
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-dependent allosteric activation and inhibition on the same molecular scaffold: the copper sensor CopY from Streptococcus pneumoniae†
Hendrik
Glauninger
a,
Yifan
Zhang
ab,
Khadine A.
Higgins
ac,
Alexander D.
Jacobs
a,
Julia E.
Martin
a,
Yue
Fu
ab,
H. Jerome
Coyne, 3rd
a,
Kevin E.
Bruce
d,
Michael J.
Maroney
e,
David E.
Clemmer
a,
Daiana A.
Capdevila
*a and
David P.
Giedroc
 *ab
*ab
aDepartment of Chemistry, Indiana University, Bloomington, IN 47405-7102, USA. E-mail: giedroc@indiana.edu; dacapdev@iu.edu; Tel: +1-812-856-3178 Tel: +1-812-856-6398
bDepartment of Molecular and Cellular Biochemistry, Indiana University, Bloomington, IN 47405, USA
cDepartment of Chemistry, Salve Regina University, Newport, RI 02840, USA
dDepartment of Biology, Indiana University, Bloomington, IN 47405, USA
eDepartment of Chemistry, University of Massachusetts, Amherst, MA 01003, USA
First published on 9th November 2017
Abstract
Resistance to copper (Cu) toxicity in the respiratory pathogen Streptococcus pneumoniae is regulated by the Cu-specific metallosensor CopY. CopY is structurally related to the antibiotic-resistance regulatory proteins MecI and BlaI from Staphylococcus aureus, but is otherwise poorly characterized. Here we employ a multi-pronged experimental strategy to define the Spn CopY coordination chemistry and the unique mechanism of allosteric activation by Zn(II) and allosteric inhibition by Cu(I) of cop promoter DNA binding. We show that Zn(II) is coordinated by a subunit-bridging 3S 1H2O complex formed by the same residues that coordinate Cu(I), as determined by X-ray absorption spectroscopy and ratiometric pulsed alkylation-mass spectrometry (rPA-MS). Apo- and Zn-bound CopY are homodimers by small angle X-ray scattering (SAXS); however, Zn stabilizes the dimer, narrows the conformational ensemble of the apo-state as revealed by ion mobility-mass spectroscopy (IM-MS), and activates DNA binding in vitro and in cells. In contrast, Cu(I) employs the same Cys pair to form a subunit-bridging, kinetically stable, multi-metallic Cu·S cluster (KCu ≈ 1016 M−1) that induces oligomerization beyond the dimer as revealed by SAXS, rPA-MS and NMR spectroscopy, leading to inhibition of DNA binding. These studies suggest that CopY employs conformational selection to drive Zn-activation of DNA binding, and a novel Cu(I)-mediated assembly mechanism that dissociates CopY from the DNA via ligand exchange-catalyzed metal substitution, leading to expression of Cu resistance genes. Mechanistic parallels to antibiotic resistance repressors MecI and BlaI are discussed.
Introduction
Transition metals are harnessed by enzymes as critical components of both their catalytic function and structural stability. It is estimated that approximately a quarter of all proteins require metals as essential cofactors.1 The same reactivity responsible for their enzymatic utility also serves to make transition metals potentially toxic, and as a result, all cells encode machinery that regulates the bioavailability of transition metals in cells via both uptake and efflux mechanisms.2 Metalloregulation of the transcription of genes encoding metal uptake and export transporters is particularly important for colonization and adaptation of pathogens in the vertebrate host. This adaptation features microbial defense mechanisms that either limit the availability of required transition metals in a process termed nutritional immunity,3 or induce metal intoxication,4 depending on the microenvironmental niche.5 In the case of Cu, there is emerging evidence to suggest that Cu is actively used to kill pathogenic invaders.6–9 Prokaryotic metalloregulatory proteins control the expression of genes responsible for the maintenance of appropriate intracellular metal levels via allosteric regulation of the DNA operator–promoter binding by metals, and thus are metal-regulated transcriptional “switches”.10–12Streptococcus pneumoniae is a commensal Gram-positive respiratory pathogen that colonizes the upper respiratory tract in humans and is a primary cause globally of bacterial pneumonia,13 with ≈30% of severe S. pneumoniae infections possessing resistance to at least one clinically important antibiotic. In the serotype 2 Streptococcus pneumoniae D39 strain, CopY is a Cu(I)-sensing metalloregulatory repressor that regulates the expression of the cop operon, encoding CopY, the membrane-anchored copper chaperone CupA,14 and the Cu-effluxer CopA.13 The cop operon contributes to virulence of S. pneumoniae in a lung infection mouse model where the ΔcupA and ΔcopA strains are attenuated for virulence; furthermore, the operon is induced in the nasopharynx and the lungs and as such, a ΔcopA strain exhibits poor colonization of the nasopharynx.13 Transcriptional analysis by real time quantitative PCR (qRT-PCR)13 and a transcriptomic analysis of a ΔcopY deletion strain14 collectively reveal that the cop operon is autoregulated by CopY. However, the details of regulatory mechanism used by Spn CopY in the pneumococcus remain unclear.
CopY is representative of a small family of copper-specific metalloregulatory proteins, initially characterized in Enterococcus hirae.15,16 CopY is a Cu(I)-sensing repressor that binds to one or more cop box sequences in the promoter and transcriptionally regulates the expression of downstream genes in response to cellular Cu(I) toxicity. The DNA-binding domain is a canonical winged-helical motif, as determined by the solution structure of the N-terminal domain of Lactococcus lactis CopR.17 CopY is proposed to be member of methicillin resistance/β-lactamase (MecI/BlaI) family repressors, based both on the sequence similarity of the N-terminal DNA binding domain and similar 5′-TACAxxTGTA cop box palindromic operator sequences (Fig. 1A).18 The C-terminal regulatory domain functions as the dimerization domain,19 is of unknown structure, and is thought to coordinate both Zn(II) and Cu(I).
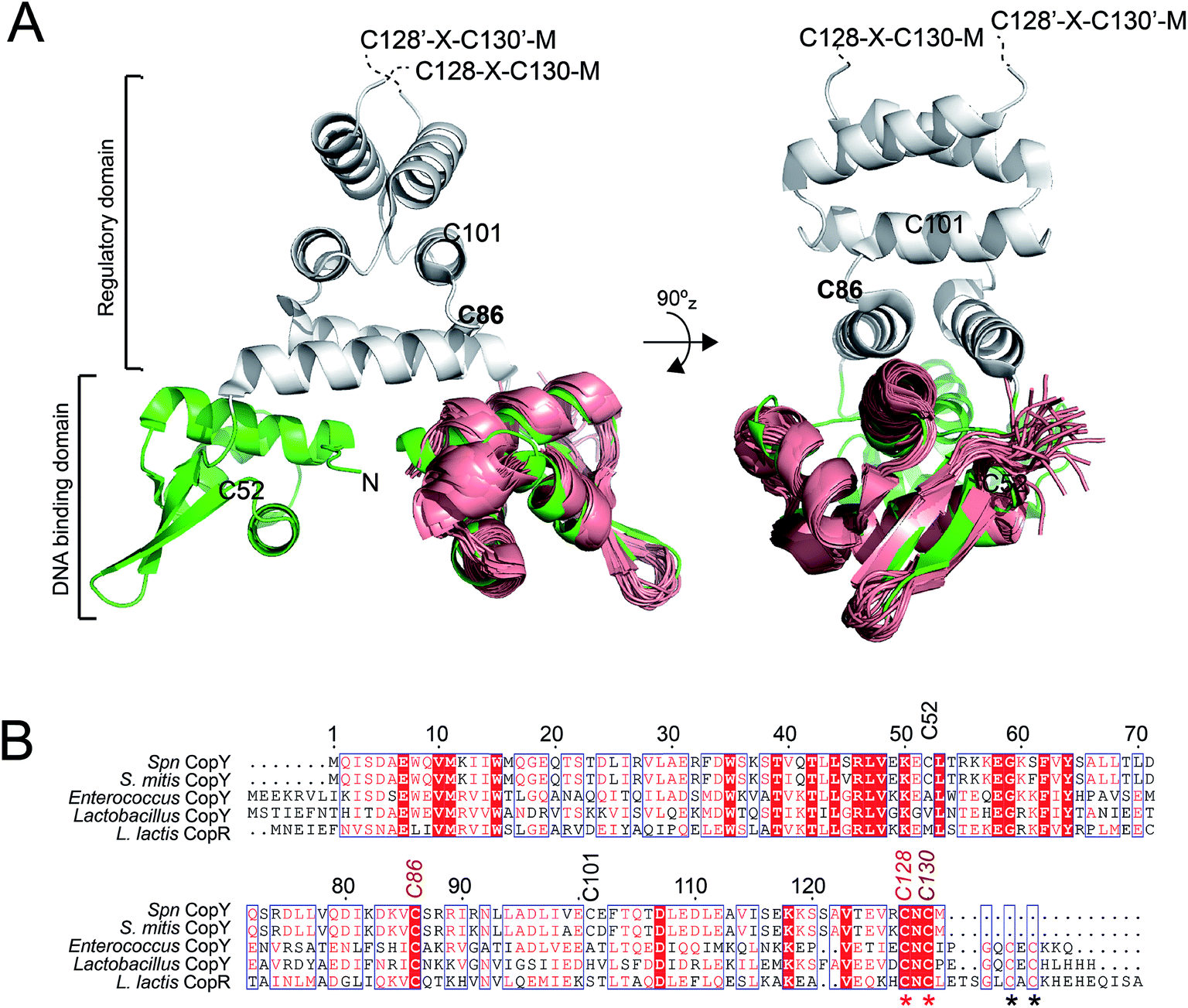 | ||
| Fig. 1 Dimeric CopY structural model. (A) Ribbon representation of the superposition of the solution structure bundle of the L. lactis CopR DNA binding domain (salmon)17 on the crystallographic structure of S. aureus BlaI (green, DNA-binding domain; grey, regulatory domain).24 The approximate positions of the five Spn CopY Cys are indicated, with the C128–C130 pair just beyond the solved structure of the BlaI determined by a multiple sequence alignment (not shown). (B) Multiple sequence alignment of CopY proteins from different bacteria. The conserved Cys residue proposed to be involved in copper and zinc binding are highlighted with asterisks. Note that S. pneumoniae CopY only contains two of the four Cys residues near the C-terminus. The C-terminus of the L. lactis CopR DNA binding domain model (see panel A) is E74 (T69 in Spn CopY), with the last residue of the β2 strand R70 (S65 in Spn CopY). K119 in the Spn CopY sequence defines the C-terminus of the BlaI model24 shown in panel A. | ||
CopY regulates copper efflux and the oxidative stress response in a small number of bacteria from closely related Firmicutes, derived from the Lactococcus, Streptococcus and Enterococcus spp. (Fig. 1B).20 CopYs conserve one or two C-terminal CxC (C, Cys; x, any amino acid) motifs that are projected to coordinate Cu(I) or Zn(II). Zn(II) is reported to function as an allosteric activator of DNA binding required for full repression of the cop operon in the absence of Cu(I) stress. As Cu(I) levels rise in the cell, two Cu(I) have been shown to displace each Zn(II) to form a luminescent Cu2–S4 cluster that impairs DNA binding.28 This mechanism of allosteric Cu(I)-induced inhibition and Zn(II)-induced activation of DNA binding is unique among metal efflux regulators, in that the binding of an alternative or non-cognate metal generally has a neutral or similar impact on DNA binding affinities relative to the cognate metal.21 Although early studies of CopY have qualitatively characterized the functional outcomes of binding of Zn(II) vs. Cu(I), the extent to which these coordination complexes differ from one another is unknown. This information is needed to elucidate the underlying mechanisms of metal-dependent allosteric activation vs. inhibition of DNA binding by CopY and how this impacts cop operon expression.
In this work, we present a comprehensive multi-pronged analysis of the different metal-bound states of CopY, particularly in terms of coordination chemistry and metal-induced global structural and dynamical changes. We uncover novel insights into the allosteric activation and inhibition of DNA binding of S. pneumoniae CopY by Zn(II) and Cu(I), respectively, and place these studies in the context of CopY-like repressors that regulate methicillin and β-lactam antibiotic resistance in other human microbial pathogens.22–26
Results and discussion
Coordination chemistry of Spn CopY
The addition of Cu(I) to a solution of apo-CopY with monitoring of Cu–S charge transfer absorption (240 nm (ref. 14 and 29)) reveals saturation at a stoichiometry of 1 per protomer or 2 per dimer (Fig. S1†). The Cu(I) complex is not strongly luminescent at room temperature in aqueous buffers (data not shown), which contrast the findings for 4-Cys containing CopYs and suggests that the cluster is probably solvent exposed and not found within a compact protein environment.30 In order to determine the equilibrium constant for Cu binding (KCu), we next anaerobically titrated apo-C101A CopY into a mixture of Cu(I) and BCS (β2,Cu = 1019.8 M−2)31 and monitored disassembly of the preformed Cu(I)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BCS2 complex which absorbs at 483 nm (Fig. 2A). The solid lines represent a simultaneous fit to three different experiments obtained at different Cu(I) and BCS concentrations to two identical Cu(I) sites model on the dimer. These data give logKCu = 16.6 (±0.1). This value is ≈1–2 orders of magnitude weaker than other Cu(I) binding repressors that have been previously characterized, including CsoR and CueR.8,32,33 This result makes the prediction that the concentration of free cytoplasmic Cu(I) in S. pneumoniae might be higher than in other pathogens that harbor a Cu(I) sensor from the CsoR or MerR families (see Conclusions).12,21 Interestingly, the Spn CopY Cu(I) binding affinity is comparable to the high affinity site on the N-terminal metal-binding domain of Spn CopA14 but larger than KCu for Cu chaperone CupA that harbors the functionally essential low affinity Cu(I) binding site (logKCu = 14.8).14 This is consistent with a model proposed for other Cu chaperone-Cu-sensing repressor pairs in which the repressor is capable of stripping Cu(I) from the Cu chaperone suggesting that the copper chaperones, transporters and transcriptional repressors have coevolved to maintain this hierarchy in metal affinities.28,34
:BCS2 complex which absorbs at 483 nm (Fig. 2A). The solid lines represent a simultaneous fit to three different experiments obtained at different Cu(I) and BCS concentrations to two identical Cu(I) sites model on the dimer. These data give logKCu = 16.6 (±0.1). This value is ≈1–2 orders of magnitude weaker than other Cu(I) binding repressors that have been previously characterized, including CsoR and CueR.8,32,33 This result makes the prediction that the concentration of free cytoplasmic Cu(I) in S. pneumoniae might be higher than in other pathogens that harbor a Cu(I) sensor from the CsoR or MerR families (see Conclusions).12,21 Interestingly, the Spn CopY Cu(I) binding affinity is comparable to the high affinity site on the N-terminal metal-binding domain of Spn CopA14 but larger than KCu for Cu chaperone CupA that harbors the functionally essential low affinity Cu(I) binding site (logKCu = 14.8).14 This is consistent with a model proposed for other Cu chaperone-Cu-sensing repressor pairs in which the repressor is capable of stripping Cu(I) from the Cu chaperone suggesting that the copper chaperones, transporters and transcriptional repressors have coevolved to maintain this hierarchy in metal affinities.28,34
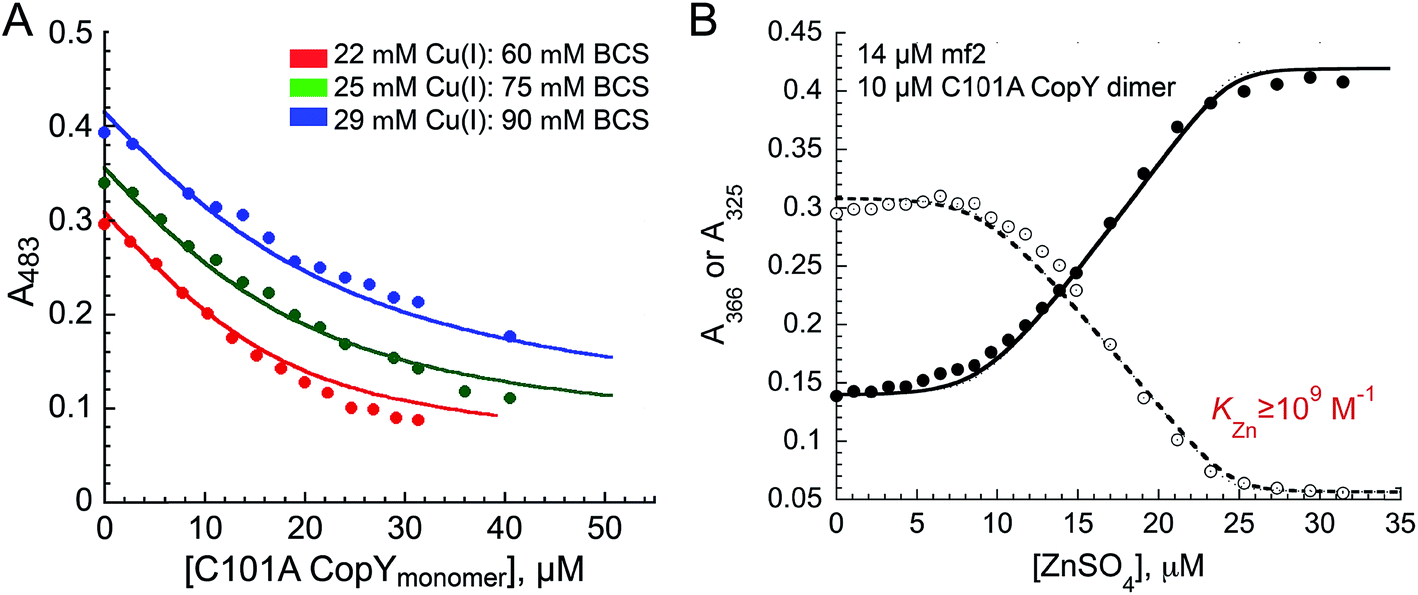 | ||
| Fig. 2 Metal binding affinities of C101A CopY. (A) Representative binding curves obtained from an anaerobic Cu(I) chelator competition to measure the Cu(I) binding affinity of CopY. Apo CopY (monomer units) was titrated into a mixture of Cu(I) and BCS at pH 7.0 in three independent experiments. Closed symbols, A483 values for the Cu(I)–(BCS)2 complex. The continuous line represents the results of a global analysis of all experiments to a simple 1:1 binding model assuming Cu binding sites in each protomer of the homodimer are identical and independent, such that KCu1 = KCu2 = KCu. Red, 22 μM CuCl and 60 μM BCS; green, 25 μM CuCl and 75 μM BCS; blue: 29 μM CuCl and 90 μM BCS, with logKCu = 16.6 (±0.1). (B) Representative Zn(II) binding curve obtained from a chelator competition assay. ZnSO4 was titrated into a solution of apo-C101A CopY (12.0 μM dimer) and mag-fura-2 (mf2) (16 μM) at pH 7.0. Open symbols, A366; closed symbols, A325 for the Zn(II)-mf2 complex. The continuous line represents the results of a global fit to 1:1 (1 Zn:1 dimer) binding model. Only a lower limit of KZn could be obtained from this experiment, KZn ≥ 109 M−1, since the binding is essentially stoichiometric. | ||
We next titrated Zn(II) into a solution of apo-C101A CopY and the modest affinity chelator mag-fura-2 (mf2; KZn = 5.0 × 107 M−1) (see Fig. 2B for a representative experiment), since previous studies on other CopY repressors reveal that active DNA-operator binding form is Zn-CopY.16,28,35 These experiments show that the stoichiometry for Zn(II) binding is 1 Zn per dimer or 0.5 Zn per protomer, with only a lower limit of KZn ≥ 109 M−1 obtained from these experiments. These results reveal that Zn and Cu are likely coordinated by ligands derived from both subunits, contrary to what has been previously proposed for the 4 Cys-containing CopYs35 and further suggest that two Cys per protomer are necessary and sufficient to mediate CopY-dependent biological regulation by Cu or Zn.
X-ray absorption spectroscopy
These metal titrations define the stoichiometry and affinities of Cu(I) and Zn(II) binding to apo-CopY; however, the metal coordination and ligand identity remain unknown. Thus, we next investigated the coordination structure of the Cu2 (1:1 protomer; 2 per dimer) and Zn1 (0.5:1; 1 per dimer) complexes by X-ray absorption spectroscopy (XAS) on the Cu (Fig. 3A) and Zn (Fig. 3B) edges. For the Cu(I) complex, the XANES spectrum is fully compatible with a trigonally coordinated Cu previously reported for 4-Cys containing CopYs.36 The 1s → 4p transition has a normalized absorption amplitude of 0.51 at 8982.8 eV for Cu2 CopY–Br and 0.52 at 8984.1 for Cu2 CopY–Cl. The EXAFS spectrum is well-described by three sulfur scatterers at 2.27 Å and a single Cu–Cu interaction at ≈2.7 Å (Table 1), in general agreement with previous work on E. hirae CopY.28 These spectra are compatible with a Cu2–S4 binuclear cluster model shown in which the C-terminal Cys pair (C128, C130 on both protomers) donates the four sulfur ligands to the complex. Despite the fact that single Cu–Cu interaction can also be modeled as a Cu–Br interaction in samples acquired in NaBr-containing buffers (Fig. S2; Table S2†), the Cu–Br bond vector at 2.65 Å (Table S2†) is far too long and we see the same spectrum in the presence of NaCl or NaBr. Thus, we favor the cluster model shown. We note that these spectra cannot be readily distinguished from higher nuclearity clusters of Cu and S, e.g., Cu4·S6 “adamantane-like” structures common in cysteine sulfur-rich peptides that lack stable secondary structure.37,38 Since Spn CopY harbors only 2 Cys per protomer, the formation of higher nuclearity clusters would induce oligomerization beyond the dimer, and this would be functionally relevant since only the dimer is expected to bind DNA (vide infra).
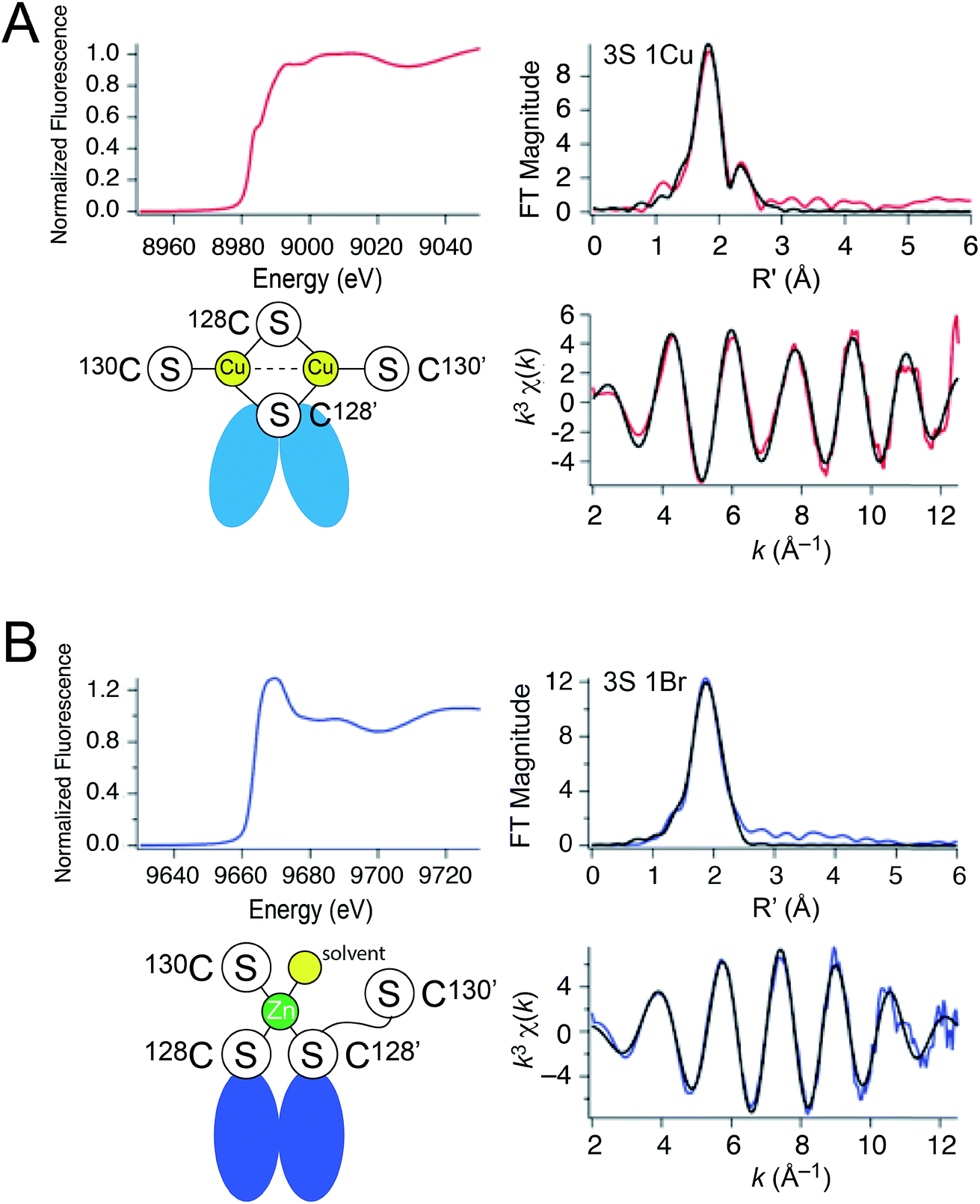 | ||
| Fig. 3 X-ray absorption spectroscopy of (A) Cu2 CopY in NaCl-containing buffer (Cu2 CopY–Cl) and (B) Zn1 CopY acquired in NaBr-containing buffer (Zn1 CopY–Br) C101A CopY. In each panel the X-ray absorption near-edge spectrum (XANES) is shown in the upper left, the extended X-ray absorption fine structure (EXAFS) and k-space spectra shown in upper right, and lower right, respectively, with cartoon models of each coordination structure consistent with the spectroscopy (red continuous lines in the EXAFS and k-space spectra, respectively; black, best fits to the data) shown in the lower left. The Cu(I) XAS spectra are consistent with the same chemical environment around each of the two bound Cu(I) ions containing 3 Cu–S bonds (d = 2.27 Å) and one Cu–Cu scatterer (d = 2.70 Å), features consistent with the binuclear cluster model shown. The Zn(II) XAS is consistent with a subunit-bridging cysteine-rich site conforming to a 3S 1H2O tetrahedral complex containing 3 Zn–S bonds (d = 2.29 Å). See Table 1 for a summary of the best fit parameters and Tables S1–S3† for fits for all possible coordination models. | ||
| Sample | CN | Shell | r (Å) | σ 2 (×10−3 Å−2) | ΔEo (eV) | % R |
|---|---|---|---|---|---|---|
| a Spectra and fits for the first Cu(I) sample entry and the Zn(II) entry are shown in Fig. 3 and S2. | ||||||
| Cu(I), NaBr | 3 | 3 S | 2.269(4) | 5.0(3) | −6.8(7) | 1.44 |
| 1 Cu | 2.703(5) | 3.9(5) | ||||
| Cu(I), NaCl | 3 | 3 S | 2.265(5) | 5.1(3) | −5(1) | 2.82 |
| 1 Cu | 2.71(1) | 7(1) | ||||
| Zn(II), NaBr | 4 | 3 S | 2.29(1) | 3.4(5) | −12(2) | 0.77 |
| 1 Br | 2.48(1) | 4(1) | ||||
The Zn1 CopY complex, in contrast, shows a single broad maximum in the post-edge XANES region with normalized intensity of 1.3, consistent with a tetrahedral complex composed of soft ligand donor atoms (Fig. 3B).39 The EXAFS analysis is characterized by one or more strong scatterers with no significant outer shell contributions, indicating the absence of ligands such as His. We acquired these spectra in the presence of NaBr rather than NaCl in an effort to probe the Zn(II) site for the presence of a ligand (Br− or H2O) from the solvent, since Cl− is not readily distinguished from S. These data acquired in NaBr-containing buffers are best described by a tetrahedral 3S 1Br complex (Table S3†), in which the conserved C128 and C130 from each of the two subunits are the anticipated thiolate ligands, with one coordination site occupied by Br−. This result suggests a solvent-accessible or open coordination site on the Zn(II). These data provide the first evidence that a change in the coordination number and geometry distinguishes CopY allosteric activation from allosteric inhibition.
Ratiometric pulsed alkylation mass spectrometry (rPA-MS) of CopY metallostates
The XAS data suggest that one of the two C-terminal Cys (C128 or C130) from one protomer within the dimer is not coordinated in the Zn(II)-bound state resulting in an open coordination site on the metal (Fig. 3B). In order to confirm this and identify which Cys is not coordinated, we measured the differential reactivity of Cys residues toward an electrophile, e.g., N-ethylmaleimide (NEM), as a reporter of the stability of single metal–ligand coordination bonds,40,41 with metal coordination attenuating the reactivity of the cysteine thiolate anion. We subjected various metallostates of CopY to a pulse of excess d5-N-ethymaleimide (d5-NEM) for time t, following by a chase with a vast excess of protiated NEM (H5-NEM) under denaturing conditions, thereby encoding a mass shift of 5.0 Da. Proteins are then digested with a suitable protease, and deuterated and protiated peptides resolved by MALDI-TOF mass spectrometry (Table 2).| Peptide | Amino acid sequence | Mass (Da) | Cys | Modification | Mass of modified peptides (Da) | |
|---|---|---|---|---|---|---|
| Calc'd | Obs'd | |||||
| a Carried out as described in Materials and methods with representative data shown in Fig. 4. | ||||||
| 83-95 | DKV![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) SRRIRNLLA SRRIRNLLA |
1543.88 | C86 | H5-NEM | 1668.93 | 1668.94 |
| d 5-NEM | 1673.97 | 1673.99 | ||||
| 120-131 | SSAVTEVRNM |
1299.55 | C128, C130 | H5-NEM/H5-NEM | 1549.64 | 1549.71 |
| d 5-NEM/H5-NEM | 1554.69 | 1554.73 | ||||
| d 5-NEM/d5-NEM | 1559.73 | 1559.72 | ||||
MALDI-TOF data as a function of pulse time, t, for the apo, Zn1 and Cu2 states reveals C128 and C130 are both highly reactive in the apo-state (Fig. 4A), with nearly full deuteration observed at pulse time of 30 s, and that this reactivity is fully quenched in the Cu2 complex (Fig. 4C). In contrast, the Zn complex is kinetically or thermodynamically less stable than the Cu2 complex but these Cys are protected relative to the apo-state, revealing detectable singly-deuterated (d5/H5) and double-deuterated (d5/d5) NEM-containing peptides at intermediate pulse times, t = 240 s (Fig. 4B). This result reveals that Cu binding is fully protective on the cysteine ligands and Zn fails to protect the four cysteine residues involved in Cu(I) coordination.
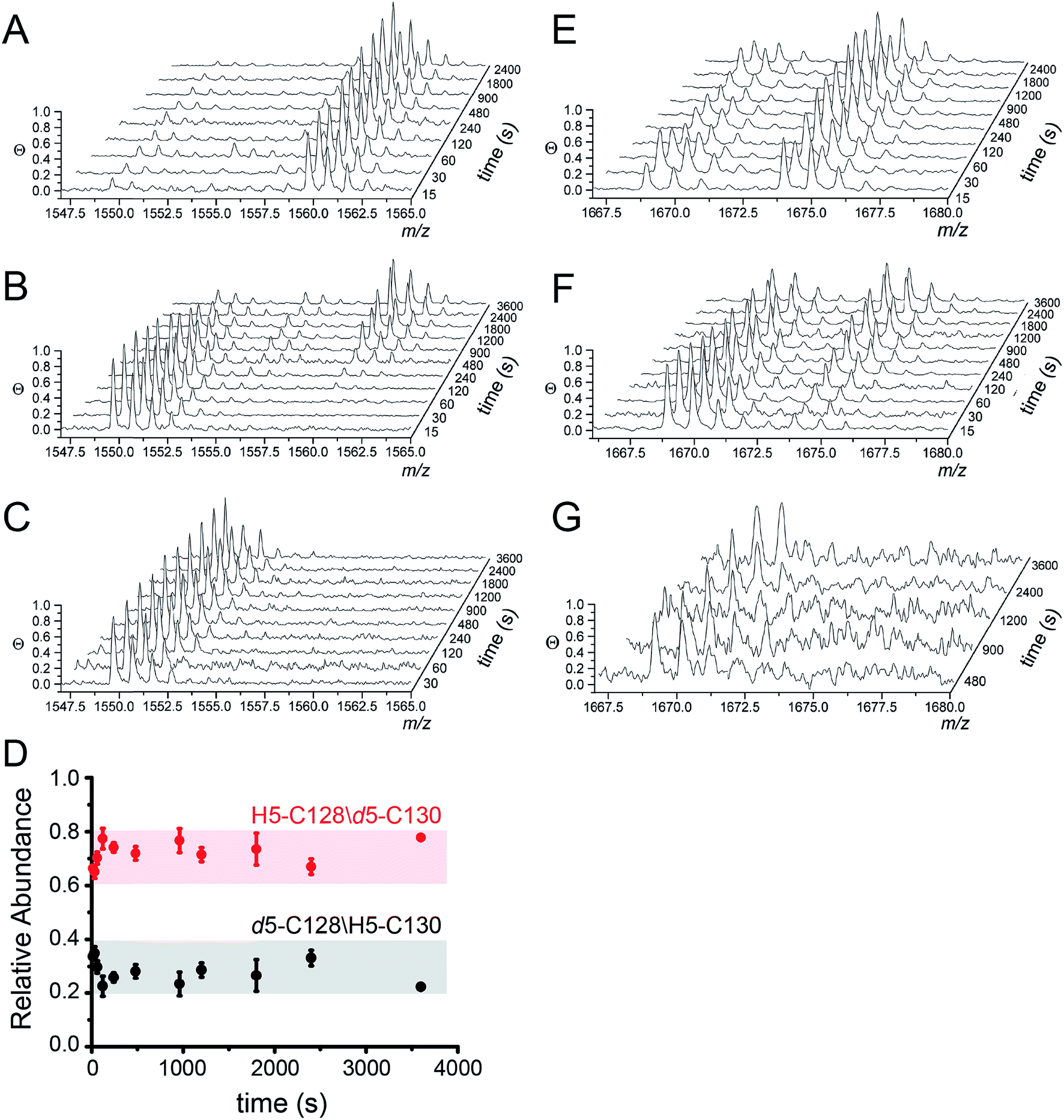 | ||
| Fig. 4 Ratiometric pulsed alkylation-mass spectrometry analysis of Spn C101A CopY in the apo-state (A and E), the Zn1 (per dimer) state (B and F) and the Cu2 (per dimer) (C and G) allosteric states. rPA-MS time course profiles for AspN-derived peptide 120-131 (panels A–C) and LysC-derived peptide 83-95 containing C86 (panels E–G) (see Table 2 for exact masses). (D) Relative quantification of d5-NEM alkylation events at C128 vs. C130 in the Zn1 state as a function of d5-NEM pulse time in the doubly alkylated d5/H5 peptide 120-131 quantified as described in Fig. S3.† Note that C130 is more reactive than C128 at all pulse times. | ||
The results from rPA-MS are consistent with the XAS but do not indicate which Cys residues are more strongly bound to the Zn. This can be determined by subjecting the singly-deuterated (d5/H5) peptide to tandem mass spectrometry (MS/MS) (Fig. S3†). Analysis of these data reveals derivatization of both C128 and C130 in the resulting fragment ions, with the C130-d5-NEM y- and b-ions consistently accumulating to higher levels that the C128-d5-NEM ions (Fig. 4D). These data therefore support a model in which C128 from both protomers is strongly coordinated to the Zn(II), while only one of the two C130 side chains is coordinated in the CopY homodimer (see Fig. 3B).41
Additionally, rPA-MS can potentially report on changes in cysteine reactivity in cysteines that are not involved in metal binding but are differentially exposed to solvent. The reactivity of C86 is also somewhat sensitive to metal binding, and is differentially reactive in the Zn- vs. Cu-bound states (Fig. 4F and G). Zn slightly attenuates the reactivity of C86 relative to apo-CopY, while Cu(I) appears nearly fully protective. These results suggest that in addition to differences in the first coordination shell, Cu(I) and Zn(II) may well trigger distinct conformational changes in the CopY dimer.
DNA-binding properties of the apo-, Zn(II) and Cu(I)-coordinated CopY
With the distinct coordination chemistries of the Zn and Cu-bound states of CopY established by our metal affinity, XAS and rPA-MS experiments, we next aimed to relate these metal binding differences to the cop operator DNA binding affinities. Previous work on E. hirae CopY suggests that Zn(II) coordination is essential for DNA binding, while Cu(I) allosterically inhibits DNA binding28,35 with the latter inducing Cu-dependent transcriptional derepression.15 We reconstituted Zn1 and Cu2 C101A CopYs and measured their DNA binding affinities for a DNA duplex containing a single cop box sequence using either a fluorescence anisotropy (FA)-based assay (Fig. 5A)42 or an electrophoretic mobility shift assay (EMSA) (Fig. 5B). The FA-based assays reveal that Zn(II) indeed functions as an allosteric activator of DNA binding, but only increases the apparent affinity ≈4-fold at equilibrium relative to the apo-state, with Ka ≈ 107 M−1 under these conditions (pH 7.0, 0.23 M NaCl, 25 °C). The EMSA experiments largely recapitulate the FA-based assays, except that the apo-CopY complex appears significantly more kinetically labile than the Zn-CopY complex, with only small amounts of DNA complex formation up to 1 μM protein (Fig. 5B). We could observe no complex formation for the Cu(I)-bound state, consistent with allosteric inhibition of cop box DNA binding by Cu(I).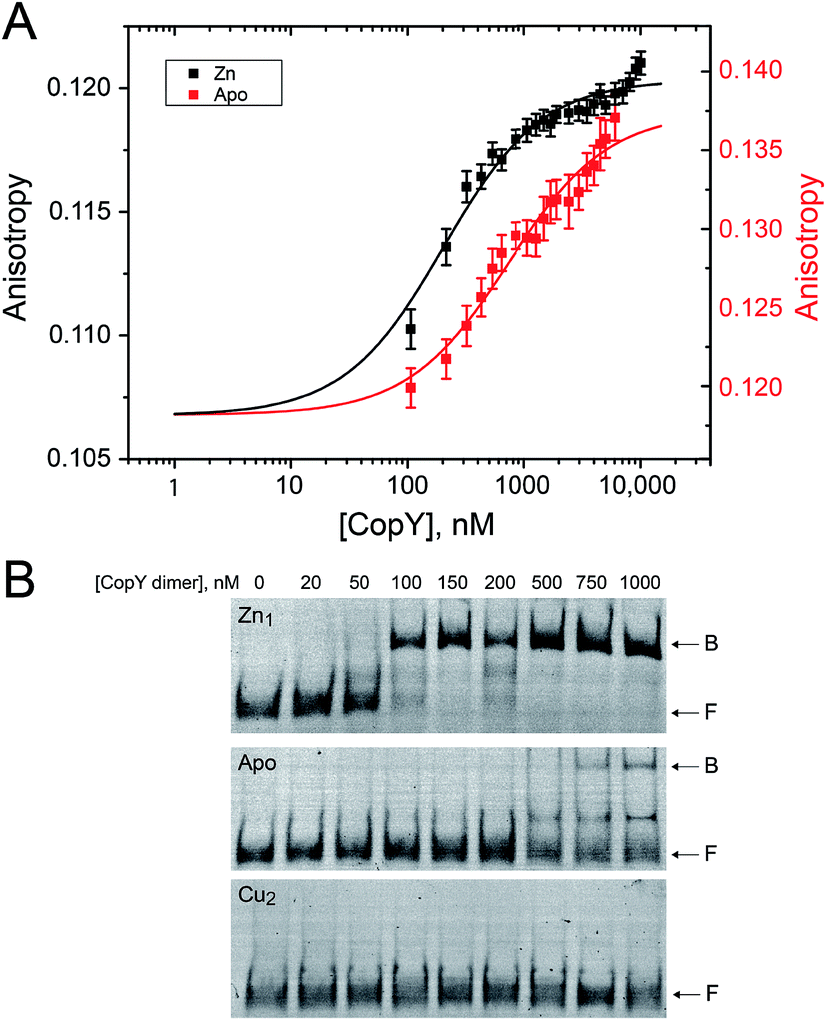 | ||
| Fig. 5
cop operator DNA binding activities of various C101A CopY metallostates. (A) Fluorescence anisotropy (FA)-based DNA binding experiment for Zn1 (black) and apo-states (red) of C101 CopY, with the continuous line a fit to 1:1 non-dissociable dimer-DNA binding model. Ka = 1.0 (±0.2) × 107 M−1 and Δr = 0.014 (±0.03) for Zn1 CopY and Ka = 2.8 (±0.6) × 106 M−1 and Δr = 0.017 (±0.03) for apo-CopY, for ΔGc = 0.6 (±0.1) kcal mol−1. Conditions: 0.23 M NaCl, 10 mM HEPES, pH 7.0, 2 mM TCEP, 40 μM ZnSO4 or 2 mM EDTA, 25 °C. (B) Representative electrophoretic mobility shift analysis (EMSA). Top, Zn1 C101 CopY dimer; middle, apo-state; bottom, Cu2 C101A CopY. B, bound; F, free DNA. The equilibrium Zn1 CopY-dimer DNA binding affinity, Ka, estimated from these data is 2.0 × 107 M−1, consistent with the FA-binding data in panel A. Solution conditions: 25 mM HEPES, pH 7.0, 200 mM NaCl (chelexed), 5 mM MgCl2, 50 μg mL−1 BSA, 25 °C. Apo-CopY samples contained 5 mM EDTA. | ||
Functional characterization of CopY mutants in cells
In order to validate our S. pneumoniae CopY coordination models and cop operator DNA binding experiments, we constructed S. pneumoniae D39 strains harboring mutant CopYs and measured their resistance to cellular copper toxicity (Fig. S4†) and transcriptional regulatory activity (Fig. 6). The addition of 0.2 mM Cu(II) to the growth medium in mid-log cells results in a ≈100-fold induction of copA expression, confirming the Cu-inducibility of the cop operon (Fig. 6, WT). In contrast, a ΔcopY strain is characterized by high level, constitutive copA expression that is unchanged by the addition of Cu. Mutant pneumococcal strains harboring C52A, C101A and C52A/C101A copY alleles are also strongly Cu-inducible, to an extent similar to the wild-type strain, findings consistent with the non-essentiality of non-conserved cysteines in CopY, C52 and C101. The strain harboring C86A CopY is also Cu-inducible consistent with no direct role in Cu sensing, but gives higher levels of basal copA expression (Fig. 6). The differential reactivity or solvent accessibility of C86 in the Zn vs. apo allosteric states (see Fig. 4) may be related to this partial loss of repressor function and, thus may be reporting on the importance of the conformation of this linker region for allosteric communication (see Fig. 1). Finally, the quadruple C52A/C101A/C128A/C130A mutant shows no significant Cu regulation, consistent with the requirement that the C-terminal Cys pair (C128, C130) be available to coordinate the allosteric activator Zn, required to obtain full repression of copA expression in the absence of Cu stress in cells. All strains grow with WT-like growth kinetics and are unaffected by copper stress, in contrast to the copA deletion strain, as expected (Fig. S4†).14 Thus, unregulated cop operon expression is not deleterious to pneumococcal growth under these growth conditions.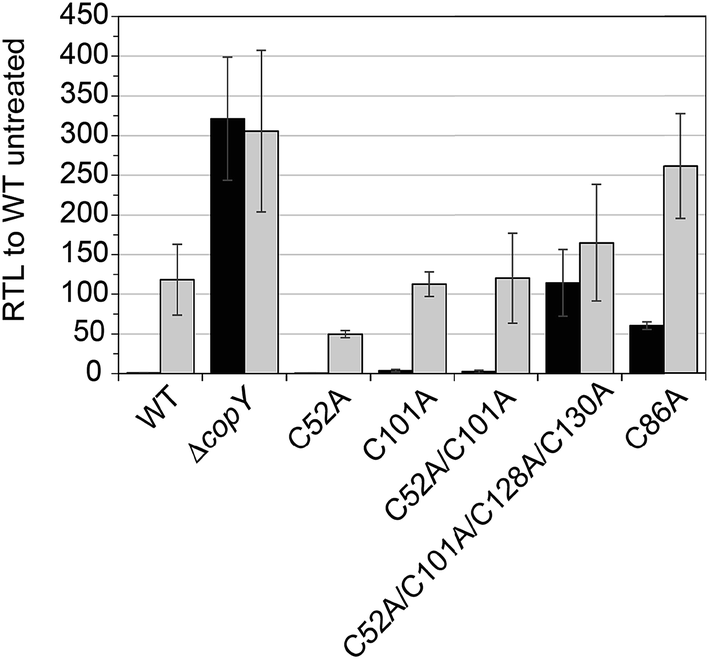 | ||
| Fig. 6 Relative transcript levels (RTL) from qRT-PCR analysis of copA transcription. Exponentially growing wild-type (WT), ΔcopY and copY mutant allelic cells were diluted into BHI and allowed to grow to approximately 0.2 OD620. Cultures were then spiked with (grey) or without (black) 200 μM Cu and incubated for 1 h before harvesting cells and RNA extraction. The average and s.d. of three biological replicates are shown. | ||
Structural analysis of CopY metallostates
While the above data clearly show that Zn(II) and Cu(I) form distinct coordination complexes that oppositely impact cop operator DNA binding affinity in vitro and in cells, the structural basis for these effects is unknown. In fact, there is no structure available for any full length CopY, with the only high resolution structure of the N-terminal DNA binding domain of L. lactis CopR (Fig. 1B).17 We therefore investigated the solution structural properties of the apo-, Zn1 (1:1 per dimer) and Cu2 (2:1 per dimer) complexes using 1H–15N NMR spectroscopy, small angle X-ray scattering (SAXS) and ion mobility-mass spectrometry (IM-MS), discussed in turn.
NMR studies of wild-type and C52A/C101A CopY metallostates
We first used NMR spectroscopy to characterize the different metallostates of Spn CopY, since this method potentially provides both high resolution and site-specific information on both structure and dynamics. 1H–15N TROSY or 1H–15N HSQC spectra were acquired for both wild-type and C52A/C101A CopYs (30.9 kDa; Fig. 7). A comparison of the spectra obtained for the apo- and Zn-saturated forms of the repressor reveal that the apoprotein is characterized by considerable conformational dynamics, as very few cross peaks are observed in these spectra. The addition of 0.5 mol protomer·equiv. Zn(II) sharpens these spectra significantly, and reveals that the structures of the wild-type and C52A/C101A CopYs are substantially identical, consistent with the fact that these two non-conserved Cys are dispensable for CopY function in cells (Fig. 6). However, the relative intensities of the cross peaks vary dramatically even in Zn1 CopY and not all anticipated cross peaks are visible, indicative of substantial dynamics in specific regions of the molecule. This severely limits efforts to obtain sequence-specific resonance assignments even with extensively deuterated samples (spectra not shown), which were ultimately unsuccessful. Strikingly, anaerobic addition of 1 mol protomer·equiv. (2 per dimer) Cu(I) to either the apo-CopY or a preformed Zn1 CopY dimer results in significant spectral line broadening, far more so than is observed in the apoprotein (Fig. S5†).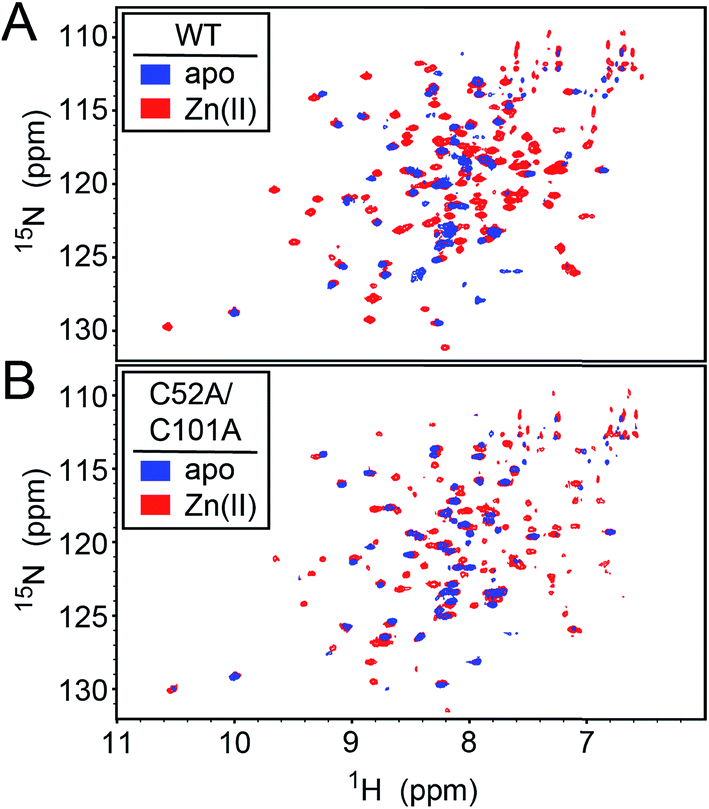 | ||
| Fig. 7 1H–15N HSQC spectra of intact wild-type (WT) (A) and C52A/C101A (B) Spn CopY in the apo-state (blue contours) and the Zn1 allosteric state (red contours). Spectra were acquired at 30 °C in 20 mM HEPES, 0.2 M NaCl, 5 mM TCEP, pH 6.0. Additional NMR spectra of Cu-bound CopY and the C-terminal regulatory domain fragment (68-131) are shown in Fig. S5.† | ||
In an effort to improve spectral quality, we also acquired 1H–15N HSQC spectra for what we anticipated would correspond to the C-terminal, metal binding regulatory domain, encompassing residues 68-131 (Fig. S5†). Unfortunately, these spectra, like that of the intact CopY, yielded very little additional information, again due to severe spectral line broadening. The largest number of cross peaks are again obtained with the Zn(II)-bound regulatory domain, and the majority of the observable cross peaks can be superimposed on those found in the Zn1-CopY dimer spectrum. Severe spectral line broadening induced specifically by Cu(I) is fully consistent with some combination of extensive conformational dynamics within a Cu(I)-bridged CopY dimer and oligomerization beyond the dimer to higher order assemblies, also bridged by Cu–S bonds.
Small angle X-ray scattering (SAXS) analysis
We next employed small angle X-ray scattering (Fig. 8) in an effort to understand changes in the global shape that occur upon metal coordination. SAXS scattering curves were obtained for the apo-, Zn1 and Cu2 states, and apo and Zn1-states show Guinier plots indicative of monodispersity and a similar radius of gyration (Rg, Fig. 8A and B; Table 3). In contrast, the Guinier plot of the Cu2 state shows clear evidence of a heterogeneous sample containing higher order oligomers (Fig. 8C) and as a result, detailed structural analysis of this state was not further carried out. The apo and Zn1-states are readily distinguished from one another in the raw scattering profiles (to q = 0.5 Å−1) as well as in the PDDF plots (p(r) vs. r) (Fig. 8E), with the scattering of the Zn bound state consistent with the prediction based on the previously published crystal structures of BlaI and MecI (Fig. 8D and E). Furthermore, the molecular envelope calculated as an ab initio bead model by DAMMIF for the Zn-bound state shows that structure in solution is consistent with a model of CopY based on previously published structures of BlaI and MecI (Fig. 8F), while fully recapitulating the experimental scattering curve (Fig. 8D). Moreover, a qualitative analysis of the Kratky plots suggests that the apo- and Cu2 forms of CopY are less structured overall than the Zn bound form (Fig. 8A–C), consistent with the reduced number of cross peaks in the 1H–15N HSQC spectra of these states relative to the Zn-bound CopY dimer. | ||
| Fig. 8 Small angle X-ray scattering (SAXS) analysis of C101A CopY in various allosteric states. Guinier plots and associated linear fits (where possible) of the composite raw scattering curves (panel D) obtained from the apo- (red) (A), Zn1 (black) (B) and Cu2 (blue) (C) C101 CopYs, with associated Kratky plots shown in the inset. (D–F) Quantitative analysis of Zn1 C101 CopY. (D) composite scattering curve (logI vs. q) for Zn1 CopY (black), with a curve calculated from the best-fit ab initio model (F) (green), shown alongside those for apo- (red) and Cu2 (blue) CopYs. (E) Pairwise distribution histogram plot from the data shown in panels A (apo CopY; red) and B (Zn1 CopY; black), with a best-fit DAMMIF ab initio model shown for the Zn1 CopY (E).54 The hydrodynamic parameters obtained for apo- and Zn1 CopYs from these data are shown in Table 3. | ||
| apo CopY | Zn1 CopY | |
|---|---|---|
| a The SAXS data for the Cu(I) binding metallostate was not further analyzed due to the extreme nonlinearity in the Guinier plots (Fig. 8C). b Derived from Guinier fitting.52 c Derived from GNOM analysis.51 d Molecular weight calculated using the MoW2 server,53 with theoretical molecular weight calculated from protein sequence (dimer) shown in parentheses. | ||
| R g (Å)b (Guinier) | 26.3 ± 1.1 | 24.3 ± 1.0 |
| R g (Å)c (GNOM) | 24.3 ± 0.4 | 22.5 ± 0.6 |
| D max (Å) | 76 | 77 |
| MW (kDa)d | 24.7 (30.9) | 26.0 (30.9) |
| MW discrepancy | 20.2% | 15.9% |
Ion mobility-mass spectrometry (IM-MS)
One interpretation of the SAXS and NMR data is that the apo-form of CopY is partially or transiently unfolded, particularly in the C-terminal regulatory domain. To test this, we analyzed the CopY metallostates by ion mobility-mass spectrometry (IM-MS), an experimental technique that can be used to examine partially unfolded states, as recently described for ubiquitin.43 We therefore acquired m/z spectra (Fig. 9A) and ion mobility distributions (Fig. 9B and S6†) for the apo-, Zn1- and Cu2 complexed forms of CopY that collectively provide insights into monomer stability and mobility profiles of these allosteric states, using methods analogous to those previously applied to the Cu-sensing repressor CsoR.44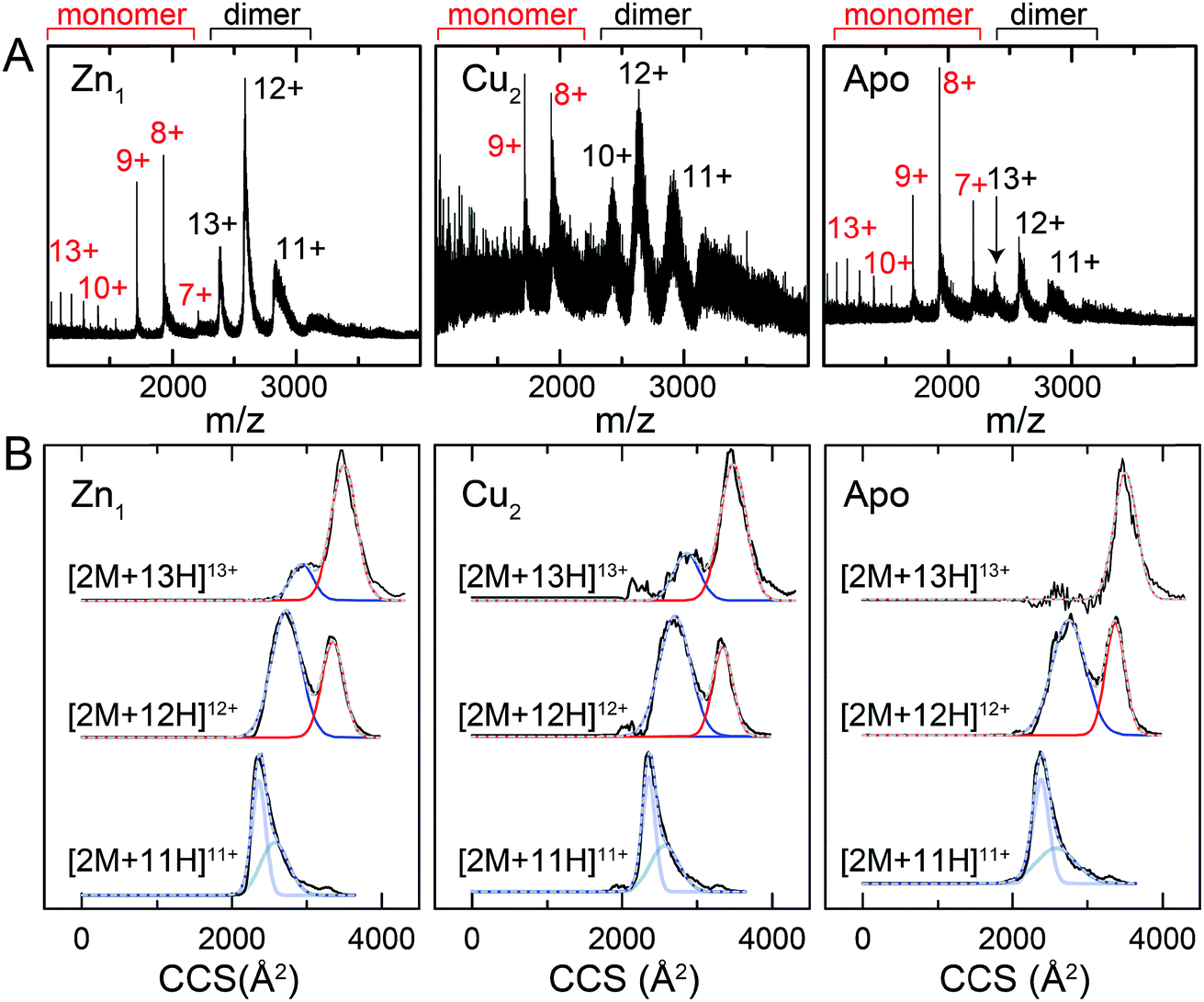 | ||
| Fig. 9 Ion mobility-mass spectrometry (IM-MS) of C101A Spn CopY in various allosteric states. Mass (m/z) spectra (A) and background subtracted mobility distributions for [2M + 11H]11+, [2M + 12H]12+ and [2M + 13H]13+ dimer charge states (B) for the Zn1 state (left) and Cu2 state (middle) and apo state (right). Monomer (highlighted in red) and dimer (black) regions are shown in panel A. In panel B, the experimental data are in black, and each distribution envelope is fit to the minimum number of Gaussians (red or blue) required to satisfactorily describe the full envelope (grey discontinuous line through the envelope). See Fig. S6† for complete 2D drift plots for these CopY metallostates. | ||
The mass spectral data (Fig. 9A and S6†) are characterized by three different regions: an area of highly charged monomer from 1000 to 1600 m/z ([M + 15H]15+ to [M + 10H]10+), a region of low charged monomer from ≈1600 to 2200 m/z ([M + 9H]9+ to [M + 7H]7+) and a region of dimer from 2200 m/z and greater (principally [2M + 13H]13+ to [2M + 11H]11+). The relative abundance of the dimer peaks compared to the low-charged monomer peaks reports on the stability of the CopY dimer. For example, in apo-CopY, the low charge states of the monomer, from [M + 9H]9+ to [M + 7H]7+, dominate the distribution relative to the three readily visible dimer peaks, [2M + 13H]13+, [2M + 12H]12+ and [2M + 11H]11+. On the other hand, the coordination of Zn(II) to the intersubunit 3S 1H2O site results in an increase in the dimer (D) peaks relative to the monomer (M) peaks, with significant suppression of the [M + 8H]8+ and [M + 7H]7+ charge states. All the monomer (M) peaks are clearly metal-free (as evidenced by the same M masses in each CopY preparation) and this may well derive from electrospray ionization-mediated dissociation of the Zn1-CopY complex during transition to the gas phase.45 This suggests that Zn binds only to the dimeric CopY, fully consistent with a subunit-bridging coordination model (Fig. 3; vide infra). The Cu2 CopY complex (Fig. 9A) m/z spectra are qualitatively similar to that of the Zn-CopY state, except that these spectra are characterized by a very low signal-to-noise (S/N) ratio. Low S/N can either be traced to a low concentration relative to other higher oligomer forms of CopY that predominate in these mixtures (vide infra), and/or relatively poor desolvation of the Cu-bound dimer when electrosprayed. Based on the SAXS and NMR analysis, we favor the former interpretation.
Mobility distributions for the [2M + 13H]13+, [2M + 12H]12+ and [2M + 11H]11+ charge states derived from the apo and the metallated CopY dimers are shown in Fig. 9B. Overall, the apo-, Zn1 and Cu2 states show two major features that correspond to an extended and compact set of conformations, ≈2500 and ≈3200 Å2, respectively. The relative abundance of each conformation depends primarily on the charge state as a result of the increase in coulombic repulsion, i.e., the compact conformation is more prevalent in the lower charge states. This columbic repulsion may also explain the small increase in collision cross-section for each conformation with increasing number of charges. Although these gas-phase conformations are not necessarily present in solution, the SAXS and NMR data suggest that CopY exists as a heterogeneous ensemble of different structures, particularly in the apo-state. An extended conformation (≈3400 Å2) is indeed more prevalent in the apo-state distributions compared to the Zn1-state (Fig. 9B), a finding more evident for the [2M + 13H]13+ charge state where the apo-state is exclusively extended. Overall, Zn coordination by the CopY dimer stabilizes a more compact form that likely resembles the SAXS model (see Fig. 8F) and is structurally compatible with DNA binding. In the case of the Cu2-metallostate, the dimer is likely not the most abundant oligomeric state in solution, but we could not find any evidence of higher oligomerization states in these ion mobility spectra. These results support the idea that Zn(II) coordination to the subunit-bridging site restricts access to an extended conformation that is likely partially unfolded and, in this way, enhances DNA binding affinity of the Zn-bound repressor relative to the apo-repressor.
Conclusions
In this work, we have determined the Cu and Zn binding affinities, stoichiometries and coordination structures of Spn CopY using chelator competition assays, X-ray absorption spectroscopy and rPA-MS. We have also used NMR spectroscopy, SAXS and IM-MS to define the differences in protein structure and dynamics between the metal-free apo state, allosterically activated (relative to DNA binding) Zn(II) bound CopY, and the allosterically inhibited Cu(I)-bound CopY. Our findings reveal that Spn CopY binds up to one dimer mol·equiv. of Zn(II) or up to two dimer mol·equiv. of Cu(I), respectively. Both metal complexes are subunit-bridging, and involve exclusively coordination by the C-terminal C128–x–C130 pair. There is no spectroscopic evidence for coordination by the C-terminal M131 residue or C86, with the coordination models consistent with mononuclear coordinately unsaturated 3S 1H2O Zn(II) complex and a binuclear Cu2–S4 Cu(I) or multinuclear Cun–Sm complex (where n < m). Although XAS data consistent with a binuclear Cu2–S4 cluster have been previously reported for E. hirae CopY,28 our SAXS show clear evidence of a heterogeneous sample containing higher order oligomers. This is further supported by the low signal-to-noise in our IM-MS spectra on the Cu2 CopY complex, the significant decrease in the number of observable cross peaks in the NMR spectra of this complex, as well as the rPA-MS findings, which reveal that all the solvent-exposed Cys are essentially fully protected by Cu(I) coordination. Although our results cannot clearly distinguish the degree to which Cu-bound oligomers form beyond that of the tetramer, this is the first direct evidence that the metallostructures must be subunit-bridging in any CopY. Further, this work suggests that Cu-mediated oligomerization beyond the dimer is an important aspect of this regulatory process (Fig. 10A).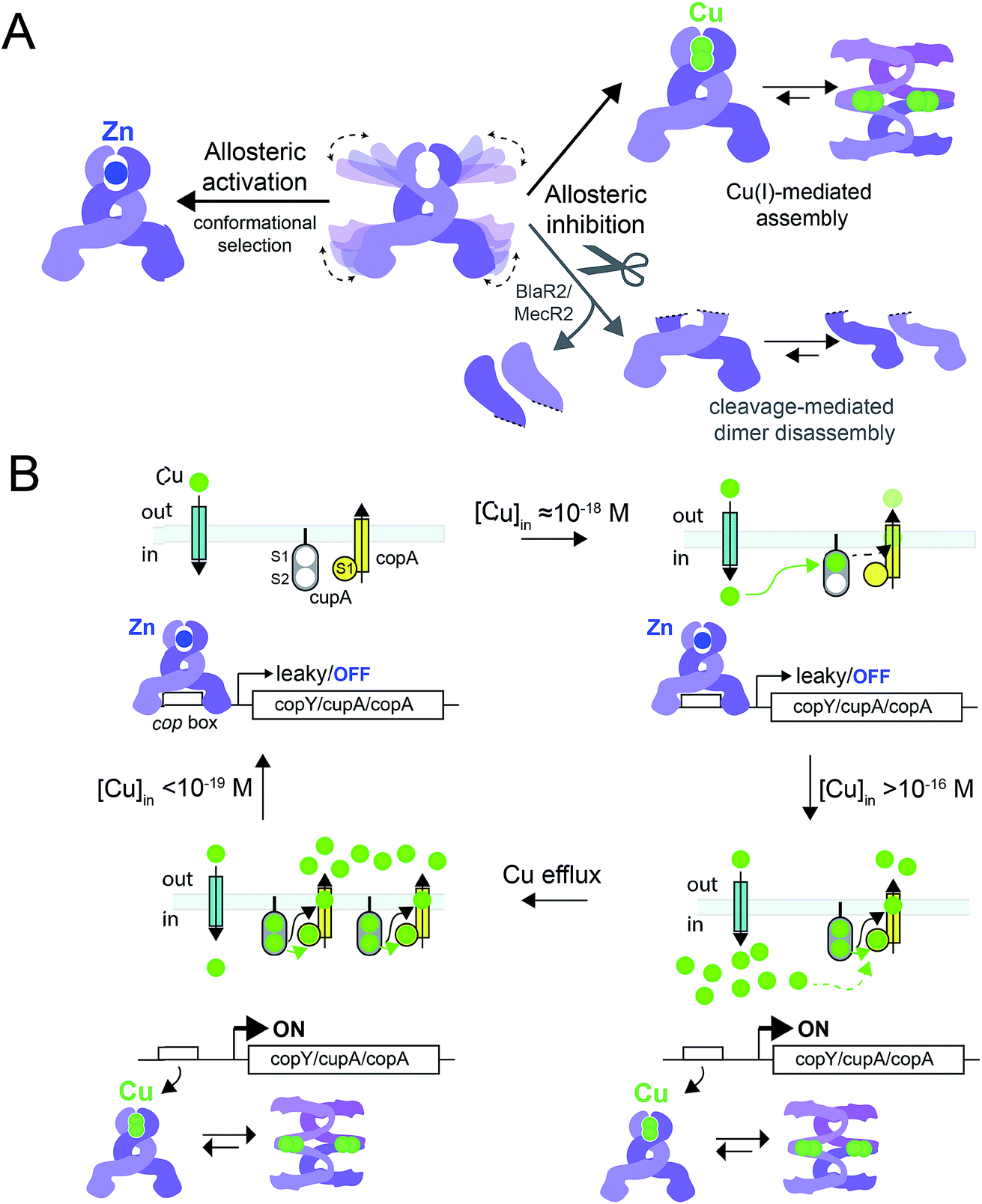 | ||
| Fig. 10 (A) Allosteric model of Spn CopY highlighting allosteric activation of DNA operator binding by apo-CopY by Zn, and allosteric inhibition of DNA binding by Cu(I), consistent with the work presented here. We compare metal-dependent CopY regulation with proteolysis-induced regulation of DNA (MecR2; BlaR2) binding by dimeric MecI/BlaI (see text for details). Higher order oligomeric states are represented only as tetramers for clarity. (B) Threshold model for Cu(I) sensing and detoxification in S. pneumoniae. The Zn and Cu(I) binding affinities, stoichiometries and coordination structures determined here are placed into the context of previously published work on the membrane-bound Cu chaperone, CupA, and the P-type ATPase efflux pump, CopA.14,55 The Zn(II) binding affinity of CopY is such that there is sufficient free Zn in the cell, governed by the relative affinities of the Zn uptake repressor, AdcR,56 and the Zn efflux regulator, SczA,5 to ensure that any CopY expressed under uninduced conditions will be Zn bound and bound to the cop box. As free Cu(I) rises to greater than 10−16 M, this is sufficient to trigger Cu binding by CopY (logKCu 16.3) leading to transcriptional derepression; as Cu levels continue to rise, the low affinity S2 Cu site on CupA is filled (logKCu 14.8), which along with CopA is required for cellular Cu resistance. CupA and CopA collaboratively function to lower cellular Cu, and at [Cu] ≤ 10−19 M, the regulatory system resets. | ||
The average Cu(I) binding affinity of Spn CopY, although lower than those measured for other Cu(I)-sensing metalloregulators,32,46 is consistent with a “threshold” model of cellular Cu resistance defined by a set-point of ≈10−16 M free metal in the pneumococcus (Fig. 10B). In this model, the Cu affinity of CopY is tuned such that it is weaker than the high affinity S1 site in metallochaperone CupA, comparable to that of N-terminal metal-binding domain of CopA, but greater than that of functionally critical S2 site in CupA.14 In this way, the Cu resistance system is transcriptionally switched “on” at ≈10−16 M bioavailable Cu(I), which exceeds the capacity of the existing CupA and CopA molecules produced as a result of “leaky” transcription to buffer Cu(I).47 This results in the synthesis of many molecules of CopY, CupA, and CopA, which in turn triggers CupA–CopA-mediated Cu export, and a subsequent drop in cellular Cu to less than 10−18 M Cu. A report that appeared during the review of this manuscript which investigates the role of CupA on CopY function in pneumococcal cells under conditions of copper toxicity adds additional detail to this threshold model.48
How CopY incorporates allosteric activation of DNA binding by Zn(II) can be explained by a conformational selection mechanism based on the ion mobility experiments where the population of an extended, partially unfolded, weakly DNA-binding conformation(s) present in the apo state is significantly quenched upon Zn binding. Ligand-dependent activation of DNA binding has been attributed to conformational selection for several transcription regulators that repress or activate gene expression,49,50 with the catabolite activator protein in E. coli the best characterized example. This mechanism has been previously suggested for MecI/BlaI based on the different orientations of the DNA binding motifs in BlaI structures from two different organisms (Fig. S8†).25,26 However, this idea has not been further evaluated since the regulation of DNA binding of MecI/BlaI is thought to occur mainly via selective proteolytic cleavage in the regulatory domain that destabilizes the dimer and dissociates the repressor–operator complex (Fig. 10A).22,23 In the case of CopY, the weaker DNA binding affinity of the apoprotein is enhanced by Zn coordination at the C-terminal CxC pair, which not only stabilizes the CopY homodimer (Fig. 9) but also selects a compact conformation that it is poised to bind DNA (Fig. 8F). Moreover, the NMR structure of Lactococcus lactis CopR17 (Fig. 1B) suggests that the relative orientations of the DNA binding helices relative to the rest of the winged helical domain are similar to that found in the intact MecI structure from S. aureus23 which are not oriented such that the reading heads “fit” into consecutive major grooves (Fig. S7†). This reinforces the idea that conformational plasticity and marginal stability of the regulatory domain must be important aspects of allosteric activation of DNA operator binding of CopY by Zn (Fig. 10A).
Allosteric inhibition by Cu, on the other hand, relies on the formation of a kinetically stable, multi-metallic Cu·S cluster(s) that may well destabilize the dimer by forming higher order oligomers, likely by means of Cu·S·Cu bridges. As discussed above, the mechanism of derepression proposed for MecI posits a proteolysis-dependent change in the stability of the dimer near the residue corresponding to C101 (Fig. 1A); however, in this case, the signaling event (proteolysis) leads to dissociation of dimers to weakly DNA-binding monomers (Fig. 10A).22,23 On the contrary, Cu leads to the formation of CopY oligomers that are aggregation-prone (Fig. 8C, 10A). It has been shown previously that Cu induces derepression of E. coli MarR-regulated genes by a tetramer assembly mechanism;51 however, in this case, the oligomers that form are mediated by oxidation of a single Cys in the DNA binding domain to a disulfide, forming crosslinked tetramers and thereby occluding the DNA-binding helices from interacting with the DNA operator. In our CopY model, stable Cu–S coordination bonds may drive assembly beyond the dimer, particularly at excess Cu (Fig. 10B). Our Cu(I)-mediated assembly-inhibition model also readily rationalizes the observation that CopYs contain variable numbers of clustered Cys residues in the C-terminus (Fig. 1B), leading to distinct metal stoichiometries and nuclearities of their multinuclear Cu(I)-thiolate complexes.28 All Cu(I) needs to do is drive polynuclear Cu–S cluster formation, sufficient to induce structural and/or dynamical changes in the C-terminal regulatory domain, leading to oligomerization and ultimately DNA dissociation. Zn(II), in contrast, must be subunit-bridging in a way that stabilizes the dimer and quenches conformational heterogeneity, thereby activating DNA binding. Our finding of a coordinately unsaturated Zn complex likely lowers the kinetic barrier and enhances the rate of metal-exchange between the more weakly bound Zn and more strongly bound Cu(I), thus facilitating transcriptional derepression of the CopY-regulated Cu-resistance genes by increased cellular copper. We propose that nature exploits a marginally kinetically or thermodynamically stable all-α-helical C-terminal regulatory domain on a common N-terminal DNA binding domain as a key feature of CopY and BlaI/MecI-family repressors to effect allostery and biological outputs in distinct ways.
Methods
See complied ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the US National Institutes of Health from grants R01 GM042569 and R35 GM118157 (to D. P. G.) and the Pew Charitable Trusts (to D. A. C.). We also thank Dr My Le for early help in analysis of the SAXS data, Mr Goran Tumbic and Dr Katie Edmonds for helpful discussions of the IM-MS and NMR data, respectively, and Dr Lixin Fan of the Small-Angle X-ray Scattering Core Facility, National Cancer Institute, Frederick, MD for acquiring the SAXS data.References
- W. Maret, Metallomics, 2010, 2, 117–125 RSC.
- C. A. Wakeman and E. P. Skaar, Curr. Opin. Microbiol., 2012, 15, 169–174 CrossRef CAS PubMed.
- M. I. Hood and E. P. Skaar, Nat. Rev. Microbiol., 2012, 10, 525–537 CrossRef CAS PubMed.
- K. Y. Djoko, C.-l. Y. Ong, M. J. Walker and A. G. McEwan, J. Biol. Chem., 2015, 290, 18954–18961 CrossRef CAS PubMed.
- J. E. Martin, K. A. Edmonds, K. E. Bruce, G. C. Campanello, B. A. Eijkelkamp, E. B. Brazel, C. A. McDevitt, M. E. Winkler and D. P. Giedroc, Mol. Microbiol., 2017, 104, 636–651 CrossRef CAS PubMed.
- K. Y. Djoko, C. L. Ong, M. J. Walker and A. G. McEwan, J. Biol. Chem., 2015, 290, 18954–18961 CrossRef CAS PubMed.
- R. A. Festa and D. J. Thiele, PLoS Pathog., 2012, 8, e1002887 CAS.
- Y. Fu, F. M. Chang and D. P. Giedroc, Acc. Chem. Res., 2014, 47, 3605–3613 CrossRef CAS PubMed.
- C. White, T. Kambe, Y. G. Fulcher, S. W. Sachdev, A. I. Bush, K. Fritsche, J. Lee, T. P. Quinn and M. J. Petris, J. Cell Sci., 2009, 122, 1315–1321 CrossRef CAS PubMed.
- D. P. Giedroc and A. I. Arunkumar, Dalton Trans., 2007, 29, 3107–3120 RSC.
- D. A. Capdevila, J. Wang and D. P. Giedroc, J. Biol. Chem., 2016, 291, 20858–20868 CrossRef CAS PubMed.
- D. A. Capdevila, K. A. Edmonds and D. P. Giedroc, Essays Biochem., 2017, 61, 177–200 CrossRef PubMed.
- S. Shafeeq, H. Yesilkaya, T. G. Kloosterman, G. Narayanan, M. Wandel, P. W. Andrew, O. P. Kuipers and J. A. Morrissey, Mol. Microbiol., 2011, 81, 1255–1270 CrossRef CAS PubMed.
- Y. Fu, H.-C. T. Tsui, K. E. Bruce, L.-T. Sham, K. A. Higgins, J. P. Lisher, K. M. Kazmierczak, M. J. Maroney, C. E. Dann, M. E. Winkler and D. P. Giedroc, Nat. Chem. Biol., 2013, 9, 177–183 CrossRef CAS PubMed.
- D. Strausak and M. Solioz, J. Biol. Chem., 1997, 272, 8932–8936 CrossRef CAS PubMed.
- P. Cobine, W. A. Wickramasinghe, M. D. Harrison, T. Weber, M. Solioz and C. T. Dameron, FEBS Lett., 1999, 445, 27–30 CrossRef CAS PubMed.
- F. Cantini, L. Banci and M. Solioz, Biochem. J., 2009, 417, 493–499 CrossRef CAS PubMed.
- R. Portmann, K. R. Poulsen, R. Wimmer and M. Solioz, BioMetals, 2006, 19, 61–70 CrossRef CAS PubMed.
- K. O. Pazehoski, P. A. Cobine, D. J. Winzor and C. T. Dameron, Biochem. Biophys. Res. Commun., 2011, 406, 183–187 CrossRef CAS PubMed.
- D. Magnani, O. Barré, S. D. Gerber and M. Solioz, J. Bacteriol., 2008, 190, 536–545 CrossRef CAS PubMed.
- H. Reyes-Caballero, G. C. Campanello and D. P. Giedroc, Biophys. Chem., 2011, 156, 103–114 CrossRef CAS PubMed.
- R. Garcia-Castellanos, A. Marrero, G. Mallorqui-Fernandez, J. Potempa, M. Coll and F. X. Gomis-Ruth, J. Biol. Chem., 2003, 278, 39897–39905 CrossRef CAS PubMed.
- R. Garcia-Castellanos, G. Mallorqui-Fernandez, A. Marrero, J. Potempa, M. Coll and F. X. Gomis-Ruth, J. Biol. Chem., 2004, 279, 17888–17896 CrossRef CAS PubMed.
- M. K. Safo, Q. Zhao, T. P. Ko, F. N. Musayev, H. Robinson, N. Scarsdale, A. H. Wang and G. L. Archer, J. Bacteriol., 2005, 187, 1833–1844 CrossRef CAS PubMed.
- J. Boudet, V. Duval, H. Van Melckebeke, M. Blackledge, A. Amoroso, B. Joris and J. P. Simorre, Nucleic Acids Res., 2007, 35, 4384–4395 CrossRef CAS PubMed.
- C. Sala, A. Haouz, F. A. Saul, I. Miras, I. Rosenkrands, P. M. Alzari and S. T. Cole, Mol. Microbiol., 2009, 71, 1102–1116 CrossRef CAS PubMed.
- D. Strausak and M. Solioz, J. Biol. Chem., 1997, 272, 8932–8936 CrossRef CAS PubMed.
- P. A. Cobine, G. N. George, C. E. Jones, W. A. Wickramasinghe, M. Solioz and C. T. Dameron, Biochemistry, 2002, 41, 5822–5829 CrossRef CAS PubMed.
- T. Liu, A. Ramesh, Z. Ma, S. K. Ward, L. Zhang, G. N. George, A. M. Talaat, J. C. Sacchettini and D. P. Giedroc, Nat. Chem. Biol., 2007, 3, 60–68 CrossRef CAS PubMed.
- D. R. Winge, C. T. Dameron, G. N. George, I. J. Pickering and I. G. Dance, in Bioinorganic Chemistry of Copper, ed. K. D. Karlin and Z. Tyeklár, Springer, Dordrecht, 1993, pp. 110–123 Search PubMed.
- Z. Xiao, J. Brose, S. Schimo, S. M. Ackland, S. La Fontaine and A. G. Wedd, J. Biol. Chem., 2011, 286, 11047–11055 CrossRef CAS PubMed.
- A. Changela, K. Chen, Y. Xue, J. Holschen, C. E. Outten, T. V. O'Halloran and A. Mondragon, Science, 2003, 301, 1383–1387 CrossRef CAS PubMed.
- Z. Ma, D. M. Cowart, B. P. Ward, R. J. Arnold, R. D. DiMarchi, L. Zhang, G. N. George, R. A. Scott and D. P. Giedroc, J. Am. Chem. Soc., 2009, 131, 18044–18045 CrossRef CAS PubMed.
- A. K. Chaplin, B. G. Tan, E. Vijgenboom and J. A. R. Worrall, Metallomics, 2015, 7, 145–155 RSC.
- P. A. Cobine, C. E. Jones and C. T. Dameron, J. Inorg. Biochem., 2002, 88, 192–196 CrossRef CAS PubMed.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- X. Chen, H. Hua, K. Balamurugan, X. Kong, L. Zhang, G. N. George, O. Georgiev, W. Schaffner and D. P. Giedroc, Nucleic Acids Res., 2008, 36, 3128–3138 CrossRef CAS PubMed.
- K. R. Brown, G. L. Keller, I. J. Pickering, H. H. Harris, G. N. George and D. R. Winge, Biochemistry, 2002, 41, 6469–6476 CrossRef CAS PubMed.
- K. Clark-Baldwin, D. L. Tierney, N. Govindaswamy, E. Gruff, C. Kim, J. M. Berg, S. Koch and J. E. Penner-Hahn, J. Am. Chem. Soc., 1998, 120, 8401–8409 CrossRef CAS.
- J. L. Apuy, X. Chen, D. H. Russell, T. O. Baldwin and D. P. Giedroc, Biochemistry, 2001, 40, 15164–15175 CrossRef CAS PubMed.
- J. L. Apuy, L. S. Busenlehner, D. H. Russell and D. P. Giedroc, Biochemistry, 2004, 43, 3824–3834 CrossRef CAS PubMed.
- D. A. Capdevila, J. J. Braymer, K. A. Edmonds, H. Wu and D. P. Giedroc, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4424–4429 CrossRef CAS PubMed.
- T. J. El-Baba, D. W. Woodall, S. A. Raab, D. R. Fuller, A. Laganowsky, D. H. Russell and D. E. Clemmer, J. Am. Chem. Soc., 2017, 139, 6306–6309 CrossRef CAS PubMed.
- A. D. Jacobs, F. M. Chang, L. Morrison, J. M. Dilger, V. H. Wysocki, D. E. Clemmer and D. P. Giedroc, Angew. Chem., Int. Ed. Engl., 2015, 54, 12795–12799 CrossRef CAS PubMed.
- R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1999, 121, 3494–3501 CrossRef CAS.
- M. Zimmermann, O. Clarke, J. M. Gulbis, D. W. Keizer, R. S. Jarvis, C. S. Cobbett, M. G. Hinds, Z. Xiao and A. G. Wedd, Biochemistry, 2009, 48, 11640–11654 CrossRef CAS PubMed.
- Y. Fu, K. E. Bruce, H. Wu and D. P. Giedroc, Metallomics, 2016, 8, 61070 RSC.
- M. J. Neubert, E. A. Dahlmann, A. Ambrose and M. D. L. Johnson, mSphere, 2017, 2, e00372-17 CrossRef PubMed.
- S. R. Tzeng and C. G. Kalodimos, Nat. Chem. Biol., 2013, 9, 462–465 CrossRef CAS PubMed.
- A. J. Guerra, C. E. Dann and D. P. Giedroc, J. Am. Chem. Soc., 2011, 133, 19614–19617 CrossRef CAS PubMed.
- Z. Hao, H. Lou, R. Zhu, J. Zhu, D. Zhang, B. S. Zhao, S. Zeng, X. Chen, J. Chan, C. He and P. R. Chen, Nat. Chem. Biol., 2013, 10, 21–28 CrossRef PubMed.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- H. Fischer, M. D. Neto, H. B. Napolitano, I. Polikarpov and A. F. Craievich, J. Appl. Crystallogr., 2010, 43, 101–109 CAS.
- D. Franke and D. I. Svergun, J. Appl. Crystallogr., 2009, 42, 342–346 CrossRef CAS PubMed.
- P. Gourdon, X. Y. Liu, T. Skjorringe, J. P. Morth, L. B. Moller, B. P. Pedersen and P. Nissen, Nature, 2011, 475, 59–64 CrossRef CAS PubMed.
- H. Reyes-Caballero, A. J. Guerra, F. E. Jacobsen, K. M. Kazmierczak, D. Cowart, U. M. Koppolu, R. A. Scott, M. E. Winkler and D. P. Giedroc, J. Mol. Biol., 2010, 403, 197–216 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04396a |
| This journal is © The Royal Society of Chemistry 2018 |