
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–organic framework composites with luminescent pincer platinum(II) complexes: 3MMLCT emission and photoinduced dehydrogenation catalysis†
Chun-Yi
Sun
ac,
Wai-Pong
To
 a,
Faan-Fung
Hung
a,
Xin-Long
Wang
c,
Zhong-Min
Su
c and
Chi-Ming
Che
*ab
a,
Faan-Fung
Hung
a,
Xin-Long
Wang
c,
Zhong-Min
Su
c and
Chi-Ming
Che
*ab
aState Key Laboratory of Synthetic Chemistry, Institute of Molecular Functional Materials, HKU-CAS Joint Laboratory on New Materials, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: cmche@hku.hk
bHKU Shenzhen Institute of Research and Innovation, Shenzhen, Guangdong 518053, China
cDepartment of Chemistry, Northeast Normal University, Changchun, Jilin, 130024 China
First published on 1st February 2018
Abstract
Pincer platinum(II) complexes are well documented to exhibit weak intermolecular interactions in the solid state and 77 K glassy solutions, leading to emissive triplet metal–metal-to-ligand charge transfer (3MMLCT) excited states that often vanish in dilute solutions. In this work, metal–organic framework (MOF) materials are introduced to provide a “solid solution” environment for easy access to 3MMLCT excited states of pincer platinum(II) complexes. Phosphorescent composites PtII@MOFs (1–4) with matrix-dependent monomers and oligomer emission properties were obtained. These PtII@MOFs are efficient catalysts for photoinduced dehydrogenation reactions.
Introduction
Platinum(II) complexes are known to exhibit weak intramolecular and intermolecular interactions that lead to triplet metal–metal-to-ligand charge transfer (3MMLCT) and/or 3[5dσ* → 6pσ] excited states.1,2 The diradical character of such excited states with enhanced metal–metal bonding interactions renders these complexes capable of performing photocatalytic C–X bond cleaving reactions.3,4 The classic example, [Pt2(μ-P2O5H2)4]4−, is a highly active catalyst for the photoinduced dehydrogenation of alcohols to aldehydes/ketones in the absence of a sacrificial electron acceptor via its long-lived 3[5dσ*6pσ] excited state.1a Due to the uniqueness of the μ-pyrophosphito ligand, extending the photochemistry of [Pt2(μ-P2O5H2)4]4− to other platinum(II) complexes is a non-trivial task. In this regard, pincer PtII complexes are appealing alternatives because their structures can be readily modified to elicit intermolecular Pt⋯Pt interactions and hence emissive 3MMLCT excited states in concentrated solutions or in the solid state.5 However, the photophysical and photochemical properties associated with the 3MMLCT excited states of PtII complexes often vanish in dilute solutions.Metal–organic frameworks (MOFs) have emerged as a new class of highly promising porous materials.6,7 In particular, the porous environment in MOFs provides a unique platform to confine and stabilize guest species, and as a result, novel properties of the incorporated guest may emerge.8 In the literature, reports on the incorporation of PtII complexes into MOFs are sparse;9 these previously reported PtII-MOF composites were formed by the coordination of PtIICl2 (ref. 9a–d) or PtIIL2 (L2 = 2,2′-bipyridine, (OPPh3)2 or (PPh3)2)9e moieties to the bipyridine units of MOFs and in some cases they were studied as photocatalysts for hydrogen production from water.9a,c,d We envisage that incorporating luminescent pincer PtII complexes into the pores of MOFs by, for example, a cation exchange method, can be a strategy to develop the 3MMLCT photochemistry of platinum(II) complexes. This method has been shown to be effective in placing phosphorescent d6 and d8 metal complexes inside MOFs.10,11 In this work, PtII complexes with a pincer C⁁N⁁C (where C is an N-heterocyclic carbene) ligand, [Pt(C⁁N⁁C)(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC6H5)]+ (Pt1) and [Pt(C⁁N⁁C)(CN)]+ (Pt2; both Pt1 and Pt2 have PF6− as a counteranion) were synthesized and used as guest species for three MOFs with different porous structures. These PtII@MOF composites were found to display matrix-dependent emission properties with emission peak maxima (λmax) ranging from 450 to 625 nm in air and also to catalyze photoinduced dehydrogenation reactions of various organic compounds with activities higher than those of the corresponding PtII complexes in homogeneous solutions by an order of magnitude.
CC6H5)]+ (Pt1) and [Pt(C⁁N⁁C)(CN)]+ (Pt2; both Pt1 and Pt2 have PF6− as a counteranion) were synthesized and used as guest species for three MOFs with different porous structures. These PtII@MOF composites were found to display matrix-dependent emission properties with emission peak maxima (λmax) ranging from 450 to 625 nm in air and also to catalyze photoinduced dehydrogenation reactions of various organic compounds with activities higher than those of the corresponding PtII complexes in homogeneous solutions by an order of magnitude.
Results and discussion
Syntheses and characterization of PtII@MOFs
Pt1 and Pt2 (Fig. 1a and b),12 and host MOFs ZJU-28,13 MOF1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 and MOF215 featuring negatively charged frameworks,13–15 were synthesized according to previously reported procedures. The molecular sizes of Pt1 and Pt2 based on optimization from theoretical calculations are 12.15 × 8.05 and 8.05 × 7.95 Å2, respectively. The formulas of MOF1, MOF2, and ZJU-28 are [(CH3)2NH2]2[Zn(TATAT)2/3]·3DMF·H2O, [(CH3)2NH2]2[ZnNa2(μ2-H2O)2(H2O)2(TATAT)]·2DMF, and [(CH3)2NH2]3[In3(BTB)4]·12DMF·22H2O, respectively (TATAT = 5,5′,5′′-(1,3,5-triazine-2,4,6-triyl)tris(azanediyl)triisophthalate; BTB = 4,4′,4′′-benzene-1,3,5-triyl-tribenzoate). Single-crystal X-ray structure determination revealed that ZJU-28 is a framework of parallel interwoven corrugated 63 nets containing two types of 1D channel (Fig. 1c) with a maximal pore size of 14.7 × 9.8 Å2.13 MOF1 shows a 3D chiral framework featuring an alternating arrangement of hexagonal and trigonal prismatic cages (Fig. 1d) with a maximal window size of 14.3 × 11.5 Å2,14 and MOF2 has a chiral framework with metal–organic nanotubes formed by heterometallic helical rods (Fig. 1e) and a channel size of 17.0 × 23.0 Å2.15
14 and MOF215 featuring negatively charged frameworks,13–15 were synthesized according to previously reported procedures. The molecular sizes of Pt1 and Pt2 based on optimization from theoretical calculations are 12.15 × 8.05 and 8.05 × 7.95 Å2, respectively. The formulas of MOF1, MOF2, and ZJU-28 are [(CH3)2NH2]2[Zn(TATAT)2/3]·3DMF·H2O, [(CH3)2NH2]2[ZnNa2(μ2-H2O)2(H2O)2(TATAT)]·2DMF, and [(CH3)2NH2]3[In3(BTB)4]·12DMF·22H2O, respectively (TATAT = 5,5′,5′′-(1,3,5-triazine-2,4,6-triyl)tris(azanediyl)triisophthalate; BTB = 4,4′,4′′-benzene-1,3,5-triyl-tribenzoate). Single-crystal X-ray structure determination revealed that ZJU-28 is a framework of parallel interwoven corrugated 63 nets containing two types of 1D channel (Fig. 1c) with a maximal pore size of 14.7 × 9.8 Å2.13 MOF1 shows a 3D chiral framework featuring an alternating arrangement of hexagonal and trigonal prismatic cages (Fig. 1d) with a maximal window size of 14.3 × 11.5 Å2,14 and MOF2 has a chiral framework with metal–organic nanotubes formed by heterometallic helical rods (Fig. 1e) and a channel size of 17.0 × 23.0 Å2.15
 | ||
| Fig. 1 Pt1 (a) and Pt2 (b) complexes used in this work. 3D polyhedral structures of ZJU-28 (c), MOF1 (d) and MOF2 (e), with C shown in gray, O red, and N blue. | ||
PtII@MOFs with different loadings of PtII complexes, namely, Pt1@ZJU-28 (1a–1e), Pt1@MOF1 (2a–2e), Pt1@MOF2 (3a–3e) and Pt2@ZJU-28 (4a–4e), were obtained as yellow or pale yellow solids (Fig. S1, ESI†) by immersing MOF crystals in DMF or MeCN solutions of the PtII complexes at different concentrations ((0.25–10) × 10−4 M). Pt2 was found to be unstable upon incorporation into MOF1 or MOF2, which precluded studies on the Pt2@MOF1 and Pt2@MOF2 composites. The powder X-ray diffraction (PXRD) data of these composites were nearly identical to those of their matrix MOFs, indicating that the ion exchange process does not affect the crystallinity of the host materials (Fig. S2, ESI†). Inductively coupled plasma (ICP) mass spectrometric (MS) measurements (Table 1) showed that the loadings of the PtII complexes ranged from 0.08 to 8.68 wt%. Distribution of the PtII complex cation in a MOF was examined by optical microscopy, scanning electron microscope (SEM) imaging, energy dispersive X-ray (EDX) elemental mapping and N2 sorption experiments. Analysis of a cross section of a crystal of 2e under an optical microscope showed that all of the surfaces of the split crystal emitted yellow light under light irradiation at 365 nm, indicating uniform distribution of Pt1 throughout the crystal (Fig. S3, ESI†). SEM imaging and EDX elemental mapping in a cross section of a split crystal of PtII@MOFs showed the Pt element to have random distribution in the inner space of the PtII@MOFs (Fig. S4–S7, ESI†). Simulation by DFT calculation for the PtII@MOFs, taking Pt1@MOF1 (2) as an example, revealed that the PtII complex resides close to the ligand of MOF and the distance between the pyridine ring in the C⁁N⁁C ligand of the PtII complex and the ligand of the MOF is ∼3.3 Å (Fig. S8, ESI†), with the adsorption energy being 8.270 eV. N2 sorption measurements revealed that the Brunauer–Emmett–Teller (BET) surface area decreased by >26% on going from MOF1 to 2e (1112 → 818 m2 g−1, Fig. S9, ESI†), supporting the confinement of Pt1 in the pores/channels of MOF1.
| PtII@MOFs | Loading of PtII complexes (wt%) | ||||
|---|---|---|---|---|---|
| Pt1@ZJU-28 | 1a | 1b | 1c | 1d | 1e |
| 0.11 | 0.28 | 0.54 | 1.36 | 1.50 | |
| Pt1@MOF1 | 2a | 2b | 2c | 2d | 2e |
| 0.08 | 0.35 | 0.64 | 1.82 | 3.45 | |
| Pt1@MOF2 | 3a | 3b | 3c | 3d | 3e |
| 0.60 | 1.37 | 2.82 | 5.92 | 8.68 | |
| Pt2@ZJU-28 | 4a | 4b | 4c | 4d | 4e |
| 0.19 | 0.27 | 0.81 | 1.32 | 2.41 | |
Spectroscopy and photophysical measurements
The electronic absorption spectra of composites 1, 2 and 3 (Fig. 2, Table 2) with low complex concentrations showed intense absorption bands at 325, 327 and 330 nm, and moderately intense bands at 390, 405 and 400 nm, respectively. The high energy absorption bands are assigned to the absorption of the matrix MOFs and intraligand (1IL) π → π* transitions of the –CCC6H5 and C⁁N⁁C pincer ligands, whereas the lower-energy bands are assigned to the mixed singlet metal-to-ligand charge transfer (1MLCT) [dπ(Pt) → π*(C⁁N⁁C)] and the alkynyl-to-C⁁N⁁C ligand-to-ligand charge transfer (LLCT) [π(–CCC6H5) → π*(C⁁N⁁C)] transitions, both of which are characteristic absorptions of monomeric pincer PtII complexes in solution.12 Notably, a redshift of the low energy absorption band was observed in composites with a higher loading of the PtII complex. For example, composite 4a ([PtII] = 0.19%; Fig. 2d) showed absorption at only 300–400 nm. For composites 4d and 4e with higher [PtII] loadings of 1.32 and 2.41 wt%, respectively, there was a new, broad absorption band at 400–500 nm attributable to a 1MMLCT transition of aggregated species of Pt2 (ref. 12) inside the MOF (Fig. 2d). For comparison, increasing the concentration of Pt2 in solution from 5 × 10−5 M to 1 × 10−3 M did not result in a notable shift in the absorption peak maxima or the formation of a new absorption band (Fig. 2f).
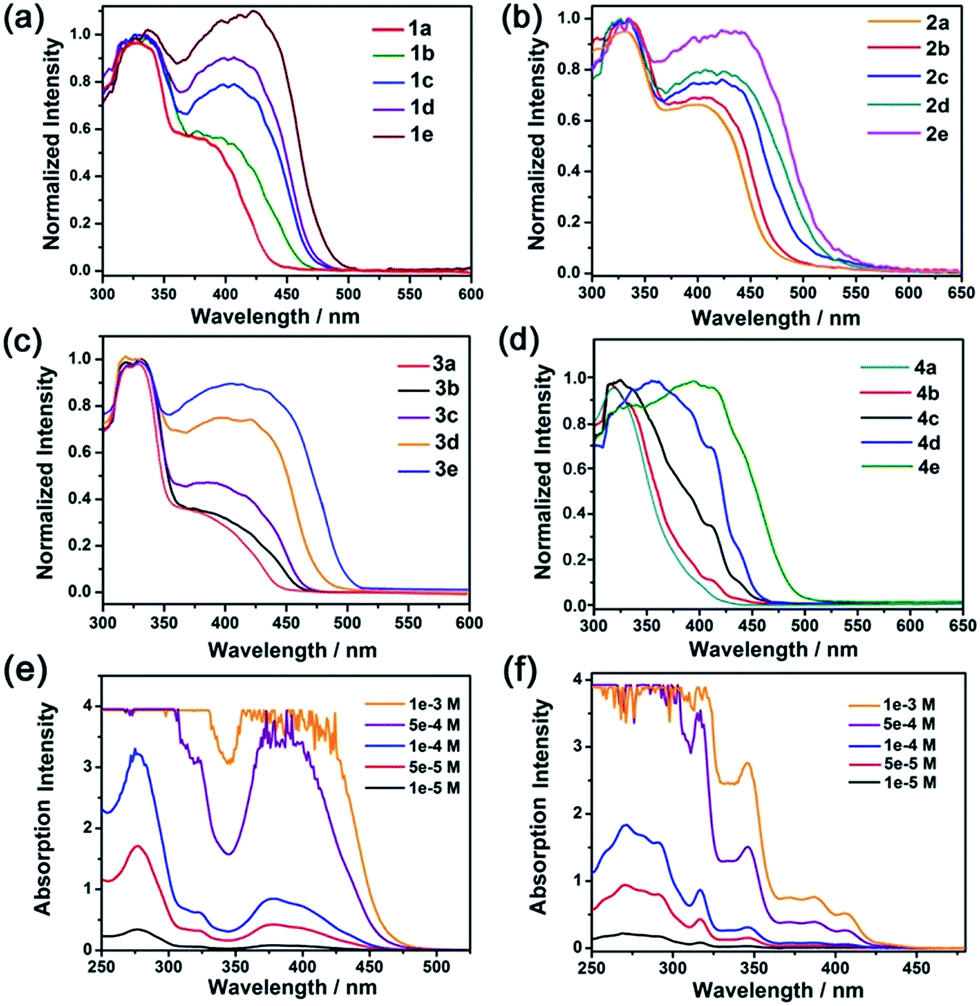 | ||
| Fig. 2 Electronic absorption spectra of PtII@MOFs (a–d) and electronic absorption spectra of Pt1 (e) and Pt2 (f) in MeCN. | ||
| Absorption | Emission | ||
|---|---|---|---|
| λ max [nm] (ε [×103 dm3 mol−1 cm−1]) | λ em [nm] (τ [μs]) | Φ [%] | |
| a PtII complexes at a concentration of 2 × 10−5 M. b Solutions for photophysical studies were degassed using five freeze–pump–thaw cycles. c Measured in open air. | |||
| Pt1 | 277 (34.2), 321 (6.47), 378 (8.47), 394 (sh 7.59), 430 (sh 3.07) | 535 (2.1)b | 36b |
| Pt2 | 271 (18.1), 280 (16.6), 291 (15.6), 317 (8.69), 346 (3.17), 372 (0.84), 387 (0.82), 406 (0.54) | 449 (0.2), 470b | 6b |
| 1b | 325, 390 (sh) | 527 (1.9)c | 25c |
| 2b | 327, 405 (sh) | 520 (1.8)c | 19c |
| 3b | 330, 400 (sh) | 519 (2.5)c | 23c |
| 4b | 316, 409 (sh) | 450, 477 (4.9)c | 23c |
| 4e | 600 (3.6)c | 81c | |
The emission properties of 1–4 were investigated. As depicted in Fig. 3, 1a displays broad emission with λmax at 530 nm (monomer emission). As the loading of Pt1 increased (1e), another emission band at 620 nm gradually developed (aggregate emission). For the composites of 2, 2a displayed broad emission with λmax at 510 nm. When the loading of Pt1 increased (2d and 2e), there was a gradual redshift of the monomer λmax accompanied by the appearance of aggregate emission as a shoulder at 590 nm. The redshift of the monomer emission is ascribed to the intermolecular interactions of PtII complexes in the ground state.16 A similar redshift of the high energy emission band by ∼20 nm was also found for 3, the framework of which contains the same organic ligand as 2. A more distinct aggregate emission was observed for 3e, presumably because of the higher concentration of Pt1, which enhances the formation of aggregate species. Similarly, Pt1 in dilute solutions ((1–5) × 10−5 M, Fig. S11, ESI†) displayed intense, unstructured emission with λmax at 530 nm and an additional emission band at 615 nm in concentrated solutions (10−4–10−3 M). For the composites of 4, a vibronic structured blue emission band17 with λmax at 450 and 470 nm was found for 4a, whereas an additional broad emission band at 600 nm became apparent in 4d, which has a much higher Pt2 loading. For 4e, which has the highest Pt2 loading (2.41 wt%), only aggregate emission was observed and the emission quantum yield of 4e was 81%. For comparison, Pt2 in dilute solution (Fig. S11b, ESI†) ((1–5) × 10−5 M) showed vibronic structured emission with λmax at 449 and 470 nm, whereas a broad emission peak at 593 nm was observed in concentrated solutions (10−4–10−3 M). In contrast to composite 4e, the monomer emission of Pt2 could still be observed in solution, even at a concentration of 1 × 10−3 M.
 | ||
| Fig. 3 Steady-state emission spectra of PtII@MOFs in open air at room temperature. | ||
Nanosecond time-resolved emission spectra of both Pt1 (5 × 10−4 M) and Pt2 (1 × 10−3 M) in MeCN (Fig. 4) exhibited gradually developing emission at ∼600 nm in addition to prompt phosphorescence at 530 nm for Pt1 and 449–470 nm for Pt2 (Fig. 4e). The growth and subsequent decay of this low energy (∼600 nm) emission is similar to the excimeric emission formed between the excited state and ground state of the PtII complex.1,18 However, different kinetic behavior for the low energy emission was observed in the PtII@MOF composites. The aggregate emissions of 1e, 2e, 3e and 4d were instantaneously generated after laser pulse excitation, similar to their corresponding monomer emission. In 4e (Fig. S12, ESI†), only one phosphorescence band with λmax at 600 nm was detected, which was consistent with the result from the steady-state measurement. The nearly simultaneous decay in the beginning of the low and high energy emission bands is indicative of the absence of excimeric emission in PtII@MOFs, and therefore, the aggregate emissions of PtII@MOFs are proposed to originate from ground state aggregate species. Comparing the electronic absorption spectra of 1–4 with their corresponding excitation spectra (Fig. S13, ESI†), which showed vastly different excitation profiles for emission at 450–530 nm and 600–620 nm, led to the attribution of their aggregate emissions to a 3MMLCT excited state of ground state aggregates of the PtII complexes19 within the pores/channels of the MOFs. The origin of the difference in emission for the aggregated emissions of PtII in solution and in the MOFs suggests that the photophysical properties of PtII complexes could be altered by employing MOFs as host materials.
 | ||
| Fig. 4 (a)–(d): Time-resolved emission spectra of PtII@MOFs in open air at room temperature. (e) and (f) Time-resolved emission spectra of the PtII complexes ([Pt1] = 5 × 10−4 M; [Pt2] = 1 × 10−3 M) in degassed MeCN solutions. | ||
Photo-catalysis
Although several PtII complexes with high energy 3IL or 3MLCT excited states (>2.5 eV) are active catalysts for photooxidation and photoinduced aerobic C–C bond formation reactions, there have been few reports on employing complexes with low energy 3MMLCT excited states for such reactions.3d,20 Composite 4e, which displays a predominant 3MMLCT excited state, was examined as a catalyst for the photoinduced α-cyanation of tertiary amines and reductive cyclization of alkyl iodides. These two reactions were performed in MeCN at room temperature (RT) under light irradiation (λ > 370 nm, Fig. 5). For the α-cyanation reaction, a product turnover number (TON) of ∼680 was achieved over 8 hours of irradiation (turnover frequency (TOF): 90.6, Table S2, ESI†) with 100% substrate conversion and 88% product yield. The catalyst could be recycled by washing with MeCN. After five cycles, the yield still reached a good value of ∼63%. No leaching of Pt2 was observed after the photochemical reaction, using ICP-MS analysis of the recovered 4e. When using Pt2 (5 × 10−4 M) as a catalyst, the product TON was found to be ∼30% of that of 4e under the same conditions. With ZJU-28 alone as a catalyst, the product TON was <10. For the photoinduced cyclization reaction, a TON of ∼155 for the desired product was achieved with 4e over 10 hours (TOF: 15.5) with 99% yield; this TON was 5-fold higher than that found with Pt2 in the corresponding homogeneous reaction. | ||
| Fig. 5 (Top) Scheme of oxidative cyanation of a tertiary amine (I) and reductive cyclization of an alkyl iodide (II). (Bottom) (a) The time course of the oxidative cyanation by 4e (Pt2@ZJU-28), Pt2 and ZJU-28; (b) the yield of the reaction in recycling experiments using 4e as the catalyst. | ||
We envisage that the 3MMLCT excited states of the PtII@MOF composites are highly reactive and can be harnessed for photoinduced C–H dehydrogenation reactions, similar to [Pt2(P2O5H2)4]4−.1a This type of reaction proceeds via inner-sphere atom abstraction by triplet excited species with vacant coordination site(s).21 The photocatalytic activity of PtII@MOFs towards the conversion of 1-phenylethanol, benzyl alcohol, isopropanol (IPA) and cyclohexene to the corresponding ketone, aldehyde and cyclohexane was evaluated using 1 and 4 as the catalysts (Fig. 6 and Table 3) and MeCN as the solvent under a N2 atmosphere and at RT.
 | ||
| Fig. 6 (Top) Photoinduced dehydrogenation reaction catalyzed by various catalysts (III). (Bottom) The TONs of acetophenone in reaction III using Pt complexes at different concentrations and 1d, 4d, 1e and 4e as the catalysts. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst | TONs of reaction IV | TONs of reaction V | TONs of reaction VIb | |||
| P1 | P2 | P3 | ||||
| a For detailed reaction conditions, please refer to the ESI. b The amount of acetone formed cannot be determined because of the overlap of its GC signal with that of cyclohexene. | ||||||
| 1d | 47.5 | 10.1 | 23.6 | 4.2 | 1.1 | |
| 1e | 30.1 | 16.6 | 31.2 | 7.5 | 1.5 | |
| 4d | 38.2 | 6.1 | 11.3 | 1.3 | 0.4 | |
| 4e | 29.4 | 9.5 | 28.5 | 2.4 | 1.0 | |
| Pt1 | 5 × 10−4 M | 10.3 | — | — | — | — |
| 5 × 10−5 M | — | — | — | — | — | |
| Pt2 | 5 × 10−4 M | 8.3 | — | — | — | — |
| 5 × 10−5 M | — | — | — | — | — | |
| ZJU-28 | — | — | — | — | — | |

Irradiation (λ > 370 nm) of an MeCN solution of 1-phenylethanol for 6 hours with 1d or 1e as the catalyst provided acetophenone with TONs of 363 and 216 (TOF: 60.5 and 36), respectively (Fig. 6, the TON for hydrogen was not determined due to its possible adsorption on the inner surface of the MOF materials). Similar photochemical reactions with Pt1 at concentrations of 0.5–5 × 10−4 M afforded trace amounts of the product. Similarly, composites 4d and 4e showed superior performance compared to Pt2 in the same photoinduced reaction. The leaching of the PtII complex from the composites was not observed after photolysis according to ICP-MS analysis (Table S1, ESI†). After catalysis, no obvious changes in the PXRD patterns were detected (Fig. S14 and S15, ESI†). The control experiment using pure ZJU-28 as the catalyst did not show obvious product formation. As composites 1d, 1e, 4d and 4e show a predominant 3MMLCT excited state upon photoexcitation, the photo-catalysis results suggest that the 3MMLCT excited states in PtII@MOF materials are responsible for the observed photocatalytic C–H bond dehydrogenation reactions. When 1-phenylethanol was replaced with benzyl alcohol, a TON of 47.5 for benzaldehyde was produced using 1d, which is ∼5-fold higher than that obtained in Pt1 solution at 5 × 10−4 M (Table 3). A negligible amount of product was detected when a low concentration (5 × 10−5 M) of Pt1 was used. This divergence in reactivity was also observed between 4d and Pt2 in solution. PtII@MOFs also reacted with IPA to furnish TONs of 6.1–16.6 of acetone upon light irradiation for 12 hours (Table 3). However, an MeCN solution of Pt1 or Pt2 did not show obvious acetone formation under similar conditions. Furthermore, after a mixture of IPA and cyclohexene was irradiated in the presence of 1d or 1e for 6 hours, cyclohexane was furnished with TONs of 23.6 and 31.2 (Table 3), respectively. The homocoupling product of the as-formed cyclohexenyl radicals (P2) and their partially hydrogenated derivative (P3) were also detected in the reaction mixture. The formation of cyclohexane is proposed to originate from the hydrogenation of cyclohexene by an in situ-generated Pt–H species, which might be formed from the abstraction of the allylic C–H atom of cyclohexene or from the reaction with IPA.1a To elucidate the origin of the hydrogen atoms, deuterated (d8) IPA was used in the reaction. Signals with a m/z of 84, 162 and 164, which correspond to non-deuterated P1, P2 and P3, respectively, could still be detected as the sole products by GC-MS, thereby excluding the possibility of IPA serving as the H-atom source. Notably, when the same reaction was conducted with Pt1 or Pt2 as the catalyst in homogeneous solution, cyclohexane was not detected. PtII@MOFs can also catalyze the photoinduced dehydrogenation of indoline and 1,2,3,4-tetrahydroquinoline (Table 4). Irradiation of an MeCN solution containing indoline and 1d or 4d produced 1H-indole (P4) with TONs of 58.6 and 44.3, respectively, which were approximately 6-fold and 17-fold higher than those obtained using Pt1 or Pt2 at a concentration of 5 × 10−4 M as the photocatalyst. When the concentration of Pt1 or Pt2 in the homogeneous reaction was reduced to 5 × 10−5 M, only a trace amount of 1H-indole was detected. For the dehydrogenation reaction of 1,2,3,4-tetrahydroquinoline, TONs of 25–27.2 of 3,4-dihydroquinoline (P5) and TONs of 8.3–12.3 of quinoline (P6) were produced using 1d and 4d (Table 4).

PtII@MOFs also catalyzed the photoinduced dehydrogenative cyclization of o-aminobenzamide with benzyl alcohol at RT. 2-Phenylquinazolin-4(3H)-one (P7) was obtained with TONs of 14.9 and 9.4 using 1d and 4d as photocatalysts, respectively (Table 5). In contrast to the heterogeneous catalyst, Pt1 and Pt2 at a concentration of 5 × 10−4 M in MeCN did not show catalytic activity in this reaction.

The improved performances of PtII@MOF catalysts such as 1d and 4d over that of Pt1 and Pt2, respectively, in the photo-catalysis described above reveals the beneficial effects of encapsulation of the PtII complexes in the pores of the MOF hosts. For the photo-induced catalytic α-cyanation of tertiary amines (reaction I) and reductive cyclization of alkyl iodide (reaction II), which are proposed to proceed via singlet oxygen11,22 and outer-sphere electron transfer23 pathways, respectively, the higher activity of PtII@MOF catalysts relative to PtII complexes is reminiscent of the better catalytic activity of AuIII@MOF than that of AuIII complexes for the two reactions.11 In the cases of the other photo-induced catalytic reactions studied in this work, that is, photo-induced dehydrogenation reactions, the enhancement of the catalytic activity upon formation of PtII@MOFs could be ascribed to the aggregation of the PtII complexes in the pores of the MOF hosts resulting in the 3MMLCT excited state. Based on the mechanism proposed for the [Pt2(μ-P2O5H2)4]4− system,1a and considering the observed hydrogenated by-products in the cyclohexene reaction, a mechanism involving light irradiation generating the 3MMLCT excited state species (Pt–Pt)* which abstracts a hydrogen atom from, for example, the α-C–H or allylic C–H bond of the alcohol, indoline, 1,2,3,4-tetrahydroquinoline or cyclohexene substrate to form a H–(Pt–Pt) species, is proposed for the PtII@MOF system; the H–(Pt–Pt) species may further abstract a hydrogen atom, forming H2(Pt–Pt) and the dehydrogenation product, and H2 is eliminated from the H2(Pt–Pt) species to regenerate (Pt–Pt). The formation of a reactive H–(Pt–Pt) species could be inferred from the formation of cyclohexane from cyclohexene. For the dehydrogenative coupling reaction (Table 5), the photo-induced dehydrogenation of benzyl alcohol catalyzed by PtII@MOF would generate benzaldehyde, which undergoes a condensation reaction with o-aminobenzamide, followed by an intramolecular nucleophilic attack on the carbon of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N imine bond by NH2 of the amide group, to give a 2-phenyl-2,3-dihydroquinazolin-4(1H)-one intermediate; dehydrogenation of this intermediate by PtII@MOF gives the final product.
N imine bond by NH2 of the amide group, to give a 2-phenyl-2,3-dihydroquinazolin-4(1H)-one intermediate; dehydrogenation of this intermediate by PtII@MOF gives the final product.
Conclusions
A series of PtII@MOFs composites displaying strong matrix-dependent phosphorescence were prepared via a cation exchange method. The cages and nanotubes of the MOFs function as concentrators for the PtII complexes and induce aggregation inside the MOFs, leading to 3MMLCT emission. With the diradical character of the 3MMLCT excited state, the PtII@MOFs showed superior performance in photoinduced C–C bond formation, dehydrogenation and dehydrogenative cyclization reactions compared to the corresponding PtII complexes in solution. This simple approach for preparing highly photocatalytically active MOF composites offers a new entryway to new classes of phosphorescent and heterogeneous photofunctional materials with useful applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Basic Research Program of China (No. 2013CB834802), the University Grants Committee Areas of Excellence Scheme (AoE/P-03/08), the NSFC/RGC Joint Research Scheme (N_HKU 752/08), the National Science Foundation of China (No. 21471027, 21601032), and the Fundamental Research Funds for the Central Universities (2412016KJ041).Notes and references
- (a) D. M. Roundhill, H. B. Gray and C.-M. Che, Acc. Chem. Res., 1989, 22, 55–61 CrossRef CAS; (b) C.-M. Che, V. W.-W. Yam, W.-T. Wong and T.-F. Lai, Inorg. Chem., 1989, 28, 2908–2910 CrossRef CAS; (c) V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1991, 30, 4446–4452 CrossRef CAS; (d) H.-K. Yip, C.-M. Che, Z.-Y. Zhou and T. C. W. Mak, J. Chem. Soc., Chem. Commun., 1992, 1369–1371 RSC.
- (a) K. M.-C. Wong and V. W.-W. Yam, Coord. Chem. Rev., 2007, 251, 2477–2488 CrossRef CAS; (b) H. B. Gray, S. Záliš and A. Vlček, Coord. Chem. Rev., 2017, 345, 297–317 CrossRef CAS.
- (a) C.-M. Che, W.-M. Lee, K.-C. Cho, P. D. Harvey and H. B. Gray, J. Phys. Chem., 1989, 93, 3095–3099 CrossRef CAS; (b) D. Li, C.-M. Che, H.-L. Kwong and V. W.-W. Yam, J. Chem. Soc., Dalton Trans., 1992, 3325–3329 RSC; (c) G. Revol, T. McCallum, M. Morin, F. Gagosz and L. Barriault, Angew. Chem., Int. Ed., 2013, 52, 13342–13345 CrossRef CAS PubMed; (d) Z. Li, Y. Han, Z. Gao and F. Wang, ACS Catal., 2017, 7, 4676 CrossRef CAS.
- (a) C.-M. Che and S.-W. Lai, Coord. Chem. Rev., 2005, 249, 1296–1309 CrossRef CAS; (b) W.-P. To, T. Zou, R. W.-Y. Sun and C.-M. Che, Philos. Trans. R. Soc., A, 2013, 371, 20120126 CrossRef PubMed; (c) K. Li, G. S. M. Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC; (d) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed.
- (a) L.-Z. Wu, T.-C. Cheung, C.-M. Che, K.-K. Cheung and M. H. W. Lam, Chem. Commun., 1998, 1127–1128 RSC; (b) C.-M. Che, W.-F. Fu, S.-W. Lai, Y.-J. Hou and Y.-L. Liu, Chem. Commun., 2003, 118–119 RSC; (c) E. J. Rivera, C. Figueroa, J. L. Colón, L. Grove and W. B. Connick, Inorg. Chem., 2007, 46, 8569–8576 CrossRef CAS PubMed; (d) K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424–434 CrossRef CAS PubMed; (e) K. Mori, K. Watanabe, K. Fuku and H. Yamashita, Chem.–Eur. J., 2012, 18, 415–418 CrossRef CAS PubMed; (f) M. Mauro, A. Aliprandi, C. Cebrián, D. Wang, C. Kübel and L. De Cola, Chem. Commun., 2014, 50, 7269–7272 RSC.
- (a) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC; (b) F. A. A. Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC; (c) H.-C. Zhou, J. R. Long and O. M. Yaghi, Special Issue: Metal-Organic Frameworks, Chem. Rev., 2012, 112(2) CrossRef PubMed; (d) H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836–868 CrossRef CAS PubMed; (e) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (f) H.-C. Zhou and S. Kitagawa, Themed Issue on Metal-Organic Frameworks (MOFs), Chem. Soc. Rev., 2014, 43(16) RSC.
- (a) O. K. Farha, A. Ö. Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed; (b) J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS PubMed; (c) H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O’Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed; (d) H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- (a) Y. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS PubMed; (b) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed; (c) J. Yu, Y. Cui, H. Xu, Y. Yang, Z. Wang, B. Chen and G. Qian, Nat. Commun., 2013, 4, 2719 Search PubMed; (d) Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- (a) T. Zhou, Y. Du, A. Borgna, J. Hong, Y. Wang, J. Han, W. Zhang and R. Xu, Energy Environ. Sci., 2013, 6, 3229–3234 RSC; (b) S. Øien, G. Agostini, S. Svelle, E. Borfecchia, K. A. Lomachenko, L. Mino, E. Gallo, S. Bordiga, U. Olsbye, K. P. Lillerud and C. Lamberti, Chem. Mater., 2015, 27, 1042–1056 CrossRef; (c) T. Toyao, M. Saito, S. Dohshi, K. Mochizuki, M. Iwata, H. Higashimura, Y. Horiuchi and M. Matsuoka, Res. Chem. Intermed., 2016, 42, 7679–7688 CrossRef CAS; (d) C.-C. Hou, T.-T. Li, S. Cao, Y. Chen and W.-F. Fu, J. Mater. Chem. A, 2015, 3, 10386–10394 RSC; (e) Y. Atoini, E. A. Prasetyanto, P. Chen, D. Jonckheere, D. De Vos and L. De Cola, Supramol. Chem., 2017, 29, 758–767 CrossRef CAS.
- (a) C.-Y. Sun, X.-L. Wang, X. Zhang, C. Qin, P. Li, Z.-M. Su, D.-X. Zhu, G.-G. Shan, K.-Z. Shao, H. Wu and J. Li, Nat. Commun., 2013, 4, 2717 Search PubMed; (b) W. Zhang, B. Li, H. Ma, L. Zhang, Y. Guan, Y. Zhang, X. Zhang, P. Jing and S. Yue, ACS Appl. Mater. Interfaces, 2016, 8, 21465–21471 CrossRef CAS PubMed.
- C.-Y. Sun, W.-P. To, X.-L. Wang, K.-T. Chan, Z.-M. Su and C.-M. Che, Chem. Sci., 2015, 6, 7105–7111 RSC.
- S. Y.-L. Leung, E. S.-H. Lam, W. H. Lam, K. M.-C. Wong, W.-T. Wong and V. W.-W. Yam, Chem.–Eur. J., 2013, 19, 10360–10369 CrossRef CAS PubMed.
- J. Yu, Y. Cui, C. Wu, Y. Yang, Z. Wang, M. O’Keeffe, B. Chen and G. Qian, Angew. Chem., Int. Ed., 2012, 51, 10542–10545 CrossRef CAS PubMed.
- C.-Y. Sun, C. Qin, C.-G. Wang, Z.-M. Su, S. Wang, X.-L. Wang, G.-S. Yang, K.-Z. Shao, Y.-Q. Lan and E.-B. Wang, Adv. Mater., 2011, 23, 5629–5632 CrossRef CAS PubMed.
- C.-Y. Sun, X.-L. Wang, C. Qin, J.-L. Jin, Z.-M. Su, P. Huang and K.-Z. Shao, Chem.–Eur. J., 2013, 19, 3639–3645 CrossRef CAS PubMed.
- (a) S.-W. Lai, M. C.-W. Chan, T.-C. Cheung, S.-M. Peng and C.-M. Che, Inorg. Chem., 1999, 38, 4046–4056 CrossRef CAS; (b) S. C. F. Kui, S. S.-Y. Chui, C.-M. Che and N. Zhu, J. Am. Chem. Soc., 2006, 128, 8297–8309 CrossRef CAS PubMed.
- W. Lu, M. C. W. Chan, K.-K. Cheung and C.-M. Che, Organometallics, 2001, 20, 2477–2486 CrossRef CAS.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed.
- (a) J. A. Bailey, V. M. Miskowski and H. B. Gray, Inorg. Chem., 1993, 32, 369–370 CrossRef CAS; (b) V. Sicilia, J. Forniés, J. M. Casas, A. Martín, J. A. López, C. Larraz, P. Borja, C. Ovejero, D. Tordera and H. Bolink, Inorg. Chem., 2012, 51, 3427–3435 CrossRef CAS PubMed.
- (a) X.-H. Li, L.-Z. Wu, L.-P. Zhang, C.-H. Tung and C.-M. Che, Chem. Commun., 2001, 2280–2281 RSC; (b) Y. Yang, D. Zhang, L.-Z. Wu, B. Chen, L.-P. Zhang and C.-H. Tung, J. Org. Chem., 2004, 69, 4788–4791 CrossRef CAS PubMed; (c) D. Zhang, L.-Z. Wu, L. Zhou, X. Han, Q.-Z. Yang, L.-P. Zhang and C.-H. Tung, J. Am. Chem. Soc., 2004, 126, 3440–3441 CrossRef CAS PubMed; (d) K. Feng, R.-Y. Zhang, L.-Z. Wu, B. Tu, M.-L. Peng, L.-P. Zhang, D. Zhao and C.-H. Tung, J. Am. Chem. Soc., 2006, 128, 14685–14690 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- W.-P. To, Y. Liu, T.-C. Lau and C.-M. Che, Chem.–Eur. J., 2013, 19, 5654–5664 CrossRef CAS PubMed.
- (a) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 CrossRef CAS PubMed; (b) P.-K. Chow, G. Chang, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084–2089 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c7sc04528j |
| This journal is © The Royal Society of Chemistry 2018 |