
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total chemical synthesis of glycocin F and analogues: S-glycosylation confers improved antimicrobial activity†
Zaid
Amso
a,
Sean W.
Bisset
bc,
Sung-Hyun
Yang
a,
Paul W. R.
Harris
 acd,
Tom H.
Wright
a,
Claudio D.
Navo
e,
Mark L.
Patchett
b,
Gillian E.
Norris
bc and
Margaret A.
Brimble
*acd
acd,
Tom H.
Wright
a,
Claudio D.
Navo
e,
Mark L.
Patchett
b,
Gillian E.
Norris
bc and
Margaret A.
Brimble
*acd
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds St, Auckland 1142, New Zealand. E-mail: m.brimble@auckland.ac.nz; Fax: +64 9 3737422; Tel: +64 9 3737599
bInstitute of Fundamental Sciences, Massey University, Colombo Rd, Palmerston North 4442, New Zealand
cMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
dSchool of Biological Sciences, The University of Auckland, 3 Symonds St, Auckland 1142, New Zealand
eDept. Química, Universidad de La Rioja, Centro de Investigación en Síntesis Química, E-26006 Logroño, Spain
First published on 12th January 2018
Abstract
Glycocin F (GccF) is a unique diglycosylated bacteriocin peptide that possesses potent and reversible bacteriostatic activity against a range of Gram-positive bacteria. GccF is a rare example of a ‘glycoactive’ bacteriocin, with both the O-linked N-acetylglucosamine (GlcNAc) and the unusual S-linked GlcNAc moiety important for antibacterial activity. In this report, glycocin F was successfully prepared using a native chemical ligation strategy and folded into its native structure. The chemically synthesised glycocin appeared to be slightly more active than the recombinant material produced from Lactobacillus plantarum. A second-generation synthetic strategy was used to prepare 2 site selective ‘glyco-mutants’ containing either two S-linked or two O-linked GlcNAc moieties; these mutants were used to probe the contribution of each type of glycosidic linkage to bacteriostatic activity. Replacing the S-linked GlcNAc at residue 43 with an O-linked GlcNAc decreased the antibacterial activity, while replacing O-linked GlcNAc at position 18 with an S-linked GlcNAc increased the bioactivity suggesting that the S-glycosidic linkage may offer a biologically-inspired route towards more active bacteriocins.
Introduction
Microorganisms produce a highly diverse array of compounds that can be harnessed as anti-bacterial agents.1 Bacteriocins are ribosomally synthesized and post-translationally modified peptides (RiPPs) secreted by bacteria as a primary defence mechanism against competing bacteria.2 Bacteriocins from lactic acid bacteria have received considerable attention due to a desirable safety profile, stability across wide pH and temperature ranges, antimicrobial activity against various pathogens, and no cross-resistance with antibiotics.3–5 These features have prompted interest in bacteriocins as novel antimicrobial agents, probiotics and natural preservatives.4Glycocin F (GccF, 1) is a potent bacteriocin originally isolated from liquid culture of Lactobacillus plantarum KW30.6 GccF is a 43-residue helical peptide that contains two interlocked disulfide bonds (5Cys–28Cys and 12Cys–21Cys) and two β-linked N-acetyl-D-glucosamine (GlcNAc) moieties, connected to the side chain of 18Ser via the oxygen atom and 43Cys via the sulfur atom.7 The glycosylated cysteine, in particular, is an extremely rare post-translational modification in bacteria and has only been found in two other glycopeptides to date (sublancin and thurandacin).8–10 GccF exhibits bacteriostatic activity against a relatively wide range of Gram positive bacteria, with L. plantarum strains suspected to be its natural target.6,7 Notably, ASM1, a close homologue of GccF, and enterocin 96 are the only glycosylated bacteriocins which have been shown to be “glycoactive”,11–13 that is, the saccharide moieties are essential for biological activity. In a previous study, removal of the GlcNAc attached to 18Ser completely abolished the activity, while removal of the C-terminal fragment 42His–43Cys(β-GlcNAc) reduced the activity 44-fold.7 The C-terminal S-glycosidic linkage is of particular interest, as the natural functions of such bonds remain unclear. Further, the improved biochemical stability of S-glycoside linkages14 compared to their O-linked congeners provides considerable scope for the development of S-glycosylated bacteriocins as both preservatives and therapeutics.
Since the discovery of GccF (1), attempts to dissect its molecular mechanism and cellular targets have been hampered by the inefficiency of its isolation from cultures of L. plantarum. Chemical synthesis of complex bacteriocin glycopeptides is an attractive alternative to direct isolation or recombinant production as it offers atomic-level control over peptide sequence and modifications. However, few total syntheses of glycosylated bacteriocins have been reported to date. An example is the synthesis of 37-residue sublancin 168 by Payne et al.9via solid-phase peptide synthesis coupled with a convergent ligation methodology. Two other glycosylated bacteriocins thurandacin and enterocin 96 have been prepared using chemoenzymatic methods.8,13
We have previously reported11 the first synthesis of a biologically active form of GccF, identical in structure to the natural product except for C-terminal amidation. The synthetic C-terminal amide analogue of GccF exhibited a reduced bacteriostatic activity compared to the natural peptide derived from L. plantarum culture.11 We therefore sought to obtain the native GccF that contains the C-terminal acid to determine the role of the C-terminal carboxylate. We then sought to extend this further by chemically synthesizing ‘glyco-mutants’ of GccF to examine the chemical linkage between the peptide backbone and the GlcNAc. Our hypothesis is that replacing O-linked sugars with S-linked sugars would not greatly perturb the structural requirements of GccF, thereby preserving the bioactivity but may also result in enhanced enzymatic stability.14
Herein, the native form of glycocin F (1) was prepared and improvements on its chemical synthesis are reported. Notably, the synthetic GccF is equipotent to the isolated material in a liquid culture assay. Furthermore, several ‘glyco-mutants’ of GccF were generated, in which both of the two native glycosylation sites (18Ser and 43Cys) were substituted with S-linked β-GlcNAc (Cys(β-GlcNAc)) or O-linked β-GlcNAc (Ser(β-GlcNAc)) residues. These analogues have enabled us to better understand the contributions of each sugar linkage to the bacteriostatic activity of glycocin F and reveal the considerable potential of S-linked glycoconjugates as antimicrobial agents.
Results and discussion
Synthesis of glycocin F (1)
The synthetic strategy to prepare native glycocin F (1) was analogous to that described for the previous synthesis of C-terminally amidated glycocin F.11 In that report, we adopted a three fragment native chemical ligation (NCL) strategy as depicted in Scheme 1. In the present work we employed the 2-chlorotrityl linker for the preparation of the racemisation-prone C-terminal acid fragment 2, bearing a C-terminal cysteine (ESI Fig. 14 and Scheme 5†). The use of benzyl alcohol-based linkers was avoided as the esterification conditions for loading a cysteine residue to such linkers can lead to Cα racemisation and/or dehydroalanine by-product formation.15 2-Chlorotrityl based resins does not involve carboxylate activation and thus does not result in racemisation. In our previous synthesis of glycocin F-amide, the 4-(4-hydroxymethyl-3-methoxyphenoxy)-butyric acid (HMPB) linker was used to prepare fragment 3, causing substantial racemisation at 27His; pleasingly changing the linker to 2-chlorotrityl linker reduced this to ca. 20%.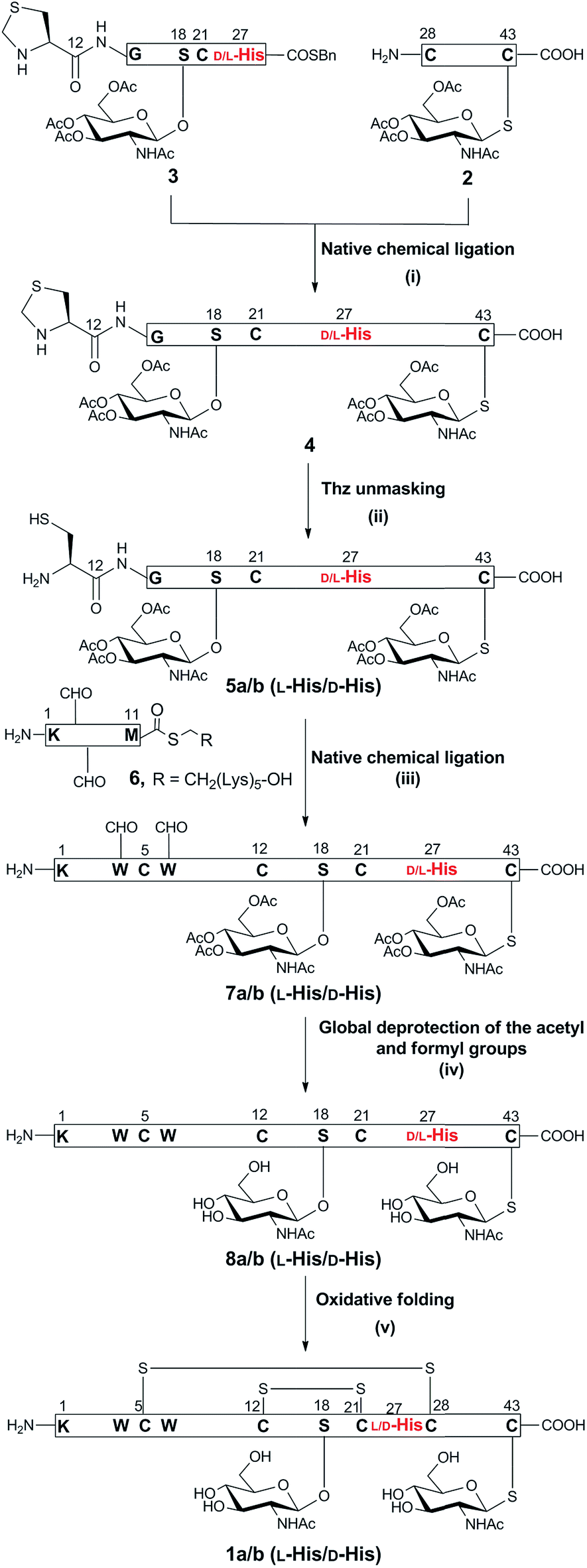 | ||
| Scheme 1 Synthesis of GccF (1) and [27D-His]-GccF, with the problematic His highlighted in red. Reagents and conditions: (i) 6 M Gn·HCl, 0.2 M Na2HPO4, 100 mM MPAA, 20 mM TCEP, pH 6.8, r.t, 2 h; (ii) 6 M Gn·HCl, 0.2 M Na2HPO4, methoxylamine·HCl, pH 4, r.t, 16 h; (iii) 6 M Gn·HCl, 0.2 M Na2HPO4, 100 mM MPAA, 20 mM TCEP, pH 6.8, r.t, 4 h; (iv) 6 M Gn·HCl, 1 M HEPES, hydrazine, 2-mercaptoethanol, 0 °C, 30 min; (v) 1.5 M Gn·HCl, 50 mM Na2HPO4, 2 mM cysteine, 0.25 mM cystine, 0.025 mM EDTA, 0.25 mM peptide, pH 8.2, 4 °C, 16 h. | ||
The necessary O-glycosylated amino acid Fmoc-Ser(β-GlcNAc(OAc)3)-OH (Fmoc = 9-fluorenylmethoxycarbonyl) and S-glycosylated amino acid Fmoc-Cys(β-GlcNAc(OAc)3)-OH building blocks were prepared as previously described (ESI Scheme 1†). Native chemical ligation was undertaken as before and the fully assembled linear peptide was folded without incident (ESI Fig. 23 and 24†). Purification by HPLC yielded epimeric glycocin F (1a) and glycocin F (1b) in 29% and 32% yield, respectively.
The presence of the two disulfide bonds in 1b was confirmed by HRESI-MS analysis (HR-ESIMS, m/z [M + 4H]4+; calculated for C226H311N57O72S7: 1041.4634, observed: 1040.8167) (ESI Fig. 26†). Glycocin F glycopeptides 1a and 1b, epimeric at 27His, were then compared with an authentic sample of glycocin F isolated from L. plantarum (ESI Fig. 25†) and assignment of the natural product was achieved by direct comparison of the HPLC retention time. Epimer 1b eluted at an identical retention time to the isolated material and therefore was assigned the L-configuration at 27His. In contrast, epimer 1a eluted earlier and was therefore assigned the D-configuration.
The secondary structure of synthetic folded GccF was determined by circular dichroism (CD) spectroscopy. Similar to the isolated GccF, the synthetic derivative 1b and epimeric analogue 1a exhibit the features expected for α-helical proteins, namely standard double negative ellipticity maxima at 210 and 221 nm, and a positive maximum near 194 nm (ESI Fig. 43†).
Second-generation synthetic strategy provides expedient access to ‘glyco-mutants’ of glycocin F
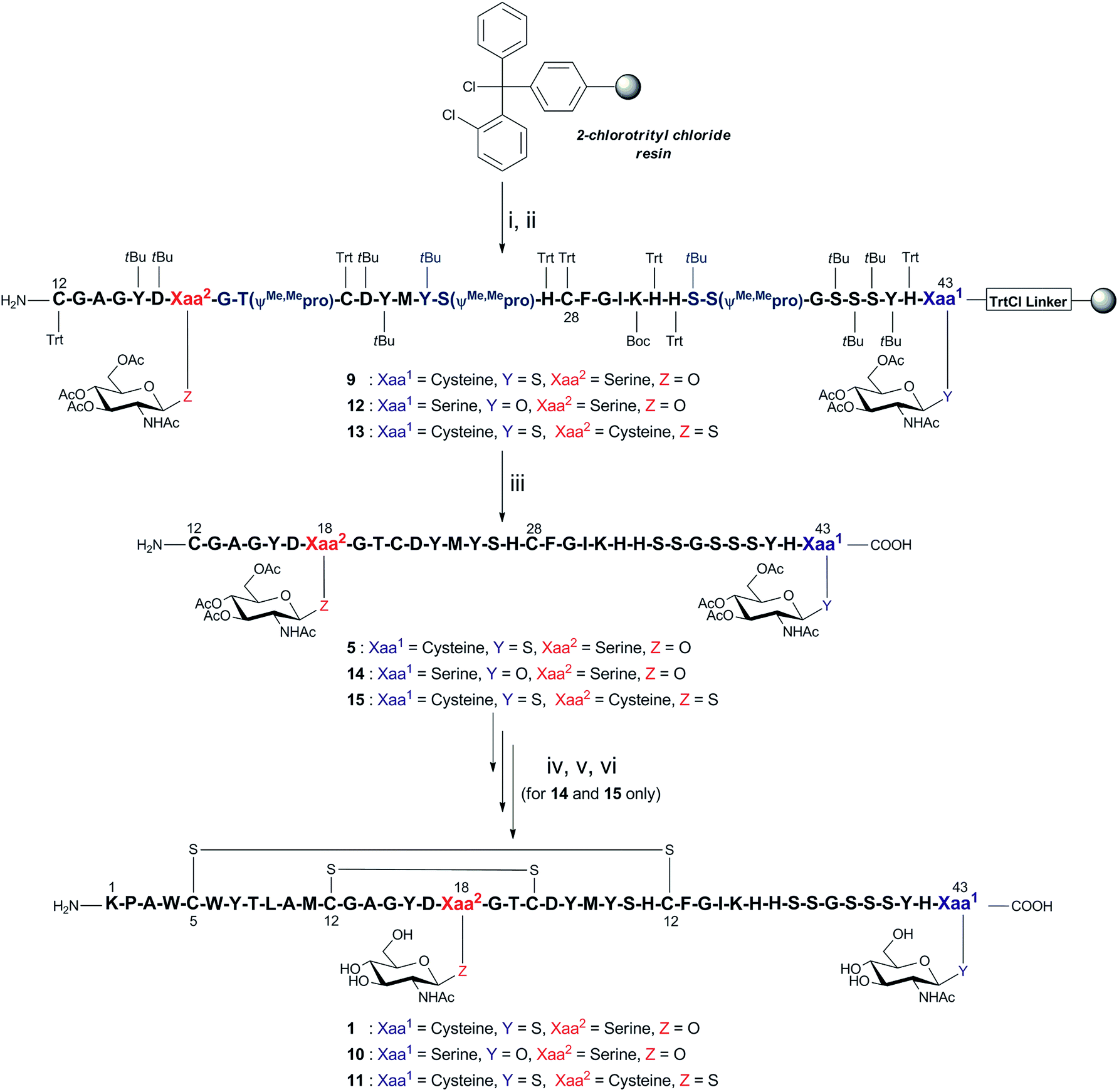 | ||
Scheme 2 Optimised synthesis of GccF fragment 9, which was then employed for the synthesis of glycol-mutants 10 and 11. Reagents and conditions: (i) Fmoc-L-Cys(β-GlcNAc(OAc)3)-OH for 9 and 13 or Fmoc-L-Ser(β-GlcNAc(OAc)3)-OH for 12, iPr2EtN, CH2Cl2, r.t, 1 h; (ii) Fmoc-SPPS (Fmoc deprotection: 20% piperidine in DMF (v/v)), r.t, (2 × 5 min); coupling: Fmoc-amino acid, HATU, iPr2EtN, DMF, r.t, 40 min except Fmoc-18Ser(β-GlcNAc(OAc)3)-OH, HATU, HOAt, TMP, DMF, r.t, overnight and Fmoc-21Cys(Trt)-OH, HATU, HOAt, TMP, CH2Cl2: DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), r.t, 2 × 1 h; (iii) 94% TFA, 2.5% EDT, 2.5% H2O, 1% iPr3SiH (v/v/v/v), r.t, 2 h; (iv)–(vi) for 14 and 15, (iv) 6 M Gn·HCl, 0.2 M Na2HPO4, 100 mM MPAA, 20 mM TCEP, pH 6.8, r.t, 4 h; (v) 6 M Gn·HCl, 1 M HEPES, hydrazine, 2-mercaptoethanol, 0 °C, 30 min; (vi) 1.5 M Gn·HCl, 50 mM Na2HPO4, 2 mM cysteine, 0.25 mM cystine, 0.025 mM EDTA, 0.25 mM peptide, pH 8.2, 4 °C, 16 h. :1, v/v), r.t, 2 × 1 h; (iii) 94% TFA, 2.5% EDT, 2.5% H2O, 1% iPr3SiH (v/v/v/v), r.t, 2 h; (iv)–(vi) for 14 and 15, (iv) 6 M Gn·HCl, 0.2 M Na2HPO4, 100 mM MPAA, 20 mM TCEP, pH 6.8, r.t, 4 h; (v) 6 M Gn·HCl, 1 M HEPES, hydrazine, 2-mercaptoethanol, 0 °C, 30 min; (vi) 1.5 M Gn·HCl, 50 mM Na2HPO4, 2 mM cysteine, 0.25 mM cystine, 0.025 mM EDTA, 0.25 mM peptide, pH 8.2, 4 °C, 16 h. | ||
Direct synthesis of fragment 5 was initiated on 2-ClTrtCl-polystyrene resin (Scheme 2), and subsequent peptide elongation was performed via Fmoc-SPPS using HATU/iPr2EtN as the coupling reagent and 20% piperidine/DMF (v/v) as the Fmoc deblocking reagent. As peptide aggregation and thus reduced coupling yields had hindered previous efforts to synthesise this long fragment, three pseudoproline dipeptides16,17 were incorporated within the peptide chain, namely 19Gly–20Thr, 25Tyr–26Ser, and 35Ser–36Ser. Previous analysis of a truncated fragment 16Gly–27His, built on 2-ClTrtCl-based resin, revealed the presence of a significant, contaminating by-product (25% relative to expected peptide) with the same mass, which was attributed to 21Cys racemisation (ESI Fig. 27†). In order to prevent this, the coupling of 21Cys was achieved using HATU and 2,4,6-trimethylpyridine (TMP) in the presence of 1-hydroxy-7-azabenzotriazole (HOAt) in CH2Cl2/DMF (v/v; 1:1), conditions known to suppress racemisation.18,19 Following Fmoc-SPPS, the fully assembled peptide 9 was recovered from the resin with simultaneous removal of side chain protecting groups using the cleavage mixture TFA/EDT/H2O/iPr3SiH (v/v/v/v; 94:2.5:2.5:1). Under the above conditions, the desired glycopeptide 5 was successfully obtained in 30% crude yield and >73% purity, with successful inhibition of 21Cys racemisation and avoiding 27His racemisation (ESI Fig. 28†).
The secondary structure of glyco-mutants 10 and 11 was determined by circular dichroism (CD) spectroscopy, which confirmed a similar α-helical structure to the native glycosin F bacteriocin (ESI Fig. 43†).
Biological evaluation of synthetic GccF (1b) and [27D-His]-GccF and “glyco-mutants”
Recent research from one of our laboratories (Norris) implicates a GlcNAc-specific phosphotransferase system (PTS) transporter in the cell membranes of susceptible Gram-positive bacteria as the likely target of glycocin F. It is hypothesised that glycocin F binds to the transmembrane domain of its receptor through the tethered GlcNAc residues and disrupts sugar-processing and regulatory activities essential for bacterial growth. Although the exact details remain unknown, a full understanding of the mechanism of action of glycocin F will afford a unique opportunity to develop an entirely new suite of PTS-targeted glycoconjugate antimicrobials for therapeutic and industrial application.To evaluate the antibacterial activity of synthetic glycocin F (1b), [27D-His]-GccF (1a) and glyco-mutants 10 and 11, the IC50 values against Lactobacillus plantarum ATCC 8014 were measured using a liquid culture assay. Synthetic GccF (1b) was used as the positive control (Table 1).
| Compound # | Modification | IC50 |
|---|---|---|
| Native | Isolated GccF | 2.0 ± 0.20 nM |
| 1b | Synthetic GccF | 1.13 ± 0.20 nM |
| 1a | [27D-His]-GccF | 0.98 ± 0.17 nM |
| 10 | 43Cys(GlcNAc) to Ser(GlcNAc) | 12.1 ± 0.20 nM |
| 11 | 18Ser(GlcNAc) to Cys(GlcNAc) | 0.60 ± 0.10 nM |
The IC50 of the bacterially produced glycocin F was found to be 2.0 ± 0.20 nM. The synthetic glycocin F (1b) exhibited an enhanced antibacterial activity relative to this batch of isolated material (IC50 = 1.13 ± 0.20 nM). Considering that the previously reported GccF-NH2 exhibited reduced bacteriostatic activity (IC50 = 1.6 nM),11 this current result provides insight into the relationship of the target glycopeptide with its target receptor: amidation of the C-terminus was in fact responsible for the reduced effect, implying that a positively charged residue within the receptor may be interacting with the negatively charged carboxylate C-terminus.
The importance of the C-terminal carboxylate for bioactivity was also verified from the IC50 value of [27D-His]-GccF (1a) (0.98 ± 0.17 nM). Interestingly, the previously reported [27D-His]-GccF-NH2 showed a significant reduction in activity (IC50 = 4.8 nM) compared to both isolated GccF and GccF-NH2, suggesting that incorporation of D-His negatively influences bioactivity. However, we herein show that the previously reported reduction in bioactivity of the [27D-His]-GccF-NH2 was in fact a consequence of the C-terminal amidation.
The antibacterial activity of the two glyco-mutants, 10 and 11 revealed an intriguing trend. The activity of glycocin F analogue 10, which contains two O-linked GlcNAc moieties, exhibited decreased activity (approximately 10-fold, IC50 = 12.1 ± 0.20 nM) compared to the native glycocin F (IC50 = 1.13 ± 0.20 nM). Notably, the activity of analogue 11, which contains two S-linked GlcNAc moieties, was increased by approximately 2-fold (IC50 = 0.60 ± 0.10 nM) relative to the native glycopeptide. Thus, in this series, the bacteriostatic activity is enhanced by replacing the O-glycoside with an S-glycoside, with the maximum activity obtained by glycopeptide 11 bearing S-linked GlcNAc at both residues 18 and 43.
This striking result is predicted to be due to the enhanced resistance of the stable S-glycosidic linkages to hydrolytic cleavage compared to O-glycosidic linkages, when exposed to the cell envelope and/or secreted glycosidases of target bacteria. Indeed, the Pratt group has recently demonstrated the resistance of S-linked GlcNAc to cleavage by human O-GlcNAcase (OGA).14 By avoiding enzymatic cleavage, the S-linked GlcNAc moieties could remain available for binding at the GccF target receptor, leading to enhanced potency and prolonged bacteriostasis.
Conclusions
The first total synthesis of native glycocin F 1, containing both S- and O-linked β-GlcNAc moieties, was accomplished using a three-fragment NCL strategy followed by oxidative folding. Structural identity to the naturally isolated glycocin F was confirmed by analytical RP-HPLC and CD spectroscopy. To avoid troublesome racemisation at 27His and to increase the overall efficiency of glycocin F synthesis, an alternative synthetic strategy was successfully developed using a two-fragment NCL strategy. This strategy was then employed for the synthesis of two ‘glyco-mutant’ analogues of glycocin F (10 and 11), each bearing a single modification at one of the residues bearing a sugar moiety. Strikingly, the bioactivity increased significantly when the sequence incorporated two S-GlcNAc moieties at positions 18 and 43 (in peptide 11).The results reported herein highlight the potential of glycocin F analogue 11 and related S-linked glycoconjugates as leads for the development of new anti-bacterial agents. Our preparation of glycocin F and analogues aims to provide fundamental insights into a novel antimicrobial mechanism of action, knowledge that is vital for the successful conversion of natural products into the ‘smart antibiotics’ needed for future therapies and industrial applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Maurice Wilkins Centre for Molecular Biodiscovery for financial support (Doctoral Scholarships to Z. A. and S. B.) and the Spanish Ministrio de Economía y Competitividad (predoctoral Fellowship to C. N.).Notes and references
- M. Hassan, M. Kjos, I. Nes, D. Diep and F. Lotfipour, J. Appl. Microbiol., 2012, 113, 723–736 CrossRef CAS PubMed.
- P. D. Cotter, R. P. Ross and C. Hill, Nat. Rev. Microbiol., 2013, 11, 95 CrossRef CAS PubMed.
- J. Cleveland, T. J. Montville, I. F. Nes and M. L. Chikindas, Int. J. Food Microbiol., 2001, 71, 1–20 CrossRef CAS PubMed.
- P. D. Cotter, C. Hill and R. P. Ross, Nat. Rev. Microbiol., 2005, 3, 777–788 CrossRef CAS PubMed.
- P. D. Cotter, R. P. Ross and C. Hill, Nat. Rev. Microbiol., 2013, 11, 95–105 CrossRef CAS PubMed.
- W. J. Kelly, R. V. Asmundson and C. M. Huang, J. Appl. Bacteriol., 1996, 81, 657–662 CrossRef CAS.
- J. Stepper, S. Shastri, T. S. Loo, J. C. Preston, P. Novak, P. Man, C. H. Moore, V. Havlicek, M. L. Patchett and G. E. Norris, FEBS Lett., 2011, 585, 645–650 CrossRef CAS PubMed.
- H. Wang, T. J. Oman, R. Zhang, C. V. G. De Gonzalo, Q. Zhang and W. A. Van Der Donk, J. Am. Chem. Soc., 2014, 136, 84 CrossRef CAS PubMed.
- Y. S. Hsieh, B. L. Wilkinson, M. R. O'Connell, J. P. Mackay, J. M. Matthews and R. J. Payne, Org. Lett., 2012, 14, 1910–1913 CrossRef CAS PubMed.
- H. Katayama, Y. Asahina and H. Hojo, J. Pept. Sci., 2011, 17, 818–821 CrossRef CAS PubMed.
- M. A. Brimble, P. J. Edwards, P. W. Harris, G. E. Norris, M. L. Patchett, T. H. Wright, S. H. Yang and S. E. Carley, Chem.–Eur. J., 2015, 21, 3556–3561 CrossRef CAS PubMed.
- G. E. Norris and M. L. Patchett, Curr. Opin. Struct. Biol., 2016, 40, 112–119 CrossRef CAS PubMed.
- R. Nagar and A. Rao, Glycobiology, 2017, 27, 766–776 CrossRef PubMed.
- C. A. De Leon, P. M. Levine, T. W. Craven and M. R. Pratt, Biochemistry, 2017, 56, 3507–3517 CrossRef CAS PubMed.
- J. Lukszo, D. Patterson, F. Albericio and S. A. Kates, Lett. Pept. Sci., 1996, 3, 157–166 CrossRef CAS.
- T. Wöhr, F. Wahl, A. Nefzi, B. Rohwedder, T. Sato, X. Sun and M. Mutter, J. Am. Chem. Soc., 1996, 118, 9218–9227 CrossRef.
- F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Angew. Chem., Int. Ed., 2010, 49, 10149–10153 CrossRef CAS PubMed.
- Y. Han, F. Albericio and G. Barany, J. Org. Chem., 1997, 62, 4307–4312 CrossRef CAS PubMed.
- Y. Zhang, S. M. Muthana, D. Farnsworth, O. Ludek, K. Adams, J. J. Barchi and J. C. Gildersleeve, J. Am. Chem. Soc., 2012, 134, 6316–6325 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis of all peptides, analytical LC and HR-MS, and circular dichroism data. See DOI: 10.1039/c7sc04383j |
| This journal is © The Royal Society of Chemistry 2018 |