
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncorporation of redox-inactive cations promotes iron catalyzed aerobic C–H oxidation at mild potentials†
Teera
Chantarojsiri‡
 ,
Joseph W.
Ziller
and
Jenny Y.
Yang
*
,
Joseph W.
Ziller
and
Jenny Y.
Yang
*
Department of Chemistry, University of California, Irvine, 92697, USA. E-mail: j.yang@uci.edu
First published on 7th February 2018
Abstract
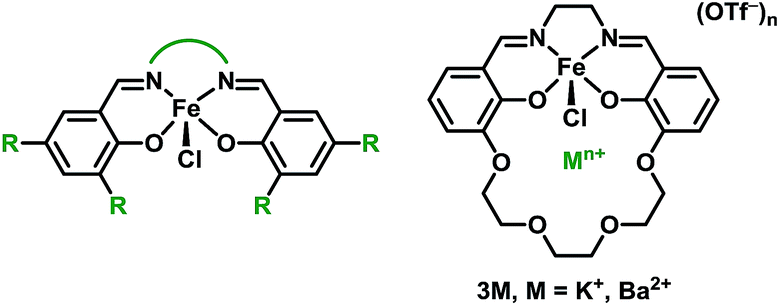
The synthesis and characterization of the Schiff base complexes Fe(II) (2M) and Fe(III)Cl (3M), where M is a K+ or Ba2+ ion incorporated into the ligand, are reported. The Fe(III/II) redox potentials are positively shifted by 440 mV (2K) and 640 mV (2Ba) compared to Fe(salen) (salen = N,N′-bis(salicylidene)ethylenediamine), and by 70 mV (3K) and 230 mV (3Ba) compared to Fe(Cl)(salen), which is likely due to an electrostatic effect (electric field) from the cation. The catalytic activity of 3M towards the aerobic oxidation of allylic C–H bonds was explored. Prior studies on iron salen complexes modified through conventional electron-donating or withdrawing substituents found that only the most oxidizing derivatives were competent catalysts. In contrast, the 3M complexes, which are significantly less oxidizing, are both active. Mechanistic studies comparing 3M to Fe(salen) derivatives indicate that the proximal cation contributes to the overall reactivity in the rate determining step. The cationic charge also inhibits oxidative deactivation through formation of the corresponding Fe2-μ-oxo complexes, which were isolated and characterized. This study demonstrates how non-redox active Lewis acidic cations in the secondary coordination sphere can be used to modify redox catalysts in order to operate at milder potentials with a minimal impact on the reactivity, an effect that was unattainable by tuning the catalyst through traditional substituent effects on the ligand.
Introduction
Transition metal complexes are ubiquitous as redox catalysts because they can both activate substrates and mediate electron transfer through multiple accessible redox states. Catalytic function is conventionally optimized through inductive effects by modifying the supporting ligand with electron-withdrawing or -donating substituents (Chart 1, left).1 Modifying ligands in this fashion impacts both the electronic structure of the metal as well as the redox properties, which can lead to an inverse correlation between the activity and operation at mild redox potentials. Scaling relationships of this type are observed for many classes of homogeneous oxidation catalyst,2,3 as well as for polymerization catalysts.4 The trade-off between catalytic activity and function at mild potentials has also been found in electrocatalysts for O2,5 CO2,6 and H+ (ref. 7) reduction, and is a major challenge in the optimization of catalysts for both high activity and energetic efficiency (low overpotential).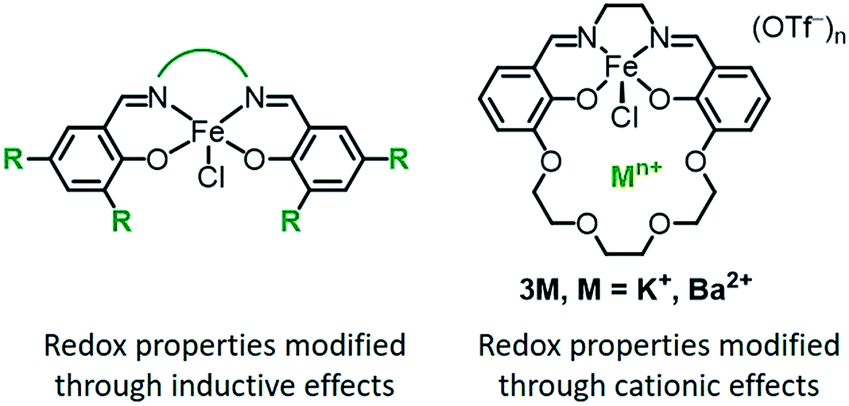 | ||
| Chart 1 | ||
Various strategies have been successfully employed to improve redox reactivity at mild potentials, such as enzyme-inspired cooperative or secondary interactions.8–36 An additional approach would utilize the secondary coordination sphere to adjust the redox properties. In nature, only three metal–ligand motifs are commonly used to mediate electron transfer (iron–sulfur clusters, hemes, and cupredoxins), yet the measured redox potentials of each cofactor span a wide range depending on their microenvironment.37 Elegant studies employing site-directed mutagenesis in artificial metalloenzymes demonstrate how secondary coordination sphere interactions, such as hydrogen-bonding or hydrophobicity, adjust the redox potential.38–40 Local electric field (electrostatic) effects have also been cited as a key component for the high catalytic activity found in enzymes41,42 and are emerging as an important tool for achieving selective or increased reactivity in organic synthesis.43–59 However, there are fewer examples of applying electrostatic interactions to enable more efficient redox catalysis.60–62
Transition metal complexes that contain crown ether-like functionalities to encapsulate alkali or alkaline earth metal ions have been reported for a variety of applications.63–92 We recently reported that the incorporation of non-redox active cations in a Co(II) Schiff base complex leads to significant positive shifts in the Co(II/I) redox potential,93 an effect that has been observed in other synthetic systems.94–101 Notably, our spectroscopic studies indicate that the shift in redox potential is likely due to an electric field potential from the cation, since the electronic structure of the metal is not significantly perturbed.93
We were interested in exploring how cation incorporation would impact the redox reactivity compared to complexes tuned through traditional inductive modifications. To this end, we examined the effect of proximal cations on iron catalysts for the aerobic allylic C–H oxidation of cyclohexene. Prior studies on iron Schiff base complexes have demonstrated a strong correlation of the Fe(III/II) redox potential (Table 3) with the overall catalytic activity.102 When inductive effects were used to modify the iron Schiff base complexes, catalysis was only observed for the tetranitro substituted complexes, which have potentials positive of −0.27 V vs. [Fe(Cp)2]+/0. For these complexes, the activity increases with the more oxidizing redox potentials.102
For this study we synthesized the iron complexes 3M, shown in Chart 1 and Scheme 1, which have either a proximal M = K+ or Ba2+ ion in the secondary coordination sphere. 3K and 3Ba are both active catalysts despite having Fe(III/II) redox potentials that would be insufficient for catalysis if they had been tuned by installing electron withdrawing groups.
 | ||
| Scheme 1 | ||
Mechanistic studies with 3M and 2M indicate that the K+ or Ba2+ ion facilitates the rate determining step. Aside from their role in adjusting the redox potential, non-redox active Lewis acidic metals are also known to play a role in the activation of dioxygen and the corresponding oxidizing intermediates.101,103–114 Thus, the incorporation of non-redox active Lewis metals in the secondary coordination sphere also provides a route toward cooperative reactivity. Increasing the cationic nature of the Fe intermediates also inhibits the major deactivation pathway, which is formation of a diiron μ-oxo complex.
These results demonstrate that the installation of a proximal cation can provide another “knob” to tune the redox reactivity. In contrast to adjusting the ligand field strength through electronic effects in the primary coordination sphere, modifications to the secondary coordination sphere are effectively used to improve the redox activity at a milder potential.
Results and discussion
Synthesis and characterization
The preparation of the ligands 1M (M = K+ or Ba2+, Scheme 1) has previously been reported.93,102 The corresponding Fe(II) and Fe(III)Cl complexes, 2M and 3M, were synthesized by metallation of the ligand with Fe(OAc)2 in methanol (MeOH) and anhydrous FeCl3 in ethanol (EtOH), respectively. The products were recrystallized by the vapor diffusion of diethyl ether (Et2O) into an acetonitrile (CH3CN) solution of the complex. 2M reacts with air in MeOH or CH3CN to form the diiron μ-oxo species (4M), while the Fe(III)Cl complexes (3M) are air-stable. The 4M complexes were isolated by recrystallization (vapor diffusion of Et2O into concentrated CH3CN solution). The purity and formulation of the complexes were confirmed using elemental analysis and mass spectrometry. The corresponding complexes Fe(salen) and Fe(Ph2salenCl4) (ligands for compound A and C, respectively, in Table 3), which have a similar primary coordination environment but lack proximal cations, were synthesized according to previously published procedures in order to compare their reactivity with 3M, as they have similar redox potentials.102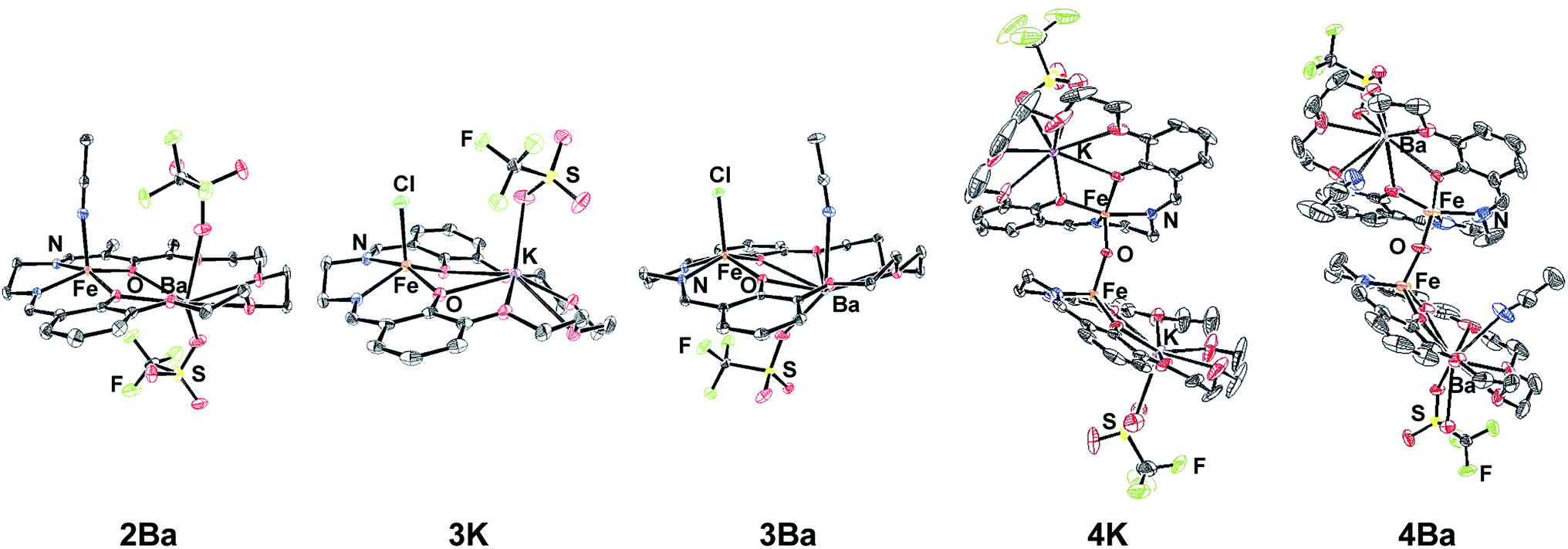 | ||
| Fig. 1 Solid-state structures of 2Ba, 3K, 3Ba, 4K, and 4Ba. Hydrogen atoms and non-coordinating anions are omitted for clarity. 4K contains two outer sphere triflate ions. Ellipsoids are drawn at 50% probability. | ||
| Compound | Fe–M distance (Å) | τ 5 | Fe–Cl distance (Å) | Fe–O distance (Å) | E 1/2 (FeIII/II) (V) |
|---|---|---|---|---|---|
| a The structure of 4K contains K+ that is disordered over 2 positions. | |||||
| 2K | — | — | — | — | −0.29 |
| 2Ba | 3.6592(4) | 0.11 | — | — | −0.09 |
| Fe(salen) | — | — | — | — | −0.73 |
| 3K | 3.6596(6) | 0.36 | 2.218 | — | −0.69 |
| 3Ba | 3.8115(3) | 0.13 | 2.228 | — | −0.53 |
| [Fe(Cl)(salen)] (A)102,115 | — | 0.20 | 2.238 | — | −0.76 (ref. 102) |
| 4K | 3.704(8), 3.751(6) | 0.30 | — | 1.787 | — |
| 4Ba | 3.780 | 0.18 | — | 1.771 | — |
| [Fe2(μ-O)(salen)2]116 | — | 0.27, 0.38 | — | 1.788, 1.785 | — |
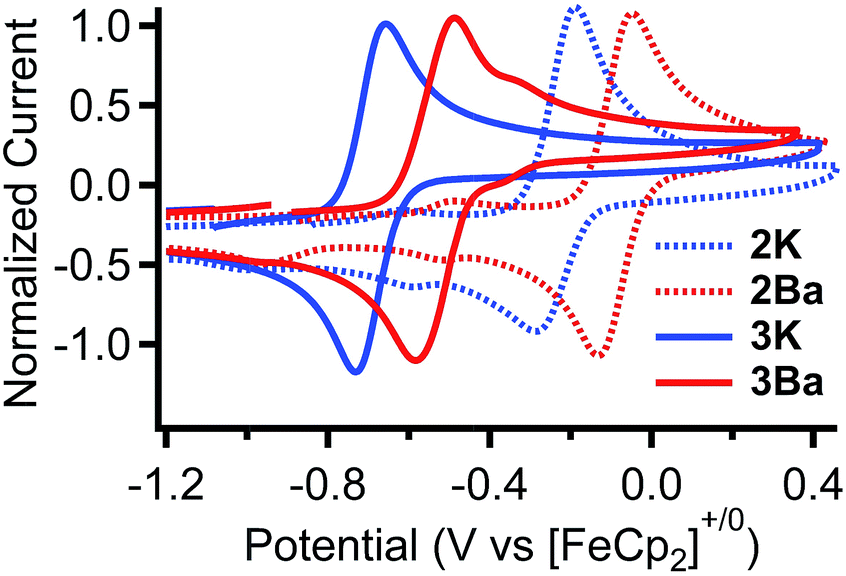 | ||
| Fig. 2 Cyclic voltammograms of the Fe(III/II) redox couple for 2K, 2Ba, 3K, and 3Ba, in 0.2 M nBu4NPF6 in CH3CN with a glassy carbon disc as the working electrode, glassy carbon rod as the counter electrode, and Ag+/Ag in 0.2 M nBu4NPF6 in CH3CN solution as the pseudo reference electrode, and ferrocene as an internal standard. | ||
The standard potentials for the Fe(III/II) couple for 2K and 2Ba are 440 and 640 mV more positive than that for Fe(salen). The standard potentials for 3K and 3Ba are 70 and 230 mV more positive compared to that for Fe(salen)Cl. Moreover, the potential for 3Ba approaches that for FeCl(Ph2salenCl4), the derivative with four electron withdrawing chloride functionalities in the ligand backbone.
Catalytic oxidation activity
The aerobic oxidation activity of 3K and 3Ba with cyclohexene was tested using a similar protocol to prior studies with iron porphyrin and iron salen complexes.102 Oxygen-saturated CH3CN solutions of cyclohexene and 3K or 3Ba were mixed (benzene was added as an internal standard). The reaction mixtures were kept under 1 atm of O2 for 24 hours before being analyzed using 1H NMR spectroscopy to quantify the concentration of the cyclohexene hydroperoxide, as well as the alcohol and ketone products. Prior studies used gas chromatography for the product analysis, so the more reactive hydroperoxide intermediate was unobservable.The overall activity and product distributions are shown in Fig. 3 and Table 2. 3K and 3Ba provide total turnover numbers of 17.4 and 46.6, respectively, for 2-cyclohexen-1-ol and 2-cyclohexenone (Fig. 3 and Tables 2 and 3). The reactivity of Fe(Cl)(salen) and Fe(Cl)(Ph2salenCl4) (compounds A and C in Table 3), which have similar redox potentials to 3K and 3Ba, respectively, was also examined and the compounds showed negligible activity towards aerobic C–H allylic oxidation (entries 1 and 2, Fig. 3 and Table 2). The hydroperoxide intermediate is observed with Fe(Cl)(salen), indicating that the latter is unreactive towards promoting subsequent oxidation reactions, a supposition supported in our mechanistic studies (vide infra).
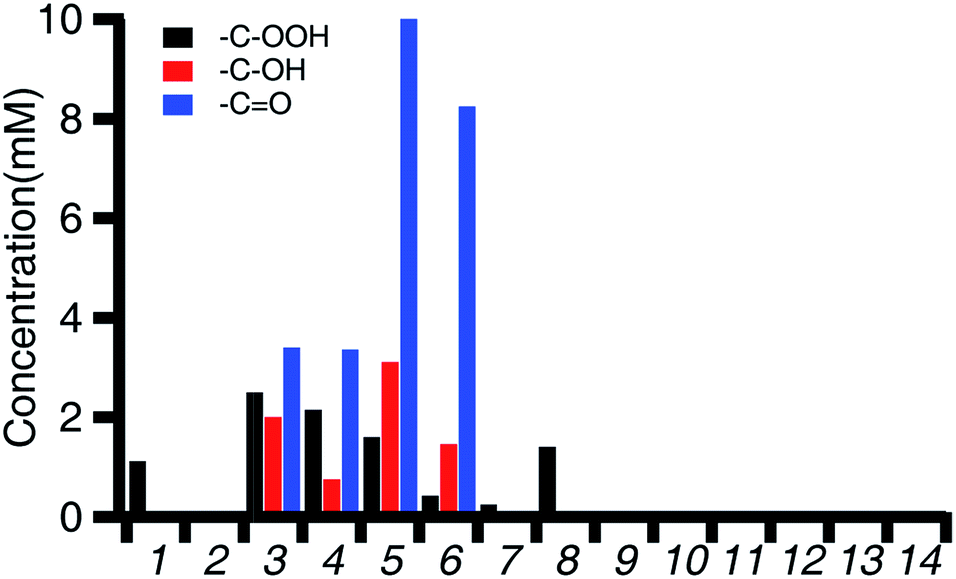 | ||
| Fig. 3 Concentration of the products detected by 1H NMR after 24 hours for the 0.5 M solutions of cyclohexene under 1 atm of O2 and the specified conditions. Conditions for each entry can be found in Table 2. | ||
| Entry | Conditions | Cyclohexenol (mM) | Cyclohexenone (mM) | Total turnover (alcohol/ketone) |
|---|---|---|---|---|
| 1 | Fe(Cl)(salen) (A) | 1.1 | 0 | 0 |
| 2 | Fe(Cl)(Ph2salenCl4) (C) | 0 | 0 | 0 |
| 3 | 3K | 2.0 | 3.4 | 17.4 |
| 4 | 3K + TBACl | 0.8 | 3.4 | 14.9 |
| 5 | 3Ba | 3.1 | 10.0 | 46.4 |
| 6 | 3Ba + TBACl | 1.5 | 8.2 | 35.9 |
| 7 | Fe(Cl)(salen) (A) + Ba(18-crown-6)(OTf)2 | 0.2 | 0 | 0 |
| 8 | Fe(Cl)(salen) (A) + Ba(OTf)2 | 0 | 0 | 0 |
| 9 | FeCl3 + Ba(OTf)2 | 0 | 0 | 0 |
| 10 | 3K + BHT | 0 | 0 | 0 |
| 11 | 3Ba + BHT | 0 | 0 | 0 |
| 12 | 1Ba | 0 | 0 | 0 |
| 13 | No Fe control | 0 | 0 | 0 |
| 14 | TBACl | 0 | 0 | 0 |
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | R |
|
E 1/2 Fe(III/II) vs. [FeCp2]+/0 | TONc |
| a ref. 102. b This work. c Total turnover numbers for 2-cyclohexen-1-ol and 2-cyclohexenone from a 0.5 M solution of cyclohexene after 24 hours under 1 atm O2 with the respective iron compound. | |||||
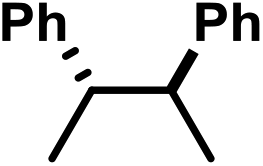
| 1 | A | H |

|
−0.81 | 0 |
| 2 | B | H |
|
−0.75 | 0 |
| 3 | 3K | — | — | −0.71 | 17 |
| 4 | 3Ba | — | — | −0.57 | 46 |
| 5 | C | Cl |
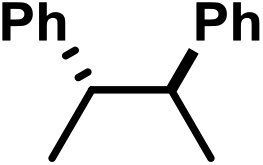
|
−0.56 | 0 |
| 6 | D | Cl |
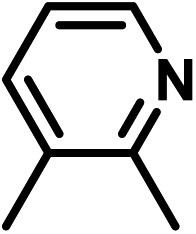
|
−0.44 | 0 |
| 7 | E | NO2 |
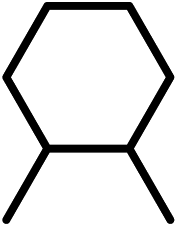
|
−0.32 | 30 |
| 8 | F | NO2 |
|
−0.255 | 165 |

The addition of external non-redox active metal salts has enhanced the oxidation activity for some transition metal catalysts, in some cases forming adducts in situ.101,103–114 For this catalyst, the incorporation of cations into the ligand framework is key for the reactivity; the addition of various Ba2+ salts to Fe(Cl)salen or FeCl3 gave no detectable quantity of product, although hydroperoxide was sometimes observed (entries 7, 8, and 9, Fig. 2 and Table 2). The absence of product under these conditions indicates that there is no intermolecular role for the non-redox active Lewis acidic metals in promoting catalysis.
The addition of excess tetrabutylammonium chloride (TBACl) to 3M had a slight inhibitory effect (entries 4 and 6, Fig. 2 and Table 2). When no iron complexes were present, with or without TBACl, or when only the ligand was present, no catalytic activity was observed. Prior studies have indicated that catalysis occurs through a radical mechanism.102,118–120 We found that addition of the radical scavenger butylated hydroxytoluene (BHT) inhibited all reactivity, supporting a similar radical-based mechanism for 3K and 3Ba.
In the proposed mechanism, the total catalytic activity of the iron macrocycles, represented by the turnover number, is determined by two factors. The first is the reduction of the Fe(III)Cl complex with the organic hydroperoxide (B in Scheme 2), which is believed to be rate limiting since the hydroperoxide is observable in millimolar concentrations during catalysis. The second is deactivation of the catalyst, which occurs though reaction of the Fe(II) intermediate with oxygen (instead of a hydroperoxide) to form the inactive diiron(III) μ-oxo complex (step C in Scheme 2). As this is a redox reaction, it is also expected to exhibit a redox-dependent rate, whereupon the complexes with lower Fe(III/II) redox potentials react more quickly with oxygen. For both the rate-limiting step and the deactivation pathway, the overall reactivity should be more favorable with higher Fe(III/II) standard potentials. These two steps are consistent with the trend observed in previously reported high-spin (S = 5/2) iron complexes, in that only complexes with Fe(III/II) couples above −0.27 V are active (Scheme 1). Incorporation of the proximal cation in 3M results in catalytic activity at significantly less oxidizing potentials.
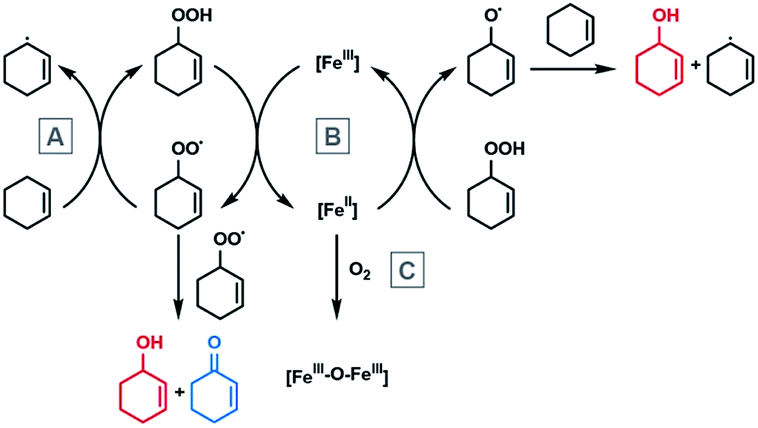 | ||
| Scheme 2 The Haber–Weiss radical-chain mechanism of cyclohexene hydroperoxide decomposition and deactivation by diiron μ-oxo formation. | ||
The reactivity of 3K and 3Ba in the rate-determining step and deactivation pathway was also investigated. For the former, the reactivity of 3K, 3Ba, and Fe(Cl)(Ph2salenCl4) (compound C in Table 3) with tert-butyl hydroperoxide (tert-BuOOH) was monitored using electronic absorption spectroscopy. Tert-BuOOH was used as a substitute for cyclohexene hydroperoxide, since the latter cannot be generated in reproducible concentrations. 3K and 3Ba showed spectroscopic changes upon the addition of tert-BuOOH,113 while no change was observed for Fe(Cl)(Ph2salenCl4) (Fig. S7†). We were unable to measure a rate constant for the reaction of 3M with tert-BuOOH, since the reduced Fe(II) complex (2M) can react with another equivalent of tert-BuOOH to regenerate 3M. However, it is evident that the 3M complexes are both reactive while Fe(Cl)(Ph2salenCl4), which has a similar redox potential to 3Ba, is unreactive. Incorporation of the cation into the ligand framework could also play a role in facilitating intramolecular electron transfer,105,121,122 as Lewis acidic metal cations can bind or otherwise interact with the lone pairs on the hydroperoxide.103
We also examined whether there was a difference in the rate of deactivation through μ-oxo formation by the Fe(II) intermediates. The rate of reaction of 2K and 2Ba with O2 was also measured using electronic absorption spectroscopy. The diiron μ-oxo complexes were independently synthesized to determine their spectroscopic signatures. While the rate of oxidation was difficult to determine because of the complicated kinetic profile (Fig. S8†), the half-lives for the oxidation of 2K and 2Ba are 18 seconds and 37 seconds at 0 °C, respectively. The slower rate of oxidation for 2Ba (and corresponding higher overall activity) is consistent with the more positive Fe(III/II) redox potential.
Conclusion
For Fe salen complexes modified through traditional inductive effects, only derivatives with four nitro groups and Fe(III/II) potentials greater than −0.27 V were sufficiently oxidizing to catalyze the aerobic oxidation of cyclohexene. However, the incorporation of K+ and Ba2+ ions in the secondary coordination sphere results in active catalysts despite having Fe(III/II) redox potentials of −0.71 and −0.57 V, respectively. Mechanistic studies indicate that incorporation of the Lewis acidic cation promotes reactivity in the rate-determining step, oxidation of an organic hydroperoxide, and inhibits deactivation through Fe2-μ-oxo formation.For catalytic reactions in which substrate activation is rate limiting, increasing the catalytic rate through inductive ligand effects often has the undesirable outcome of shifting the redox potential to more extreme values. Overcoming these scaling relationships requires utilizing alternative routes to facilitate reactivity or non-inductive methods to modify redox properties. The incorporation of a non-redox active proximal cation in the secondary coordination sphere was successfully used in this system to achieve catalytic activity at a less oxidizing potential.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. 1554744. We would like to thank Professor A. S. Borovik and V. F. Oswald for helpful discussions, assistance collecting spectroscopic data, and instrumental support.Notes and references
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- H. M. Neu, T. Yang, R. A. Baglia, T. H. Yosca, M. T. Green, M. G. Quesne, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2014, 136, 13845–13852 CrossRef CAS PubMed.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS PubMed.
- L. Chiang, L. E. Allan, J. Alcantara, M. C. Wang, T. Storr and M. P. Shaver, Dalton Trans., 2014, 43, 4295–4304 RSC.
- M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CAS.
- A. J. P. Cardenas, B. Ginovska, N. Kumar, J. Hou, S. Raugei, M. L. Helm, A. M. Appel, R. M. Bullock and M. O’Hagan, Angew. Chem., Int. Ed., 2016, 55, 13509–13513 CrossRef CAS PubMed.
- J. J. Kiernicki, M. Zeller and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 18194–18197 CrossRef CAS PubMed.
- K.-N. T. Tseng, J. W. Kampf and N. K. Szymczak, J. Am. Chem. Soc., 2016, 138, 10378–10381 CrossRef CAS PubMed.
- D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Chem. Soc. Rev., 2013, 42, 8360–8375 RSC.
- A. J. M. Miller, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2008, 130, 11874–11875 CrossRef CAS PubMed.
- R. L. Shook, S. M. Peterson, J. Greaves, C. Moore, A. L. Rheingold and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 5810–5817 CrossRef CAS PubMed.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- S. M. M. Knapp, T. J. Sherbow, R. B. Yelle, L. N. Zakharov, J. J. Juliette and D. R. Tyler, Organometallics, 2013, 32, 824–834 CrossRef CAS.
- M. Yoshida, M. Kondo, S. Torii, K. Sakai and S. Masaoka, Angew. Chem., Int. Ed., 2015, 54, 7981–7984 CrossRef CAS PubMed.
- W. W. Kramer and C. C. L. McCrory, Chem. Sci., 2016, 7, 2506–2515 RSC.
- C. L. Ford, Y. J. Park, E. M. Matson, Z. Gordon and A. R. Fout, Science, 2016, 354, 741–743 CrossRef CAS PubMed.
- H. Sun, Y. Han, H. Lei, M. Chen and R. Cao, Chem. Commun., 2017, 53, 6195–6198 RSC.
- N. B. M. Posada, M. A. Guimarães, D. S. Padilha, J. A. L. C. Resende, R. B. Faria, M. Lanznaster, R. S. Amado and M. Scarpellini, Polyhedron, 2018, 141, 30–36 CrossRef CAS.
- J. Y. Yang and D. G. Nocera, J.Am. Chem. Soc., 2007, 129, 8192–8198 CrossRef CAS PubMed.
- G. M. Jacobsen, J. Y. Yang, B. Twamley, A. D. Wilson, R. M. Bullock, M. Rakowski DuBois and D. L. DuBois, Energy Environ. Sci., 2008, 1, 167–174 CAS.
- J. Y. Yang, R. M. Bullock, W. G. Dougherty, W. S. Kassel, B. Twamley, D. L. DuBois and M. Rakowski DuBois, Dalton Trans., 2010, 39, 3001–3010 RSC.
- J. Y. Yang, R. M. Bullock, M. R. DuBois and D. L. DuBois, MRS Bull., 2011, 36, 39–47 CrossRef CAS.
- S. Samanta, K. Mittra, K. Sengupta, S. Chatterjee and A. Dey, Inorg. Chem., 2013, 52, 1443–1453 CrossRef CAS PubMed.
- B. de Souza, G. L. Kreft, T. Bortolotto, H. Terenzi, A. J. Bortoluzzi, E. E. Castellano, R. A. Peralta, J. B. Domingos and A. Neves, Inorg. Chem., 2013, 52, 3594–3596 CrossRef CAS PubMed.
- X. Zhang, Y. Zhu, X. Zheng, D. L. Phillips and C. Zhao, Inorg. Chem., 2014, 53, 3354–3361 CrossRef CAS PubMed.
- J. T. Bays, N. Priyadarshani, M. S. Jeletic, E. B. Hulley, D. L. Miller, J. C. Linehan and W. J. Shaw, ACS Catal., 2014, 4, 3663–3670 CrossRef CAS.
- H. Yamagishi, S. Nabeya, T. Ikariya and S. Kuwata, Inorg. Chem., 2015, 54, 11584–11586 CrossRef CAS PubMed.
- E. W. Dahl, T. Louis-Goff and N. K. Szymczak, Chem. Commun., 2017, 53, 2287–2289 RSC.
- M. Li, X. Wang, Y. Luo and C. Chen, Angew. Chem., Int. Ed., 2017, 56, 11604–11609 CrossRef CAS PubMed.
- A. Rana, B. Mondal, P. Sen, S. Dey and A. Dey, Inorg. Chem., 2017, 56, 1783–1793 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- T. Zhang, C. Wang, S. Liu, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- D. W. Shaffer, Y. Xie, D. J. Szalda and J. J. Concepcion, J. Am. Chem. Soc., 2017, 139, 15347–15355 CrossRef CAS PubMed.
- J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Chem. Rev., 2014, 114, 4366–4469 CrossRef CAS PubMed.
- F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC.
- P. Hosseinzadeh, N. M. Marshall, K. N. Chacón, Y. Yu, M. J. Nilges, S. Y. New, S. A. Tashkov, N. J. Blackburn and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 262–267 CrossRef CAS PubMed.
- A.-F. Miller, Acc. Chem. Res., 2008, 41, 501–510 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- S. D. Fried, S. Bagchi and S. G. Boxer, Science, 2014, 346, 1510–1514 CrossRef CAS PubMed.
- R. Maji and S. E. Wheeler, J. Am. Chem. Soc., 2017, 139(36), 12441–12449 CrossRef CAS PubMed.
- B. Chattopadhyay, J. E. Dannatt, I. L. Andujar-De Sanctis, K. A. Gore, R. E. Maleczka, D. A. Singleton and M. R. Smith, J. Am. Chem. Soc., 2017, 139, 7864–7871 CrossRef CAS PubMed.
- J. M. Um, D. A. DiRocco, E. L. Noey, T. Rovis and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 11249–11254 CrossRef CAS PubMed.
- H. Yang and M. W. Wong, J. Am. Chem. Soc., 2013, 135, 5808–5818 CrossRef CAS PubMed.
- E. Lyngvi, J. W. Bode and F. Schoenebeck, Chem. Sci., 2012, 3, 2346–2350 RSC.
- D. A. DiRocco, E. L. Noey, K. N. Houk and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 2391–2394 CrossRef CAS PubMed.
- T. J. Seguin and S. E. Wheeler, ACS Catal., 2016, 6, 2681–2688 CrossRef CAS.
- Q. N. N. Nguyen, M. W. Lodewyk, S. Bezer, M. R. Gagné, M. L. Waters and D. J. Tantillo, ACS Catal., 2015, 5, 1617–1622 CrossRef CAS.
- K. Lee, D. L. Silverio, S. Torker, D. W. Robbins, F. Haeffner, F. W. van der Mei and A. H. Hoveyda, Nat. Chem., 2016, 8, 768–777 CrossRef CAS PubMed.
- T. J. Seguin and S. E. Wheeler, Angew. Chem., Int. Ed., 2016, 55, 15889–15893 CrossRef CAS PubMed.
- A. C. Doney, B. J. Rooks, T. Lu and S. E. Wheeler, ACS Catal., 2016, 6, 7948–7955 CrossRef CAS.
- C. R. Kennedy, J. A. Guidera and E. N. Jacobsen, ACS Cent. Sci., 2016, 2, 416–423 CrossRef CAS PubMed.
- G. Xiao, G. A. Cintron-Rosado, D. A. Glazier, B.-m. Xi, C. Liu, P. Liu and W. Tang, J. Am. Chem. Soc., 2017, 139, 4346–4349 CrossRef CAS PubMed.
- M. C. Holland, S. Paul, W. B. Schweizer, K. Bergander, C. Mück-Lichtenfeld, S. Lakhdar, H. Mayr and R. Gilmour, Angew. Chem., Int. Ed., 2013, 52, 7967–7971 CrossRef CAS PubMed.
- A. L. Chan, J. Estrada, C. E. Kefalidis and V. Lavallo, Organometallics, 2016, 35, 3257–3260 CrossRef CAS.
- J. B. Smith and A. J. M. Miller, Organometallics, 2015, 34, 4669–4677 CrossRef CAS.
- C. C. Lu and J. C. Peters, J. Am. Chem. Soc., 2002, 124, 5272–5273 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- H. Shao, S. K. Muduli, P. D. Tran and H. S. Soo, Chem. Commun., 2016, 52, 2948–2951 RSC.
- S. Sung, D. Kumar, M. Gil-Sepulcre and M. Nippe, J. Am. Chem. Soc., 2017, 139, 13993–13996 CrossRef CAS PubMed.
- F. C. J. M. van Veggel, W. Verboom and D. N. Reinhoudt, Chem. Rev., 1994, 94, 279–299 CrossRef CAS.
- C. J. Baylies, T. Riis-Johannessen, L. P. Harding, J. C. Jeffery, R. Moon, C. R. Rice and M. Whitehead, Angew. Chem., Int. Ed., 2005, 44, 6909–6912 CrossRef CAS PubMed.
- A. Gund, B. K. Keppler and B. Nuber, Inorg. Chem., 1995, 34, 2788–2790 CrossRef CAS.
- S. K. Dutta, D. Gan and M. W. Perkovic, Eur. J. Inorg. Chem., 2003, 2003, 2812–2819 CrossRef.
- N. D. Lowe and C. D. Garner, J. Chem. Soc., Dalton Trans., 1993, 2197–2207, 10.1039/DT9930002197.
- T. Lazarides, A. Barbieri, C. Sabatini, F. Barigelletti, H. Adams and M. D. Ward, Inorg. Chim. Acta, 2007, 360, 814–824 CrossRef CAS.
- L. J. Charbonnière, R. F. Ziessel, C. A. Sams and A. Harriman, Inorg. Chem., 2003, 42, 3466–3474 CrossRef PubMed.
- S. Belanger, M. Gilbertson, D. I. Yoon, C. L. Stern, X. Dang and J. T. Hupp, J. Chem. Soc., Dalton Trans., 1999, 3407–3411, 10.1039/A906106A.
- L. H. Uppadine, J. E. Redman, S. W. Dent, M. G. B. Drew and P. D. Beer, Inorg. Chem., 2001, 40, 2860–2869 CrossRef CAS PubMed.
- M. Chiba, H.-B. Kim and N. Kitamura, Anal. Sci., 2002, 18, 461–466 CrossRef CAS PubMed.
- V. Wing-Wah Yam, C.-K. Li and C.-L. Chan, Angew. Chem., Int. Ed., 1998, 37, 2857–2859 CrossRef.
- V. Wing-Wah Yam, Y.-L. Pui, W.-P. Li, K. Kam-Wing Lo and K.-K. Cheung, J. Chem. Soc., Dalton Trans., 1998, 3615–3622, 10.1039/A806243I.
- C.-K. Li, X.-X. Lu, K. M.-C. Wong, C.-L. Chan, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2004, 43, 7421–7430 CrossRef CAS PubMed.
- X.-X. Lu, C.-K. Li, E. C.-C. Cheng, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2004, 43, 2225–2227 CrossRef CAS PubMed.
- M. Delgado, J. M. Ziegler, T. Seda, L. N. Zakharov and J. D. Gilbertson, Inorg. Chem., 2016, 55, 555–557 CrossRef CAS PubMed.
- M. R. Kita and A. J. M. Miller, J. Am. Chem. Soc., 2014, 136, 14519–14529 CrossRef CAS PubMed.
- Z. Cai, D. Xiao and L. H. Do, J. Am. Chem. Soc., 2015, 137, 15501–15510 CrossRef CAS PubMed.
- M. R. Kita and A. J. M. Miller, Angew. Chem., Int. Ed., 2017, 56, 5498–5502 CrossRef CAS PubMed.
- P. D. Beer, P. K. Hopkins and J. D. McKinney, Chem. Commun., 1999, 1253–1254, 10.1039/A903440D.
- S. Shrestha, C. Gimbert-Suriñach, M. Bhadbhade and S. B. Colbran, Eur. J. Inorg. Chem., 2011, 2011, 4331–4337 CrossRef CAS.
- Z. Halime, M. Lachkar, N. Matsouki, G. Charalambidis, M. di Vaira, A. G. Coutsolelos and B. Boitrel, Tetrahedron, 2006, 62, 3056–3064 CrossRef CAS.
- Y.-H. Kim and J.-I. Hong, Chem. Commun., 2002, 512–513, 10.1039/B109596J.
- J. Mårtensson, K. Sandros and O. Wennerström, J. Phys. Org. Chem., 1994, 7, 534–544 CrossRef.
- Y. Shen and B. P. Sullivan, Inorg. Chem., 1995, 34, 6235–6236 CrossRef CAS.
- Á. Gordillo, E. de Jesús and C. López-Mardomingo, Org. Lett., 2006, 8, 3517–3520 CrossRef PubMed.
- J. Fresneda, E. de Jesús, P. Gómez-Sal and C. López Mardomingo, Eur. J. Inorg. Chem., 2005, 2005, 1468–1476 CrossRef.
- M. Sawamura, Y. Nakayama, W.-M. Tang and Y. Ito, J. Org. Chem., 1996, 61, 9090–9096 CrossRef CAS.
- C. D. Hall, G. J. Kirkovits and A. C. Hall, Chem. Commun., 1999, 1897–1898, 10.1039/A904069B.
- S. Castro-Juiz, A. Fernández, M. López-Torres, D. Vázquez-García, A. J. Suárez, J. M. Vila and J. J. Fernández, Organometallics, 2009, 28, 6657–6665 CrossRef CAS.
- C. Nataro, H. M. Baseski, C. M. Thomas, B. J. Wiza and K. M. Rourke, Polyhedron, 2001, 20, 1023–1028 CrossRef CAS.
- A. H. Reath, J. W. Ziller, C. Tsay, A. J. Ryan and J. Y. Yang, Inorg. Chem., 2017, 56, 3713–3718 CrossRef CAS PubMed.
- R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12486–12489 CrossRef CAS PubMed.
- E. Y. Tsui and T. Agapie, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10084–10088 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef CAS PubMed.
- P.-H. Lin, M. K. Takase and T. Agapie, Inorg. Chem., 2015, 54, 59–64 CrossRef CAS PubMed.
- D. E. Herbert, D. Lionetti, J. Rittle and T. Agapie, J. Am. Chem. Soc., 2013, 135, 19075–19078 CrossRef CAS PubMed.
- C. P. Horwitz and Y. Ciringh, Inorg. Chim. Acta, 1994, 225, 191–200 CrossRef CAS.
- H. M. Neu, J. Jung, R. A. Baglia, M. A. Siegler, K. Ohkubo, S. Fukuzumi and D. P. Goldberg, J. Am. Chem. Soc., 2015, 137, 4614–4617 CrossRef CAS PubMed.
- S. Hong, Y.-M. Lee, M. Sankaralingam, A. K. Vardhaman, Y. J. Park, K.-B. Cho, T. Ogura, R. Sarangi, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 8523–8532 CrossRef CAS PubMed.
- A. Böttcher, M. W. Grinstaff, J. A. Labinger and H. B. Gray, J. Mol. Catal. A: Chem., 1996, 113, 191–200 CrossRef.
- S. Fukuzumi and K. Ohkubo, Chem.–Eur. J., 2000, 6, 4532–4535 CrossRef CAS.
- S.-M. Yiu, W.-L. Man and T.-C. Lau, J. Am. Chem. Soc., 2008, 130, 10821–10827 CrossRef CAS PubMed.
- Y. Morimoto, H. Kotani, J. Park, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 403–405 CrossRef CAS PubMed.
- C. G. Miller, S. W. Gordon-Wylie, C. P. Horwitz, S. A. Strazisar, D. K. Peraino, G. R. Clark, S. T. Weintraub and T. J. Collins, J. Am. Chem. Soc., 1998, 120, 11540–11541 CrossRef CAS.
- H. Guo, Z. Chen, F. Mei, D. Zhu, H. Xiong and G. Yin, Chem.–Asian J., 2013, 8, 888–891 CrossRef CAS PubMed.
- L. Dong, Y. Wang, Y. Lv, Z. Chen, F. Mei, H. Xiong and G. Yin, Inorg. Chem., 2013, 52, 5418–5427 CrossRef CAS PubMed.
- Z. Zhang, K. L. Coats, Z. Chen, T. J. Hubin and G. Yin, Inorg. Chem., 2014, 53, 11937–11947 CrossRef CAS PubMed.
- S. Qin, L. Dong, Z. Chen, S. Zhang and G. Yin, Dalton Trans., 2015, 44, 17508–17515 RSC.
- S. Zhang, Z. Chen, S. Qin, C. Lou, A. M. Senan, R.-Z. Liao and G. Yin, Org. Biomol. Chem., 2016, 14, 4146–4157 CAS.
- C. Choe, L. Yang, Z. Lv, W. Mo, Z. Chen, G. Li and G. Yin, Dalton Trans., 2015, 44, 9182–9192 RSC.
- J. Park, Y. Morimoto, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 5236–5239 CrossRef CAS PubMed.
- Y. J. Park, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 9258–9261 CrossRef CAS PubMed.
- M. Gerloch, J. Lewis, F. E. Mabbs and A. Richards, Nature, 1966, 212, 809–810 CrossRef CAS.
- M. Gerloch and F. E. Mabbs, J. Chem. Soc. A, 1967, 1900–1908, 10.1039/J19670001900.
- A. T. M. P. Coggon, F. E. Mabbs and V. N. McLachlan, J. Chem. Soc. A, 1971, 1014–1019, 10.1039/J19710001014.
- A. Böttcher, E. R. Birnbaum, M. W. Day, H. B. Gray, M. W. Grinstaff and J. A. Labinger, J. Mol. Catal. A: Chem., 1997, 117, 229–242 CrossRef.
- M. Grinstaff, M. Hill, J. Labinger and H. Gray, Science, 1994, 264, 1311–1313 CAS.
- J. A. Labinger, Catal. Lett., 1994, 26, 95–99 CrossRef CAS.
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934 CrossRef CAS PubMed.
- K. D. Karlin, Nat. Chem., 2010, 2, 711 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental conditions and synthesis, solid state structure images, crystal data and refinement, UV-visible spectra, EPR spectra, and oxidation products under rigorously dry conditions. CCDC 1580343, 1580342, 1580341, 1580340, and 1580339. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04486k |
| ‡ Present address: Department of Chemistry, Mahidol University, Bangkok, 10400 Thailand. |
| This journal is © The Royal Society of Chemistry 2018 |