and H. T. Soh, Acc. Chem. Res., 2015, 48, 911–920 CrossRef CAS PubMed.
- H. Pei, X. Zuo, D. Zhu, Q. Huang and C. Fan, Acc. Chem. Res., 2013, 47, 550–559 CrossRef PubMed.
- H. Kimura-Suda, D. Y. Petrovykh, M. J. Tarlov and L. J. Whitman, J. Am. Chem. Soc., 2003, 125, 9014–9015 CrossRef CAS PubMed.
- C. H. Fan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9134–9137 CrossRef CAS PubMed.
- Z.-g. Yu and R. Y. Lai, Chem. Commun., 2012, 48, 10523–10525 RSC.
- Y. Wu and R. Y. Lai, Anal. Chem., 2014, 86, 8888–8895 CrossRef CAS PubMed.
- C. Li, X. Li, L. Wei, M. Liu, Y. Chen and G. Li, Chem. Sci., 2015, 6, 4311–4317 RSC.
- Y. Xiao, X. Lou, T. Uzawa, K. J. Plakos, K. W. Plaxco and H. T. Soh, J. Am. Chem. Soc., 2009, 131, 15311–15316 CrossRef CAS PubMed.
- C. E. Immoos, S. J. Lee and M. W. Grinstaff, J. Am. Chem. Soc., 2004, 126, 10814–10815 CrossRef CAS PubMed.
- Y. Xiao, X. Qu, K. W. Plaxco and A. J. Heeger, J. Am. Chem. Soc., 2007, 129, 11896–11897 CrossRef CAS PubMed.
- K. J. Cash, A. J. Heeger, K. W. Plaxco and Y. Xiao, Anal. Chem., 2008, 81, 656–661 CrossRef PubMed.
- K. J. Cash, F. Ricci and K. W. Plaxco, J. Am. Chem. Soc., 2009, 131, 6955–6957 CrossRef CAS PubMed.
- C. Li, Y. Tao, Y. Yang, Y. Xiang and G. Li, Anal. Chem., 2017, 89, 5003–5007 CrossRef CAS PubMed.
- C. Li, D. Wu, X. Hu, Y. Xiang, Y. Shu and G. Li, Anal. Chem., 2016, 88, 7583–7590 CrossRef CAS PubMed.
- H. Li, P. Dauphin-Ducharme, N. Arroyo-Curras, C. H. Tran, P. A. Vieira, S. Li, C. Shin, J. Somerson, T. E. Kippin and K. W. Plaxco, Angew. Chem., Int. Ed., 2017, 56, 7492–7495 CrossRef CAS PubMed.
- N. Arroyo-Currás, J. Somerson, P. A. Vieira, K. L. Ploense, T. E. Kippin and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2017, 201613458 Search PubMed.
- H. Pei, N. Lu, Y. Wen, S. Song, Y. Liu, H. Yan and C. Fan, Adv. Mater., 2010, 22, 4754–4758 CrossRef CAS PubMed.
- M. Lin, P. Song, G. Zhou, X. Zuo, A. Aldalbahi, X. Lou, J. Shi and C. Fan, Nat. Protoc., 2016, 11, 1244–1263 CrossRef CAS PubMed.
- M. Lin, J. Wang, G. Zhou, J. Wang, N. Wu, J. Lu, J. Gao, X. Chen, J. Shi, X. Zuo and C. Fan, Angew. Chem., Int. Ed., 2015, 54, 2151–2155 CrossRef CAS PubMed.
- J. D. Slinker, N. B. Muren, S. E. Renfrew and J. K. Barton, Nat. Chem., 2011, 3, 228–233 CrossRef CAS PubMed.
- N. Lu, H. Pei, Z. L. Ge, C. R. Simmons, H. Yan and C. H. Fan, J. Am. Chem. Soc., 2012, 134, 13148–13151 CrossRef CAS PubMed.
- H. Li, N. Arroyo-Curras, D. Kang, F. Ricci and K. W. Plaxco, J. Am. Chem. Soc., 2016, 138, 15809–15812 CrossRef CAS PubMed.
- M. Zayats, Y. Huang, R. Gill, C.-a. Ma and I. Willner, J. Am. Chem. Soc., 2006, 128, 13666–13667 CrossRef CAS PubMed.
- X. Zuo, S. Song, J. Zhang, D. Pan, L. Wang and C. Fan, J. Am. Chem. Soc., 2007, 129, 1042–1043 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04663d |
| This journal is © The Royal Society of Chemistry 2018 |