
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of macrocyclic ring systems by carbonylation of trifunctional P/B/B frustrated Lewis pairs†
Long
Wang
a,
Shunxi
Dong
a,
Constantin G.
Daniliuc
 a,
Lei
Liu
b,
Stefan
Grimme
b,
Robert
Knitsch
c,
Hellmut
Eckert
cd,
Michael Ryan
Hansen
c,
Gerald
Kehr
a and
Gerhard
Erker
*a
a,
Lei
Liu
b,
Stefan
Grimme
b,
Robert
Knitsch
c,
Hellmut
Eckert
cd,
Michael Ryan
Hansen
c,
Gerald
Kehr
a and
Gerhard
Erker
*a
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstr. 40, 48149 Münster, Germany. E-mail: erker@uni-muenster.de
bMulliken Center for Theoretical Chemistry, Institut für Physikalische und Theoretische Chemie, Universität Bonn, Beringstr. 4, 53115 Bonn, Germany
cInstitut für Physikalische Chemie, Westfälische Wilhelms-Universität Münster, Corrensstr. 28/30, 48149 Münster, Germany
dInstitute of Physics in Sao Carlos, University of Sao Paulo, CEP 369, Sao Carlos, SP 13566-590, Brazil
First published on 14th December 2017
Abstract
The trifunctional P/B/B frustrated Lewis pairs 11a–c featuring bulky aryl groups at phosphorus [Dmesp (a), Tipp (b), Mes* (c)] react with H2 by heterolytic hydrogen splitting followed by cleavage of HB(C6F5)2 to give the zwitterionic six-membered heterocyclic PH phosphonium/borate products 14a–c. Compounds 11a,b react with carbon monoxide by means of a Lewis acid induced CO insertion/rearrangement sequence that eventually results in the formation of the macrocyclic dimers 17a,b. The respective carbonylation reaction of the Mes*P/B/B FLP gives the macrocyclic trimer 18c. The new products were characterized spectroscopically and by X-ray diffraction and the reaction mechanism was analyzed by DFT calculations.
Introduction
Macrocyclic compounds have very interesting structural features.1,2 Many such systems play significant roles in medicine and biology3–7 and many serve as important chemical reagents.8–12 Macrocyclic ring closure is often difficult to achieve selectively since ring closure principally competes with entropically favoured formation of the linear oligomers. Chemical synthesis of macrocycles, therefore, has relied on a variety of specific measures in order to achieve the required chemoselectivity; high dilution is one important and often used principle13–15 as is template directed synthesis. Conformational features as well as electrostatic effects may play a role.16–19 There are some reactions that seem to bear a “natural” tendency for macrocyclic ring formation.20–28We have now found that Lewis pair formation might favor intermolecular cyclooligomerization in cases where the direct internal interaction of the Lewis acid and base functionalities is effectively precluded by specific geometric restrictions. We have found that this may selectively lead to cyclodimeric and even cyclotrimeric ring systems in a rather simple experimental procedure. First examples will be presented and discussed in this account.
Results and discussion
Alkyl boranes are important building blocks in organic synthesis. Many such systems are readily available by convenient hydroboration routes.29–31 Many alkyl boranes insert carbon monoxide into the boron–carbon bond. This reaction type has been used for the preparation of CO derived ketones, aldehydes or alcohols.32 Hydroboration of alkenes with Piers' borane [HB(C6F5)2] (2) occurs readily.33,34 The resulting products, such as the respective styrene (1) hydroboration compound PhCH2CH2B(C6F5)2 (3), however, do not readily insert CO at ambient conditions (r.t., 1.5 bar CO pressure, see Scheme 1, see the ESI† for details). CO insertion was not observed even in the presence of additional B(C6F5)3. The difference is even more pronounced with vicinal P/B frustrated Lewis pairs (FLPs), such as compound 6, which readily reacts with CO, but does not form the CO insertion product into the [B]–CH2– linkage but rather undergoes cooperative 1,1-P/B addition to the carbon atom of carbon monoxide to yield the CO-bridged product 7. A number of related P/B FLPs show a similar behavior.35–40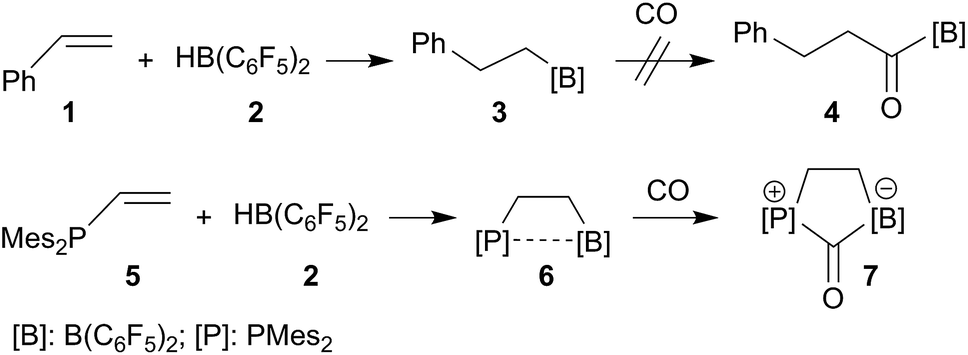 | ||
| Scheme 1 Behavior of strongly electrophilic pentafluorophenyl containing boranes toward carbon monoxide. | ||
We reasoned that this behavior might originate from the very special properties of the strong B(C6F5)2 Lewis acid unit making the alkyl migration step to carbon monoxide unfavorable. Introduction of a second B(C6F5)2 group into the system might potentially provide a way out of this behavior: specifically located it could function as an activator for the P/B bonded carbonyl unit and thus initiate the otherwise unfavorable CO insertion reaction in such systems. This turned out to be a successful concept and, in addition, it opened an easy pathway to several rather unusually structured macrocyclic ring systems. Three such examples with some of their remarkable characteristic features will be reported in this account.
We have prepared the aryldivinylphosphanes 8a–c by treatment of the respective ArPCl2 precursors41–47 with two molar equiv. of vinyl magnesium bromide. We had reported the reaction of compound 8c with one molar equiv. of HB(C6F5)2 which had given the unique zwitterionic methylene phosphonium product 10cvia internal B(C6F5)2 addition to the adjacent vinyl phosphane (see Scheme 2).48 Addition of a second equiv. of Piers' borane had given the P/B/B49a–e system 11c, for which we had observed a dynamic equilibrium of the P⋯B/B coordination by dynamic 19F NMR spectroscopy (see Scheme 2).49f We have now also generated the P/B/B systems 11a,b featuring the bulky 2,6-dimesitylphenyl (Dmesp) and 2,4,6-triisopropylphenyl (Tipp) aryl groups at phosphorus, respectively. Compound 11a also features an equilibrating dynamic structure in solution analogous to the previously described behavior of 11c. We had shown that the P/B/B system 11c splits dihydrogen in the presence of the external base tBu3P to give 12c.49f
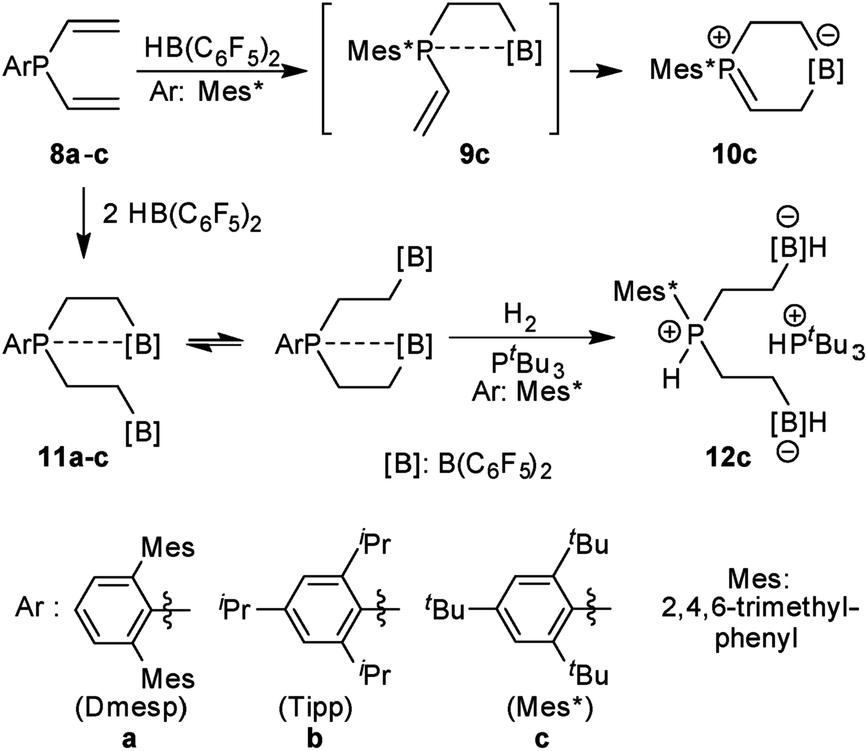 | ||
| Scheme 2 Formation and some previously reported reactions of the P/B/B FLP systems 11. | ||
We have now exposed the small series of P/B/B FLPs 11a–c to dihydrogen in the absence of the external base and found a markedly different behavior which indicated a surprising mode of participation of the extra –B(C6F5)2 Lewis acid. Typically, the in situ generated system 11a was exposed to a H2 atmosphere (1.5 bar) in dichloromethane solution for 30 min at r.t. to give a mixture of the zwitterionic heterocyclic phosphonium/borate product 14a (Ar: Dmesp) and HB(C6F5)2. The latter was removed from the mixture by the hydroboration reaction with 1-pentene converting it to pentane soluble pentyl-B(C6F5)2. It was isolated and identified as its pyridine adduct 15py (for details see the ESI†). The heterocycle 14a was eventually isolated as a white solid in 78% yield. Its X-ray crystal structure analysis (see Fig. 1) showed the presence of the chair-shaped 1,4-P/B heterocycle with tetracoordinated boron and the PH(Dmesp) phosphonium unit being part of it. In solution, compound 14a shows a typical borate 11B NMR feature at δ −14.6 and a phosphonium 31P NMR signal at δ 9.0 (1H: δ 5.56, 1JPH = 463.0 Hz). The 13C NMR spectrum shows signals of the six-membered core unit at δ 19.0 (PCH2, 1JPC = 43.1 Hz) and δ 20.8 (broad, BCH2), respectively (see Scheme 3, also see the ESI† for details).
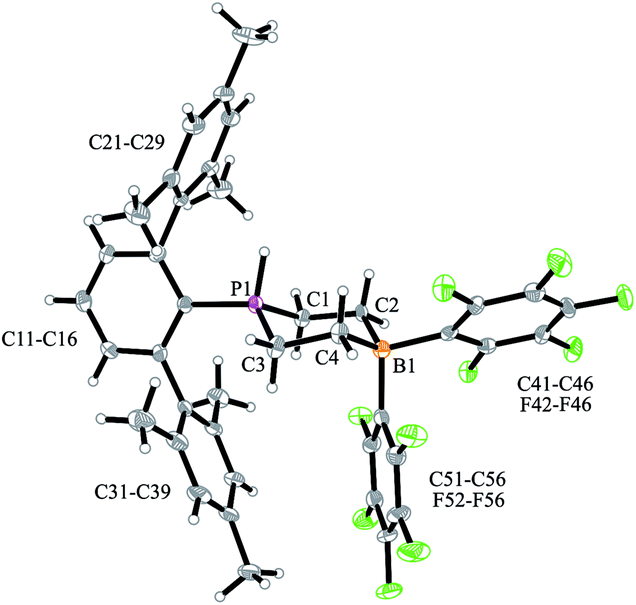 | ||
| Fig. 1 A view of the molecular structure of the P/B/B hydrogenation product 14a (thermal ellipsoids are shown with 50% probability). Selected bond lengths (Å) and angles (°): P1–C1 1.786(3), P1–C3 1.786(3), C1–C2 1.540(4), C3–C4 1.546(5), C2–B1 1.648(5), C4–B1 1.652(5), C1–P1–C3 106.7(2), C2–B1–C4 107.5(3), ΣP1CCC 339.2(5). | ||
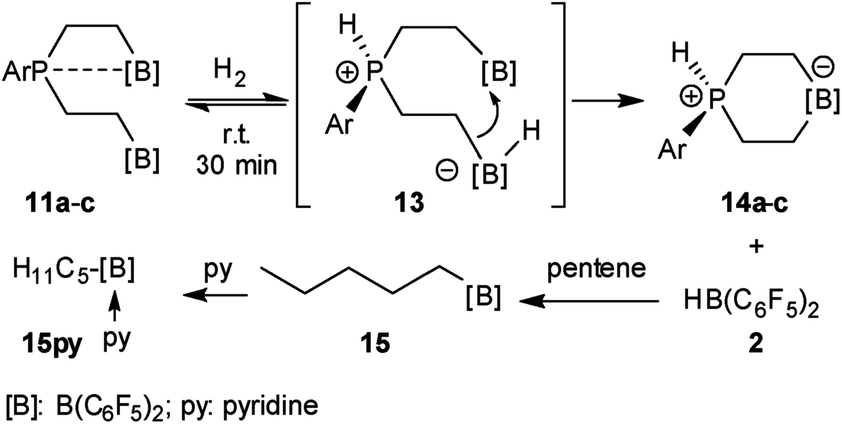 | ||
| Scheme 3 Reaction of the P/B/B FLPs 11 with dihydrogen. | ||
The P/B/B compounds 11b,c reacted analogously with dihydrogen with formation of the PH/B products 14b,c. We isolated them both in ca. 70% yield; both were characterized by spectroscopy and by X-ray diffraction (see the ESI† for details). We assume a reaction pathway (see Scheme 3) that is initiated by heterolytic splitting of dihydrogen by using a P/B pair50–58 of the P/B/B FLP 11 to give the PH+/BH−/B intermediates 13. We assume that the additional –B(C6F5)2 Lewis acid becomes actively involved and forms the six-membered P/B heterocycles 14 by a σ-bond metathesis type reaction59–61 with concomitant formation of HB(C6F5)2. The formation of the products 14 and HB(C6F5)2 (2) gave us a strong indication of the active role of the additional-B(C6F5)2 Lewis acid in the compounds 11. This we used advantageously in the reaction of the P/B/B FLPs 11a–c with carbon monoxide.
We generated the P/B/B system 11b (Ar: Tipp) in situ (24 h, r.t., see Scheme 2) and then exposed the solution to a carbon monoxide atmosphere (1.5 bar, r.t.). After 30 min reaction time a white precipitate of the cyclodimeric CO insertion product 17b had formed. It was isolated as a white solid in 54% yield (for details see the ESI†). Compound 17b is thermally quite stable in solution. However, it lost carbon monoxide upon heating for 6 h at 80 °C in benzene-d6 solution to re-form the starting material 11b.
We assume a reaction pathway as it is depicted in Scheme 4, which was supported by the results of DFT calculations69 (for details see the ESI†). All structures were optimized with a composite DFT method PBEh-3c,69 followed by single point energy calculations at the PW6B95-D3 level of theory65,67,68 with a Gaussian AO def2-TZVP basis set.64,66 The COSMO-RS (conductor-like screening model for real solvents) solvation model62,63 (with toluene as the solvent) was used to compute solvation free energies. Endergonic opening of the P⋯B linkage of 11b yields the reactive P/B/B intermediate 11open, which may undergo the typical 1,1-P/B FLP addition reaction to carbon monoxide.35,36 Carbonyl activation by the remaining pendent –B(C6F5)2 functionality70,71via16A might initiate the kinetically facile and thermodynamically feasible formation of the CO insertion product3216B. Isomerization forms the P/B Lewis pair. In 16C the direct internal interaction of the carbonyl oxygen with the pendent borane Lewis acid is geometrically precluded; the system may serve as an active C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/B frustrated Lewis pair. This leads to dimerization giving the observed macrocyclic reaction product 17b.
O/B frustrated Lewis pair. This leads to dimerization giving the observed macrocyclic reaction product 17b.
 | ||
| Scheme 4 Carbonylation reaction of the P/B/B FLPs 11a, with computed reaction Gibbs free energies at PW6B95-D3/def2-TZVP (COSMO-RS, toluene)//PBEh-3c level of the theory. The energy of 11b + CO is selected as the reference and all values are given in kcal mol−1 at 298 K. The value of the dimer 17b refers to the monomeric subunit for the purpose of comparability. | ||
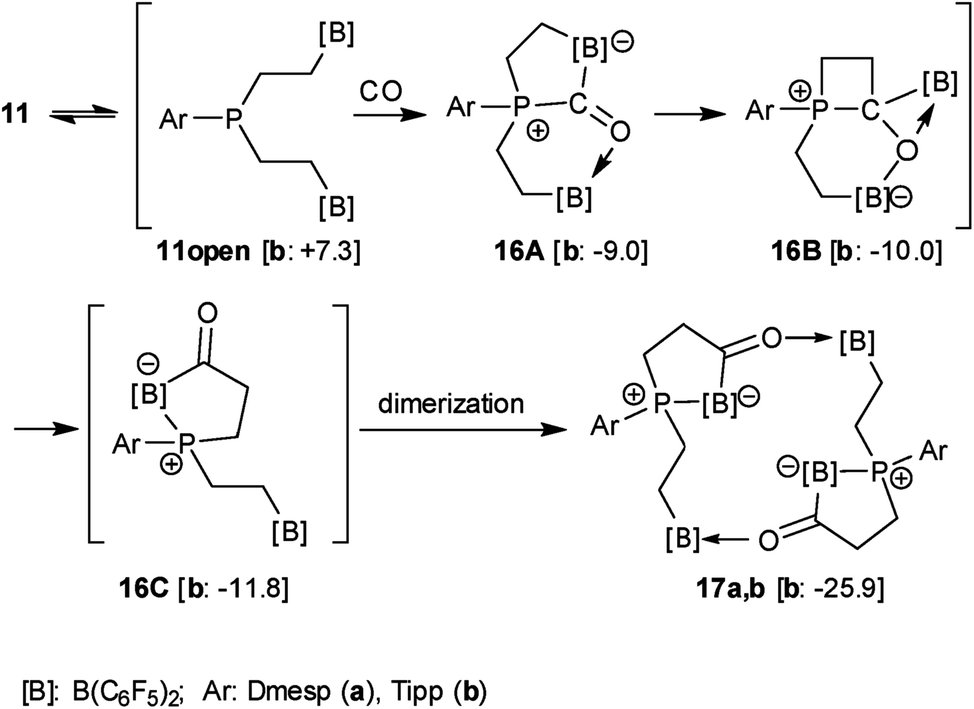
The X-ray crystal structure analysis confirmed the sixteen-membered cyclodimeric heterocyclic ring structure. The monomeric units are connected by a pair of carbonyl CO···borane interactions. The remaining boron atoms form Lewis pair interactions with their adjacent phosphane Lewis bases. In this situation two diastereoisomers are possible due to the phosphorus chirality;72,73 we found the near to C2-symmetric rac-structure in the crystal (see Fig. 2, also see the ESI† for details).
 | ||
| Fig. 2 A view of the structure of the framework of macrocyclic dimer 17b (the bulky substituents at boron and phosphorus were omitted for clarity; thermal ellipsoids are shown with 50% probability). Selected bond lengths (Å) and angles (°): P1–B1 2.063(2), B1–C3A 1.632(3), C3A–O1 1.254(2), O1–B4 1.609(2), P2–B3 2.067(2), B3–C3B 1.648(3), C3B–O2 1.255(2), O2–B2 1.618(2), B1–C3A–O1 120.8(2), C3A–O1–B4 129.6(1), B3–C3B–O2 122.7(2), C3B–O2–B2 130.8(1). | ||
In solution, compound 17b shows the NMR features of the symmetry-equivalent monomeric subunits. We monitored eight separate 1H NMR sp3-CH signals of the methylene groups, the 19F NMR signals of four different C6F5 substituents at boron and a single 31P NMR signal at δ 21.0. From a 13C labelled sample we located the 13C NMR carbonyl resonance at δ 269.0.70,71 Compound 17b shows a ![[small nu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e1dd.gif) (CO) = 1588 cm−1 [(13CO) = 1547 cm−1] carbonyl stretching band.
(CO) = 1588 cm−1 [(13CO) = 1547 cm−1] carbonyl stretching band.
The Dmesp substituted P/B/B system 11a reacts analogously with CO. We isolated the C2-symmetrical dimer 17a in 61% yield and characterized it by C,H-elemental analysis, by NMR (10B: δ 7.8, −10.0; 31P: δ 19.1) and IR spectroscopy ((CO) = 1579 cm−1) and by X-ray diffraction (for details see the ESI†). It is thermally slightly less stable than 17b. Compound 17a lost CO upon heating to 50 °C (12 h) in dichloromethane solution to give the starting material 11a.
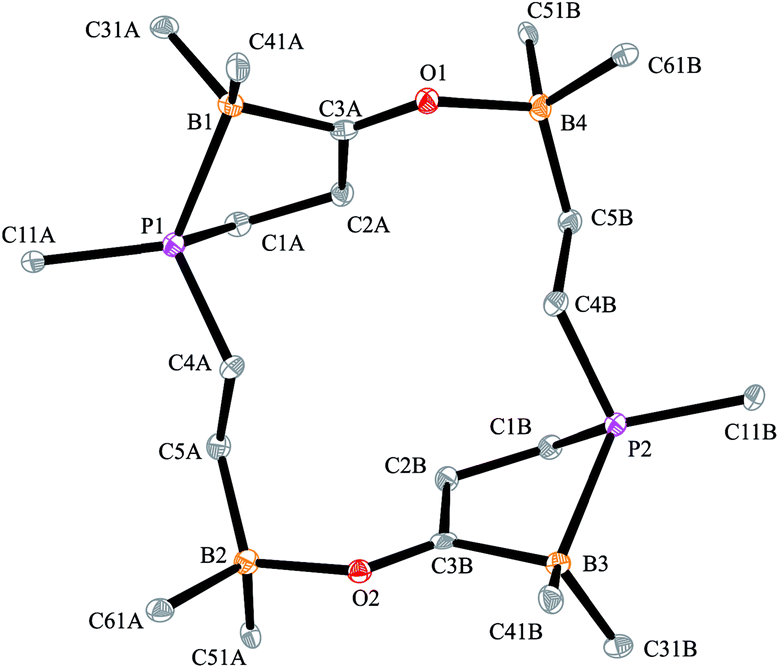
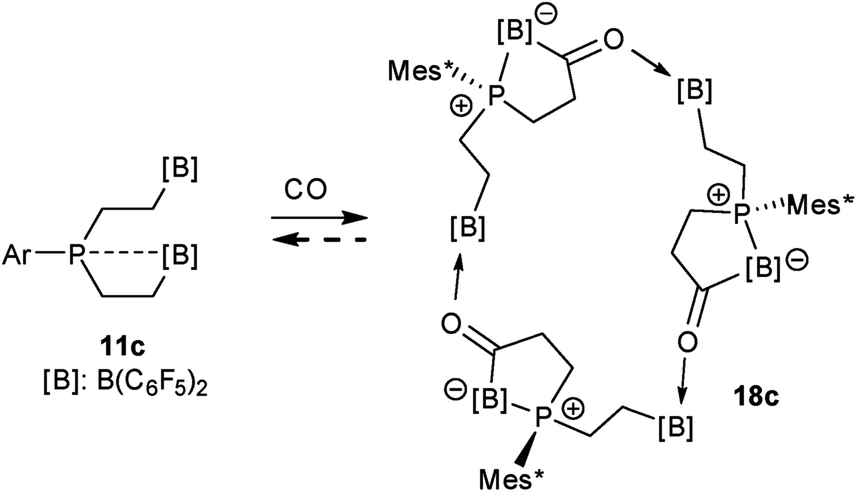
The carbonylation reaction of the Mes*P/B/B system 11c took a slightly different course. Exposure of the in situ generated Mes*P/B/B system 11c to CO (1.5 bar, r.t. 30 min) gave compound 18c (81% isolated) (see Scheme 5, for details see the ESI†). The compound was stable in the solid state but lost carbon monoxide with re-formation of the starting material 11c in solution (CD2Cl2). Therefore, the solution NMR data were monitored using in situ generated samples at low temperature (for details see the ESI†). Single crystals of compound 18c for the X-ray crystal structure analysis were obtained from a toluene solution in a carbon monoxide atmosphere at −5 °C. Compound 18c shows a macrocyclic twenty four-membered core structure. It is composed of three monomeric subunits that were probably formed by a CO insertion/rearrangement sequence analogous to the one described in Scheme 4; this was supported by DFT calculations (for details see the ESI†). The presence of the three phosphorus chirality centers would principally allow for two diastereoisomers, an all-cis-(of averaged C3-symmetry) and a cis-, trans-, trans-isomer. The latter structural situation is found in the crystal of compound 18c. Each of the three symmetry inequivalent but chemically closely related subunits features a five-membered P/B containing heterocyclic carbonyl moiety. The CO group is used for bridging to the pendent –B(C6F5)2 Lewis acid of the next monomeric subunit (see Fig. 3).
 | ||
| Scheme 5 Formation of the cyclotrimeric carbonylation product 18c. | ||
 | ||
| Fig. 3 A view of the core structure of the macrocyclic P/B/B carbonylation trimer 18c (the substituents at boron and phosphorus are omitted for clarity; thermal ellipsoids are shown with 15% probability). Selected bond lengths (Å) and angles (°): P1–B1 2.096(5), C3A–O1 1.253(5), O1–B6 1.590(6), P3–B5 2.110(5), C3C–O3 1.245(5), O3–B4 1.603(6), P2–B3 2.094(5), C3B–O2 1.239(5), O2–B2 1.625(6), B1–C3A–O1 117.7(4), C3A–O1–B6 133.7(3), B5–C3C–O3 119.6(4), C3C–O3–B4 133.4(3), B3–C3B–O2 118.3(4), C3B–O2–B2 131.9(4). | ||
Arylphosphanes usually have the C(aryl)-P vector oriented in line with the aryl plane. Very bulky arylphosphanes may deviate from this behavior (which may be expressed by the P1–C11–C14 angle as schematically shown in Fig. 4 for one of the three Mes*-P units of the trimer 18c). We note an almost co-linear arrangement for the pair of Tipp-P units in the dimer 17b. The respective P1–C11–C14 angles for the pair of crystallographically independent Tipp-P subunits were found at 174.4° and 173.1°. We find a slightly bent structure for the more bulky Dmesp-P groups in compound 17a with P1–C11–C14 angles of 171.0° and 166.2°, respectively, but we note a rather extreme bending of the Mes*-P moiety. In the Mes*-PCl2 reagent the P1–C11–C14 type angle amounts to 156°.46 In the three Mes*-P subunits in our trimer 18c we find this distortion of the (aryl)C–P moieties being increased further by ca. 10° to P1–C11–C14 values of 144.1°, 147.9°, and 146.1°, respectively. Fig. 4 shows a side-on projection of one out of the three Mes*-P moieties of compound 18c, which visualizes this strong distortional effect. This might actually have created an overall conformational situation that may have contributed to determining the observed chemistry of this system to a considerable extent, especially the specific association behaviour of the monomeric subunits forming the observed cyclotrimer 18c.
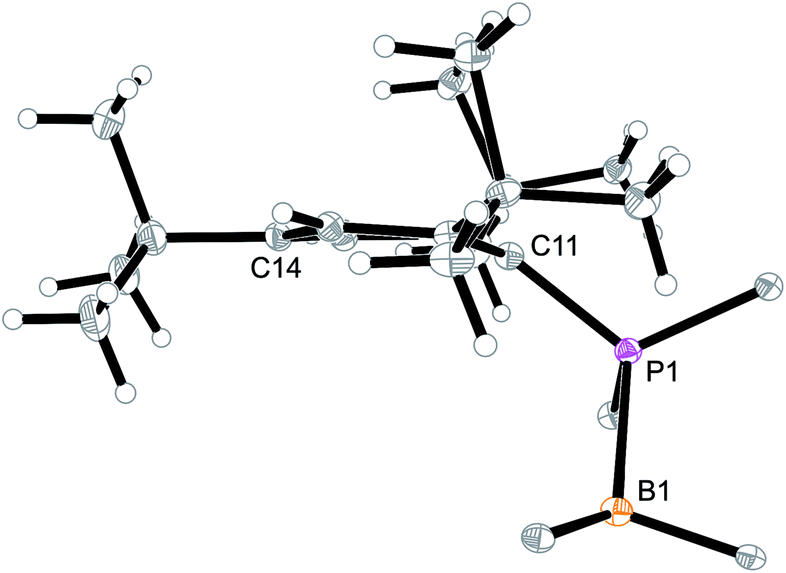 | ||
| Fig. 4 Side view of a 2,4,6-tri-tbutylphenyl-P (i.e. Mes*-P) unit of the macrocyclic trimer 18c. The P1–C11–C14 angle of this unit amounts to 144.1°. | ||
The solid-state 31P MAS NMR spectra confirmed the asymmetric (C1) structure of the cyclotrimer 18c containing three different phosphorus atoms. Consequently, three 31P NMR signals at 12, 13 and 15 ppm were observed which are broadened and additionally split by the indirect 31P–11B spin–spin coupling (1J(31P–11B) ∼ 80 Hz). As illustrated in Fig. 5(a), simultaneous 11B and 1H decoupling enhances the resolution (see Fig. 5, left, for further details including the 11B{31P} REDOR and 2D-INEPT experiments see the ESI†). Compound 18c also showed three equal intensity 31P NMR resonances in solution (Fig. 5, right). It showed 22 different 1H NMR signals (two with relative intensity two, all others with intensity one at 243 K) of the 12 pairs of diastereotopic CH2 hydrogen atoms as well as 12 methylene 13C NMR signals of the core ring carbons. There are nine separate 1H NMR t-Bu singlets and the 19F NMR signals of 12 C6F5 groups at the boron atoms of compound 18c. The 13CO derived isotopologue showed three 13C NMR carbonyl signals [δ 273.3 (d, 2JPC = 23.6 Hz), 272.0 (d, 2JPC = 29.9 Hz) and 271.9 (d, 2JPC = 29.9 Hz)] (for details see the ESI†).
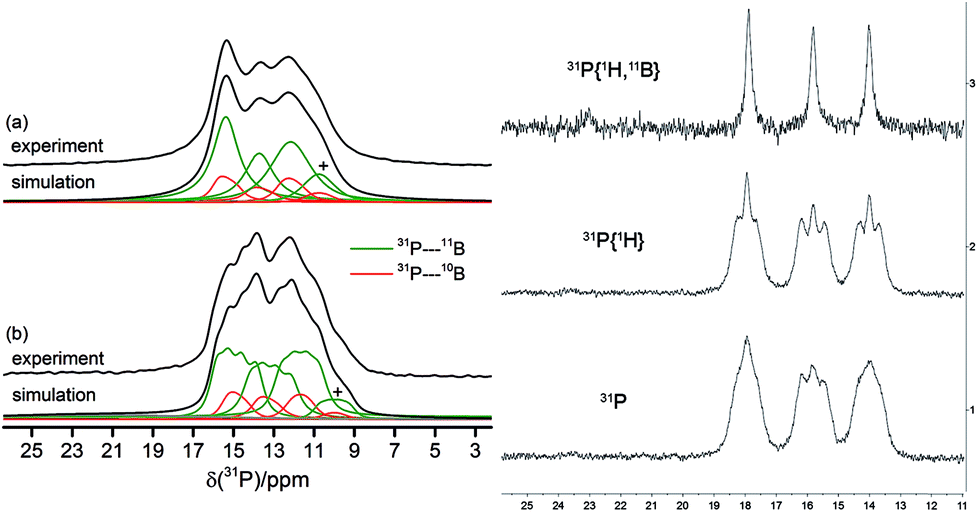 | ||
| Fig. 5 31P MAS NMR spectra obtained for the C1-symmetric macrocyclic trimer 18c. Left: (a) 1H → 31P{1H, 11B} CP/MAS NMR spectrum of 18c, (b) 31P{1H} MAS NMR spectrum and corresponding simulations, on the indirect 31P–11B and/or 31P–10B spin–spin interactions. The symbol + marks a suspected impurity. Right: 31P, 31P{1H}, and 31P{1H, 11B} NMR spectra of compound 18c in solution (CD2Cl2, 203 K). | ||
Conclusions
It seems that the presence of the additional B(C6F5)2 Lewis acid function influences the reaction of the internal ethylene-bridged P/B FLP functionality of the compounds 11 in two decisive ways: activation of the carbonyl group at the stage of the conventional cooperative P/B CO addition intermediate 16A35,36 by the reactive boron Lewis acid probably makes the CO insertion reaction into the adjacent –CH2–B(C6F5)2 group feasible. Our DFT analysis of the CO insertion step of the Mes*P/B/B system revealed an exergonic (ΔG ca. −4 kcal mol−1) formation of the intermediate 16Bc (the intermediate analogous to 16Bb in the Tipp system shown in Scheme 4). In contrast the hypothetical CO insertion reaction from compound 7 (see Scheme 1) was computed by the DFT calculations as markedly endergonic [ΔG ca. +9 kcal mol−1, rel. ΔG(7) = 0]. Once the carbonyl compound is formed by C–C bond formation it is prone to rearrangement generating a monomeric intermediate 16C featuring both an organic carbonyl function and a remote free –B(C6F5)2 Lewis acid, a combination which paves the way to formation of the unique macrocyclic oligomers 17 and 18 by Lewis adduct formation between these pairs of functional groups.Why are the macrocyclic dimers and even a cyclotrimer formed in our examples instead of the alternative linear oligomers? Actually, we do not know for sure, but we may speculate that this has to do with the special properties encountered in phosphane/borane frustrated Lewis pair chemistry. This chemistry is governed by van der Waals interactions between the bulky protagonists and it becomes increasingly apparent that conformational features strongly determine frustrated Lewis pair behavior.35,36,50 In our case it might be a combination of both factors that serves to tip the balance toward cyclooligomer formation. The conformational influence is probably indicated by the different behavior of the (Dmesp)P and (Tipp)P containing FLP pairs 11a,bvs. the Mes*P derived system 11c in the carbonylation/cyclooligomerization reaction. The former systems feature rather normal steric features of the bulky aryl-P linkage, whereas the latter shows the special conformational feature of the uncommon strongly bent P-aryl moiety.46 Our DFT analysis points to an energetic difference in the formation of the observed dimer (17b) in the Tipp substituted system vs. the cyclotrimer (18c) in the case of the Mes* containing system: in the Tipp containing system we find an energetic preference of the formation of the cyclodimer of ca. 5 kcal mol−1 over the trimer, whereas in the case of the more bulky Mes* system this becomes reversed and the cyclotrimer is favored by ca. 10 kcal mol−1 over the dimer (see the ESI† for details). The favored formation of the unusual macrocyclic trimer 18c might indeed point to a marked influence of specific conformational features introduced by the very bulky aryl Mes* substituent into this chemistry.
The formation of the macrocyclic dimers and trimers from our carbonylated P/B/B FLP systems may place some frustrated Lewis pair reactions into the group of macrocyclic ring closure procedures that show a “natural” tendency of favoring the internal bond formation in cases of a suitable general design.20–26 This unique behavior of the carbonylation chemistry of the P/B/B systems 11 emphasizes the potential that frustrated Lewis pair chemistry has for discovering surprisingly facile pathways to unusual products formed under mild reaction conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council and the Deutsche Forschungsgemeinschaft (SFB 858; Leibniz Award to S. G.) is gratefully acknowledgment.Notes and references
- K. Gloe, Macrocyclic Chemistry: Current Trends and Future Perspective, Springer, 2005 Search PubMed.
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961–2004 CrossRef CAS PubMed.
- J. Mallinson and I. Collins, Future Med. Chem., 2012, 4, 1409–1438 CrossRef CAS PubMed.
- E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco, D. Kozakov, S. Vajda and A. Whitty, Nat. Chem. Biol., 2014, 10, 723–731 CrossRef CAS PubMed.
- S. E. Allen, N. V. Dokholyan and A. A. Bowers, ACS Chem. Biol., 2016, 11, 10–24 CrossRef CAS PubMed.
- R. M. Kellogg, Angew. Chem., Int. Ed. Engl., 1984, 23, 782–794 CrossRef.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef.
- F. Davis and S. Higson, Macrocycles: Construction, Chemistry and Nanotechnology Applications, Wiley-VCH Verlag GmbH, 2011 Search PubMed.
- J. Li, D. Yim, W.-D. Jang and J. Yoon, Chem. Soc. Rev., 2017, 46, 2437–2458 RSC.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- L. Rossa and F. Vögtle, Top. Curr. Chem., 1983, 113, 1–86 CrossRef CAS.
- S. J. Rodgers, C. Y. Ng and K. N. Raymond, J. Am. Chem. Soc., 1985, 107, 4094–4095 CrossRef CAS.
- M. Malesevic, U. Strijowski, D. Bächle and N. Sewald, J. Biotechnol., 2004, 112, 73–77 CrossRef CAS PubMed.
- A. D. Cort, G. Ercolani, L. Mandolini and P. Mencarelli, J. Chem. Soc., Chem. Commun., 1993, 538–540 RSC.
- N. V. Gerbeleu, V. B. Arion and J. P. Burgess, Template Synthesis of Macrocyclic Compounds, Wiley-VCH Verlag GmbH, 1999 Search PubMed.
- J. C. Collins and K. James, Med. Chem. Commun., 2012, 3, 1489–1495 RSC.
- V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef PubMed.
- A. Fürstner and K. Langemann, J. Org. Chem., 1996, 61, 3942–3943 CrossRef.
- A. Fürstner, O. R. Thiel and G. Blanda, Org. Lett., 2000, 2, 3731–3734 CrossRef.
- A. Fürstner, O. R. Thiel and L. Ackermann, Org. Lett., 2001, 3, 449–451 CrossRef.
- C. W. Lee and R. H. Grubbs, J. Org. Chem., 2001, 66, 7155–7158 CrossRef CAS PubMed.
- A. Gradillas and J. Pérez-Castells, Angew. Chem., Int. Ed., 2006, 45, 6086–6101 CrossRef CAS PubMed.
- V. M. Marx, M. B. Herbert, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 94–97 CrossRef CAS PubMed.
- X. Shen, T. T. Nguyen, M. J. Koh, D. Xu, A. W. H. Speed, R. R. Schrock and A. H. Hoveyda, Nature, 2017, 541, 380–385 CrossRef CAS PubMed.
- W. Zhang and J. S. Moore, Angew. Chem., Int. Ed., 2006, 45, 4416–4439 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- H. C. Brown, Tetrahedron, 1961, 12, 117–138 CrossRef CAS.
- H. C. Brown and B. Singaram, Pure Appl. Chem., 1987, 59, 879–894 CAS.
- H. C. Brown and P. V. Ramachandran, Pure Appl. Chem., 1991, 63, 307–316 CrossRef CAS.
- H. C. Brown, Acc. Chem. Res., 1969, 2, 65–72 CrossRef CAS . See also: G. S. Hair, R. A. Jones, A. H. Cowley and V. Lynch, Organometallics, 2001, 20, 177–181 CrossRef.
- D. J. Parks, R. E. v. H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809–811 CrossRef CAS.
- D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492–5503 CrossRef CAS.
- M. Sajid, A. Lawzer, W. Dong, C. Rosorius, W. Sander, B. Schirmer, S. Grimme, C. G. Daniliuc, G. Kehr and G. Erker, J. Am. Chem. Soc., 2013, 135, 18567–18574 CrossRef CAS PubMed.
- L.-M. Elmer, G. Kehr, C. G. Daniliuc, M. Siedow, H. Eckert, M. Tesch, A. Studer, K. Williams, T. H. Warren and G. Erker, Chem.–Eur. J., 2017, 23, 6056–6068 CrossRef CAS PubMed.
- A. J. P. Cardenas, B. J. Culotta, T. H. Warren, S. Grimme, A. Stute, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2011, 50, 7567–7571 CrossRef CAS PubMed.
- M. Sajid, A. Stute, A. J. P. Cardenas, B. J. Culotta, J. A. M. Hepperle, T. H. Warren, B. Schirmer, S. Grimme, A. Studer, C. G. Daniliuc, R. Fröhlich, J. L. Petersen, G. Kehr and G. Erker, J. Am. Chem. Soc., 2012, 134, 10156–10168 CrossRef CAS PubMed.
- O. Ekkert, G. G. Miera, T. Wiegand, H. Eckert, B. Schirmer, J. L. Petersen, C. G. Daniliuc, R. Frohlich, S. Grimme, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 2657–2664 RSC.
- L. Wang, K. Samigullin, M. Wagner, A. C. McQuilken, T. H. Warren, C. G. Daniliuc, G. Kehr and G. Erker, Chem.–Eur. J., 2016, 22, 11015–11021 CrossRef CAS PubMed.
- A. Saednya and H. Hart, Synthesis, 1996, 1455–1458 CrossRef CAS.
- V. Chandrasekhar, P. Sasikumar, R. Boomishankar and G. Anantharaman, Inorg. Chem., 2006, 45, 3344–3351 CrossRef CAS PubMed.
- D. G. Yakhvarov, E. Hey-Hawkins, R. M. Kagirov, Y. H. Budnikova, Y. S. Ganushevich and O. G. Sinyashin, Russ. Chem. Bull., 2007, 56, 935–942 CrossRef CAS.
- C. Overländer, J. J. Tirrée, M. Nieger, E. Niecke, C. Moser, S. Spirk and R. Pietschnig, Appl. Organomet. Chem., 2007, 21, 46–48 CrossRef.
- E. Rivard and P. P. Power, Inorg. Chem., 2007, 46, 10047–10064 CrossRef CAS PubMed.
- S. Ito, H. Miyake and M. Yoshifuji, Phosphorus, Sulfur, and Silicon and the Related Elements, 2009, 184, 917–927 CrossRef CAS.
- J. Möbus, Q. Bonnin, K. Ueda, R. Fröhlich, K. Itami, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2012, 51, 1954–1957 CrossRef PubMed.
- Y. Hasegawa, G. Kehr, S. Ehrlich, S. Grimme, C. G. Daniliuc and G. Erker, Chem. Sci., 2014, 5, 797–803 RSC.
- (a) X. Zhao, A. J. Lough and D. W. Stephan, Chem.–Eur. J., 2011, 17, 6731–6743 CrossRef CAS PubMed; (b) X. Zhao and D. W. Stephan, Chem. Commun., 2011, 47, 1833–1835 RSC; (c) R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 4974–4977 CrossRef CAS PubMed; (d) E. Theuergarten, J. Schlosser, D. Schluns, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2012, 41, 9101–9110 RSC; (e) J. Ugolotti, G. Kehr, C. G. Daniliuc and G. Erker, Synthesis, 2017, 49, 53–58 CAS; (f) L. Wang, S. Zhang, Y. Hasegawa, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2017, 53, 5499–5502 RSC.
- P. Spies, G. Erker, G. Kehr, K. Bergander, R. Frohlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072–5074 RSC.
- D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 332, 1–350 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 334, 1–317 CrossRef.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- K. V. Axenov, C. M. Mömming, G. Kehr, R. Fröhlich and G. Erker, Chem.–Eur. J., 2010, 16, 14069–14073 CrossRef CAS PubMed.
- K. Chernichenko, Á. Madarász, I. Pápai, M. Nieger, M. Leskelä and T. Repo, Nat. Chem., 2013, 5, 718–723 CrossRef CAS PubMed.
- M. Sajid, G. Kehr, T. Wiegand, H. Eckert, C. Schwickert, R. Pöttgen, A. J. P. Cardenas, T. H. Warren, R. Fröhlich, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 8882–8895 CrossRef CAS PubMed.
- G.-Q. Chen, G. Kehr, C. G. Daniliuc, C. Mück-Lichtenfeld and G. Erker, Angew. Chem., Int. Ed., 2016, 55, 5526–5530 CrossRef CAS PubMed.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS PubMed.
- R. Waterman, Organometallics, 2013, 32, 7249–7263 CrossRef CAS.
- Y. Wang, W. Chen, Z. Lu, Z. H. Li and H. Wang, Angew. Chem., Int. Ed., 2013, 52, 7496–7499 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. Hansen, J. Chem. Phys., 2015, 143, 054107 CrossRef PubMed.
- H. Jacobsen, H. Berke, S. Döring, G. Kehr, G. Erker, R. Fröhlich and O. Meyer, Organometallics, 1999, 18, 1724–1735 CrossRef CAS.
- D. Vagedes, R. Fröhlich and G. Erker, Angew. Chem., Int. Ed., 1999, 38, 3362–3365 CrossRef CAS PubMed.
- M. Dutartre, J. Bayardon and S. Juge, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC.
- J. P. Verkade and L. D. Qinn, Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis, Organic Compounds and Metal Complexes, Wiley-VCH Verlag GmbH, 1987 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: additional experimental details, further spectral and crystallographic data, additional data from the solid state NMR and theoretical studies. CCDC 1549697–1549702. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04394e |
| This journal is © The Royal Society of Chemistry 2018 |