
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvercoming double-step CO2 adsorption and minimizing water co-adsorption in bulky diamine-appended variants of Mg2(dobpdc)†
Phillip J.
Milner
a,
Jeffrey D.
Martell
a,
Rebecca L.
Siegelman
 a,
David
Gygi
b,
Simon C.
Weston
c and
Jeffrey R.
Long
*ade
a,
David
Gygi
b,
Simon C.
Weston
c and
Jeffrey R.
Long
*ade
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: jrlong@berkeley.edu
bDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA
cCorporate Strategic Research, ExxonMobil Research and Engineering Company, Annandale, NJ 08801, USA
dDepartment of Chemical Engineering, University of California, Berkeley, CA 94720, USA
eMaterials Sciences Division, Lawrence Berkeley National Lab, Berkeley, CA 94720, USA
First published on 26th October 2017
Abstract
Alkyldiamine-functionalized variants of the metal–organic framework Mg2(dobpdc) (dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate) are promising for CO2 capture applications owing to their unique step-shaped CO2 adsorption profiles resulting from the cooperative formation of ammonium carbamate chains. Primary,secondary (1°,2°) alkylethylenediamine-appended variants are of particular interest because of their low CO2 step pressures (≤1 mbar at 40 °C), minimal adsorption/desorption hysteresis, and high thermal stability. Herein, we demonstrate that further increasing the size of the alkyl group on the secondary amine affords enhanced stability against diamine volatilization, but also leads to surprising two-step CO2 adsorption/desorption profiles. This two-step behavior likely results from steric interactions between ammonium carbamate chains induced by the asymmetrical hexagonal pores of Mg2(dobpdc) and leads to decreased CO2 working capacities and increased water co-adsorption under humid conditions. To minimize these unfavorable steric interactions, we targeted diamine-appended variants of the isoreticularly expanded framework Mg2(dotpdc) (dotpdc4− = 4,4′′-dioxido-[1,1′:4′,1′′-terphenyl]-3,3′′-dicarboxylate), reported here for the first time, and the previously reported isomeric framework Mg-IRMOF-74-II or Mg2(pc-dobpdc) (pc-dobpdc4− = 3,3′-dioxidobiphenyl-4,4′-dicarboxylate, pc = para-carboxylate), which, in contrast to Mg2(dobpdc), possesses uniformally hexagonal pores. By minimizing the steric interactions between ammonium carbamate chains, these frameworks enable a single CO2 adsorption/desorption step in all cases, as well as decreased water co-adsorption and increased stability to diamine loss. Functionalization of Mg2(pc-dobpdc) with large diamines such as N-(n-heptyl)ethylenediamine results in optimal adsorption behavior, highlighting the advantage of tuning both the pore shape and the diamine size for the development of new adsorbents for carbon capture applications.
Introduction
Carbon dioxide generated from burning fossil fuels at thermoelectric power plants is widely acknowledged to be a major contributor to global climate change.1 One proposed strategy to minimize global CO2 emissions is carbon capture and sequestration (CCS), in which the CO2 is separated from the other constituents of flue gas (primarily N2, O2, and H2O) and injected underground.1,2 Due to their low costs, effective performance under humid conditions, and high selectivity for CO2 over N2, aqueous amine solutions are the most technology-ready materials for carbon capture applications.3 However, these materials suffer from a number of drawbacks, including low working capacities (∼2 wt%), corrosiveness, and thermal degradation upon regeneration.3a,4 In contrast, porous solids such as zeolites are more thermally robust than aqueous amines and can exhibit lower regeneration energies,5 favorable properties that have led to their investigation for carbon capture.6 Unfortunately, CO2 adsorption in most of these materials is impaired by the water present in flue gas, which passivates the CO2 binding sites and/or leads to degradation.7 Amine-functionalized porous solids, such as amine-appended silicas, are promising alternatives that combine the best properties of both aqueous amines and porous solids while maintaining high CO2/N2 selectivities under humid conditions.6,7b,c,8 However, the co-adsorption of water with CO2 remains a significant challenge for the practical use of these adsorbents because of the parasitic energy costs incurred as water is desorbed from the bed with CO2 upon regeneration.7c,9Metal–organic frameworks are a class of porous solids, consisting of metal nodes connected by polytopic organic linkers,10 that have recently been evaluated for CO2 capture.11 In particular, amine-functionalized11c,12 and hydrophobic13 metal–organic frameworks have shown promise due to their potential ability to capture CO2 in the presence of water while minimizing water co-adsorption. However, the long-term carbon capture performance and stability of many of these materials under humid conditions has not been reliably established. Therefore, there remains a continuing need for the design and rigorous study of new stable adsorbents for carbon capture applications.
Recently, we14 and others15 have evaluated a new class of diamine-appended metal–organic frameworks prepared by post-synthetically appending alkylethylenediamines and 1,3-diaminopropanes to the open M2+ coordination sites lining the hexagonal channels of M2(dobpdc) (dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate; M = Mg, Mn, Fe, Co, Zn) frameworks (Fig. 1a). Upon diamine functionalization (Fig. 1b), these materials display step-shaped adsorption of CO2 with a step pressure that can be tuned by judicious choice of the M2+ ion and appended diamine.14a–c These sigmoidal adsorption profiles result from the cooperative formation of highly stabilized ammonium carbamate chains along the crystallographic c-axis (Fig. 1c) and enable high working capacities (>2.5 mmol g−1) with minimal temperature swings (e.g., ΔT = 60 °C).14a–c In addition, these materials generally possess modest regeneration energies and high CO2/N2 selectivities, even under humid conditions.14a,b Our prior studies of diamine-appended variants of Mg2(dobpdc) suggested that 1°,2°-alkylethylenediamines, such as N-ethylethylenediamine (e-2) and N-(iso-propyl)ethylenediamine (i-2) (Fig. 1d), are among the most promising for carbon capture from dilute gas streams, due to their low CO2 adsorption step pressures (≤1 mbar at 40 °C) and stability towards diamine loss compared to secondary,secondary (2°,2°) diamines.14a However, prior to this work it remained unclear if these diamine-appended metal–organic frameworks possess the requisite stability required for long-term application in a carbon capture process.
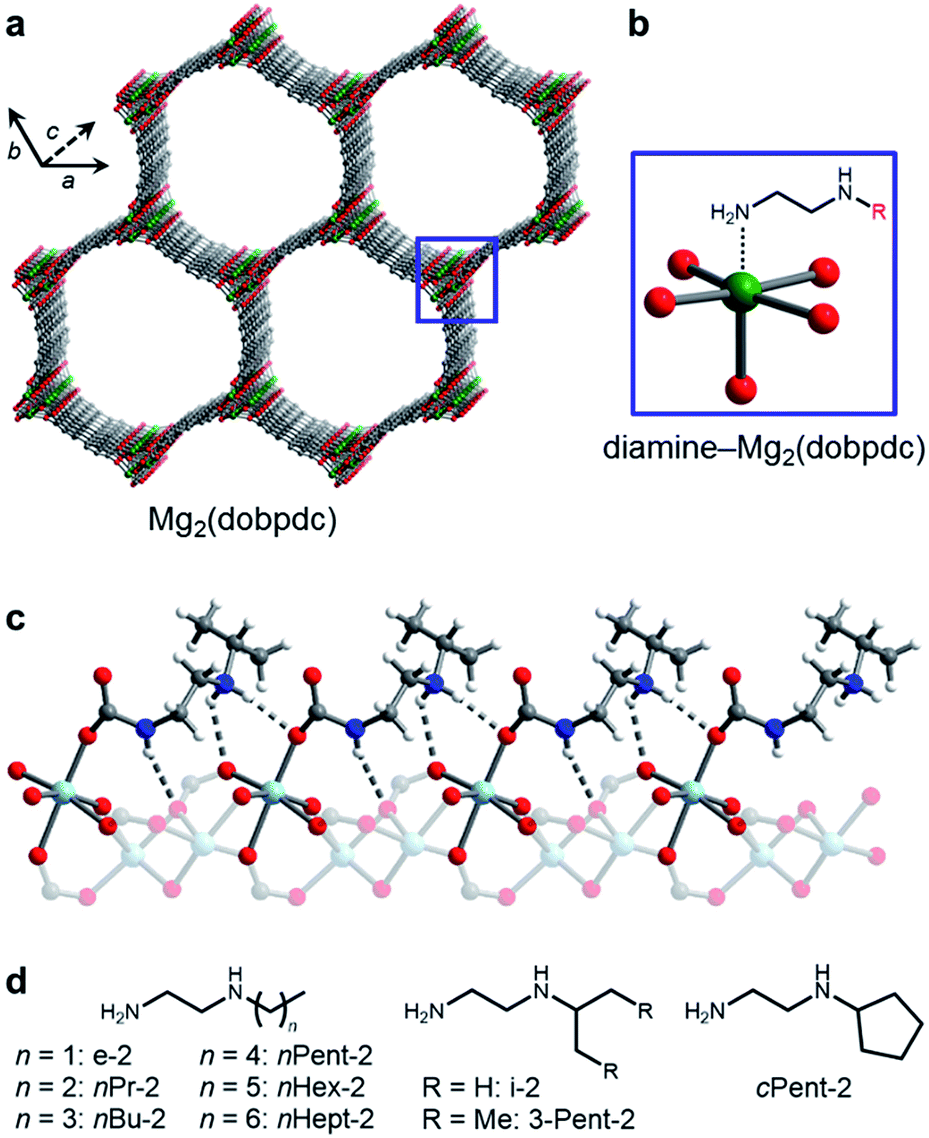 | ||
| Fig. 1 (a) Structure of the metal–organic framework Mg2(dobpdc) (dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate). Green, grey, and red spheres correspond to Mg, C, and O, respectively. (b) Addition of alkylethylenediamines to the methanol-solvated framework yields adsorbents with diamines appended to the open Mg2+ sites. (c) Upon CO2 adsorption, ammonium carbamate chains form along the c-axis, illustrated by the single crystal X-ray diffraction structure of i-2–Zn2(dobpdc)–CO2.14b Light blue, grey, red, dark blue, and white spheres correspond to Zn, C, O, N, and H, respectively. (d) The bulky linear and branched primary,secondary (1°,2°) alkylethylenediamines studied in this work. | ||
Herein, we investigate the properties of Mg2(dobpdc) appended with 1°,2°-alkylethylenediamines bearing large hydrophobic alkyl groups, with the goal of minimizing diamine volatilization and water co-adsorption (Fig. 1d). Unexpectedly, the use of these bulky diamines leads to two distinct CO2 adsorption steps, each corresponding to half of the expected capacity of 1 CO2 per diamine.14b We demonstrate that changing the base framework, a hitherto unexplored strategy, leads to a single CO2 adsorption step in all cases. Thus, frameworks appended with bulky 1°,2°-alkylethylenediamines display a single CO2 adsorption step at low pressures (≤1 mbar at 40 °C) along with minimal co-adsorption of water in thermogravimetric analysis (TGA) experiments. These properties render bulky 1°,2°-alkylethylenediamine-appended metal–organic frameworks particularly promising adsorbents for carbon capture from dilute gas streams.
Experimental
General procedures
1H NMR spectra were collected on a Bruker AMX 300 MHz NMR spectrometer and referenced to residual dimethyl sulfoxide (δ 2.50 ppm). Attenuated total reflectance (ATR) infrared (IR) spectra were collected on a Perkin-Elmer Spectrum 400 Fourier Transform (FT) IR spectrometer. Laboratory powder X-ray diffraction (PXRD) patterns were collected using a Bruker AXS D8 Advance diffractometer with CuKα radiation (λ = 1.5418 Å). All synthetic manipulations were carried out under air unless noted otherwise. All solvents and reagents, as well as the diamines N-ethylethylenediamine (e-2), N-(n-propyl)ethylenediamine (nPr-2), N-(n-butyl)ethylenediamine (nBu-2), N-(iso-propyl)ethylenediamine (i-2), N,N-diethylethylenediamine (ee-2), and N-(2-aminoethyl)pyrrolidine (pyrr-2), were purchased from commercial sources and used without further purification unless otherwise noted. The diamines N-(n-pentyl)ethylenediamine (nPent-2), N-(n-hexyl)ethylenediamine (nHex-2), N-(n-heptyl)ethylenediamine (nHept-2), N-(cyclopentyl)ethylenediamine (cPent-2), and N-(3-pentyl)ethylenediamine (3-Pent-2), were prepared according to the procedures outlined in ESI Section 1.†17 The linker H4dobpdc was purchased from Hangzhou Trylead Chemical Technology Co. The linker H4dotpdc was prepared according to the literature procedure.18 The linker H4pc-dobpdc was prepared according to the procedure outlined in ESI Section 8.† The metal–organic frameworks Mg2(dobpdc)14b and Mg2(pc-dobpdc)16 were prepared according to literature procedures.Synthesis of Mg2(dotpdc)
A 20 mL scintillation vial was charged with 4,4′′-dihydroxy-[1,1′:4′,1′′-terphenyl]-3,3′′-dicarboxylic acid (35.0 mg, 0.100 mmol, 1.00 equiv.) and Mg(NO3)2·6H2O (64.0 mg, 0.250 mmol, 2.50 equiv.). Methanol (5.5 mL) and fresh N,N-dimethylformamide (DMF) (4.5 mL) were added, and the solution was sonicated until all of the solids dissolved. The vial was wrapped in Teflon tape, sealed, and heated at 120 °C on a dry bath for 14 h, during which time a white solid precipitated from solution. The vial was cooled to room temperature, and the resulting solid was collected by filtration and washed with DMF (15 mL). The solid was then transferred to a vial filled with DMF (10 mL) and allowed to soak at 120 °C for 24 h. The supernatant was decanted and replaced with fresh DMF (10 mL), and the vial was re-heated to 120 °C. This washing process was repeated a total of three times. Methanol (10 mL) was then added to the off-white solid, and the solid was soaked in methanol at 60 °C for 3 h. The supernatant was decanted and replaced with fresh methanol (10 mL), and the vial was re-heated to 60 °C. This washing process was repeated a total of three times. Activation of the resulting powder at 250 °C under flowing N2 for 14 h, followed by activation under reduced pressure (<10 μbar) at 250 °C for 14 h, afforded activated Mg2(dotpdc) (31.8 mg, 81% yield, average of two syntheses) as a fine, pale yellow powder. The PXRD pattern, IR spectrum, and 77 K N2 adsorption isotherm are included in ESI Section 5.†Synthesis of diamine-appended variants of Mg2(dobpdc), Mg2(dotpdc), and Mg2(pc-dobpdc)14b
A 20 mL scintillation vial was charged with toluene (4 mL) and the diamine (1 mL). The methanol-solvated metal–organic framework (∼20 mg) was filtered and washed with toluene (2× 10 mL) (Note: Mg2(dobpdc) should not be allowed to dry completely in air due to potential decomposition).14e The framework was added to the diamine solution, and the vial was swirled several times and allowed to stand at room temperature for 24 h. At this time, the mixture was filtered, and the resulting powder was thoroughly washed with toluene (3× 20 mL) and allowed to dry for ∼3 min under reduced pressure, yielding ∼30 mg of the diamine-appended metal–organic framework. Larger scale samples for isothermal measurements were prepared by carrying out this procedure on 3× scale. Activation conditions, PXRD patterns, IR spectra, pure CO2 adsorption/desorption isobars, humid N2 and CO2 adsorption isobars, and thermogravimetric N2 decomposition curves for all new diamine-appended metal–organic frameworks are included in Sections 6, 7, 9, and 10 of the ESI.† Diamine loadings were determined by suspending ∼5 mg of the diamine-appended metal–organic framework in 0.5 mL of DMSO-d6, adding several drops of DCl (35 wt% in D2O), heating until the mixture became homogeneous, and analyzing the resulting solution by 1H NMR.14bPreparation of single crystals of Zn2(pc-dobpdc)
A stock solution of the ligand was prepared by dissolving H4pc-dobpdc (11.5 mg, 0.042 mmol) in N,N-dimethylacetamide (DMA) (1.4 mL). A separate stock solution was prepared by dissolving Zn(NO3)2·6H2O (31.2 mg, 0.100 mmol) in a mixture of H2O (1.4 mL) and ethanol (1.4 mL). The reaction solution was prepared by charging a 20 mL scintillation vial with 1.2 mL of the metal stock solution and 0.60 mL of the ligand stock solution (overall 10 mM H4pc-dobpdc, 2.5 equiv. Zn(NO3)2·6H2O, 1.8 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 v/v/v DMA:water:ethanol). The vial was wrapped in Teflon tape, sealed, and heated on a dry bath at 100 °C for 48 h, at which time colorless, hexagonal prism-shaped crystals had formed. A single crystal was removed from the reaction mixture for X-ray diffraction. Crystallographic tables and experimental details are included in Section 11 of the ESI.†
:1:1 v/v/v DMA:water:ethanol). The vial was wrapped in Teflon tape, sealed, and heated on a dry bath at 100 °C for 48 h, at which time colorless, hexagonal prism-shaped crystals had formed. A single crystal was removed from the reaction mixture for X-ray diffraction. Crystallographic tables and experimental details are included in Section 11 of the ESI.†
Thermogravimetric analysis and cycling measurements
Dry thermogravimetric analysis (TGA) experiments were conducted using a TA Instruments TGA Q5000. Humid TGA experiments were conducted using a TA Instruments TGA Q50. The incident gas stream was humidified by passing it through two water bubblers in series, leading to an estimated water content of 1.3% (∼30% relative humidity at 25 °C), as determined by comparison to previously obtained water isotherms.14a Isobaric measurements were carried out using a ramp rate of 2 °C min−1. The cylinder of 15% CO2 in N2 was purchased from Praxair. Samples were activated under flowing N2 for 20–30 min until the mass stabilized; exact activation conditions for each diamine-appended material were determined by careful analysis of the dry N2 thermal decomposition profiles and are included in the ESI.† Masses are uncorrected for buoyancy effects. Dry N2 decomposition experiments were carried out using a heating ramp rate of 1.5 °C min−1. A flow rate of 25 mL min−1 was used for all TGA experiments.Gas adsorption measurements
Adsorption isotherms with N2 and CO2 were obtained by volumetric methods using a Micromeritics ASAP 2020 gas adsorption analyzer. All gases were 99.998% purity or higher. Isotherms collected at 25, 35, 40, and 45 °C were measured using a circulating water bath to control the temperature. Samples were regenerated at 100 °C under reduced pressure (<10 μbar) for 2–4 h between isotherms. The isotherm data points were considered equilibrated after <0.01% pressure change occurred over a 15 s interval.Calculations of differential enthalpies of adsorption
The CO2 isotherms of Mg2(dotpdc) and Mg2(pc-dobpdc) were fit using the dual-site Langmuir–Freundlich equation (eqn (1)), where q is the amount of CO2 adsorbed in mmol g−1, qsati is the saturation capacity of each site i in mmol g−1, bi is the Langmuir parameter for each site i in bar−1, P is the pressure in bar, and vi is the Freundlich parameter for each site i. The isotherm fits were used to solve for the exact pressures (pq) corresponding to specific CO2 loadings (q) at different temperatures (T). The Clausius–Clapeyron relationship (eqn (2)) was used to calculate the differential enthalpies of adsorption (Δhads) based on the slopes of the linear trendlines fit to ln(pq) vs. 1/T at constant values of q.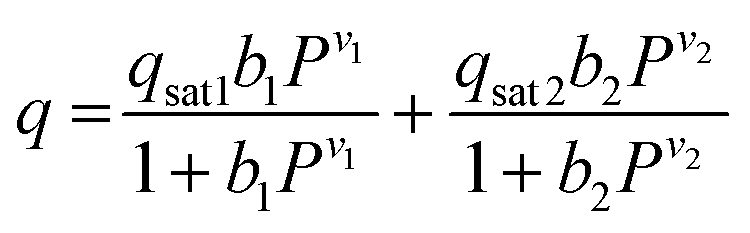 | (1) |
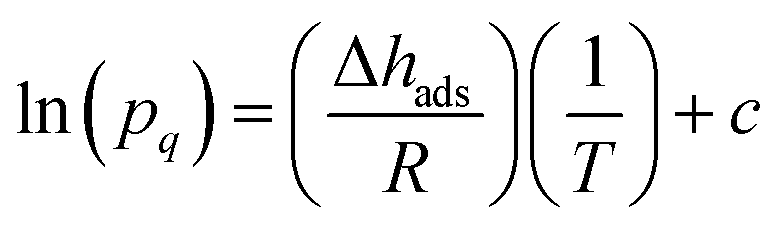 | (2) |
Results and discussion
Synthesis of 1°,2°-alkylethylenediamines and grafting to Mg2(dobpdc)
Our initial studies of diamine-appended variants of Mg2(dobpdc) suggested that 1°,2°-alkylethylenediamines (Fig. 1d) warranted further study for carbon capture applications due to their improved thermal stabilities compared to 2°,2°-alkylethylenediamines, lower step pressures compared to 1°,3°-alkylethylenediamines, and minimal hysteresis upon CO2 desorption.14b For example, we previously found that appending e-2 and i-2 to Mg2(dobpdc) produced adsorbents exhibiting step-shaped adsorption of CO2 at low partial pressures (≤1 mbar) at 40 °C.14b These favorable properties result from both the strong primary amine–metal bond in the amine phase as well as the formation of tightly bound ammonium carbamate chains with extensive hydrogen-bonding to the framework (Fig. 1c).14b Based on these results, we reasoned that increasing the size of the alkyl group on the 2° amine should reduce diamine loss upon long-term adsorption/desorption cycling without interfering in the cooperative CO2 adsorption mechanism.To probe the effect of increasing the size of the alkyl group on the secondary amine, we synthesized a series of Mg2(dobpdc) variants functionalized with 1°,2°-alkylethylenediamines bearing linear alkyl substituents, including ethyl (e-2), n-propyl (nPr-2), n-butyl (nBu-2), n-pentyl (nPent-2), n-hexyl (nHex-2), and n-heptyl (nHept-2), as well as branched alkyl groups, including iso-propyl (i-2), cyclopentyl (cPent-2), and 3-pentyl (3-Pent-2) (Fig. 1d). Although e-2, i-2, nPr-2, and nBu-2 are commercially available, the other 1°,2°-alkylethylenediamines shown in Fig. 1d are not, and so a simple synthetic protocol was developed for the preparation of diamines substituted with nPent, nHex, nHept, cPent, and 3-Pent groups (see ESI Section 1† for details).17
Nearly all of the bulky 1°,2°-alkylethylenediamines in Fig. 1d could be grafted to Mg2(dobpdc) with high loadings (>90% occupancy of the Mg2+ sites), as determined from 1H NMR spectra collected after digestion with DCl in DMSO-d6 (Table S1†). Unfortunately, all attempts to graft the largest diamine in this series, nHept-2, led to low loadings (∼69%), likely due to the inability of the pore to readily accommodate the large n-Heptyl groups. As expected, the grafting of increasingly large diamines to Mg2(dobpdc) led to a gradual decrease in the Langmuir surface areas determined from 77 K N2 adsorption isotherms, from a maximum of 3780 m2 g−1 for activated Mg2(dobpdc) to a minimum of 503 m2 g−1 for nHex-2–Mg2(dobpdc) (Fig. S5†). Consistent with our previous results,14b the temperature at which the maximum rate of diamine loss occurred upon thermolysis increased concomitantly with the molecular weight of the diamine, from 280 °C for N-methylethylenediamine (m-2)14b to 344 °C for nHex-2 (Fig. S4†). Accordingly, Mg2(dobpdc) variants appended with the largest diamines are expected to exhibit the greatest stability to diamine volatilization during CO2 adsorption/desorption cycling.
Observation of two-step CO2 adsorption
We next examined the CO2 adsorption/desorption profiles of the 1°,2°-alkylethylenediamine-appended Mg2(dobpdc) variants. Thermogravimetric analysis (TGA) measurements conducted under flowing CO2 enabled high-throughput analysis of a large series of adsorbents. In these measurements, high CO2 adsorption step temperatures correspond to low CO2 step pressures in isothermal measurements. The results of these experiments are included in full in Fig. S6† and are summarized in Fig. 2.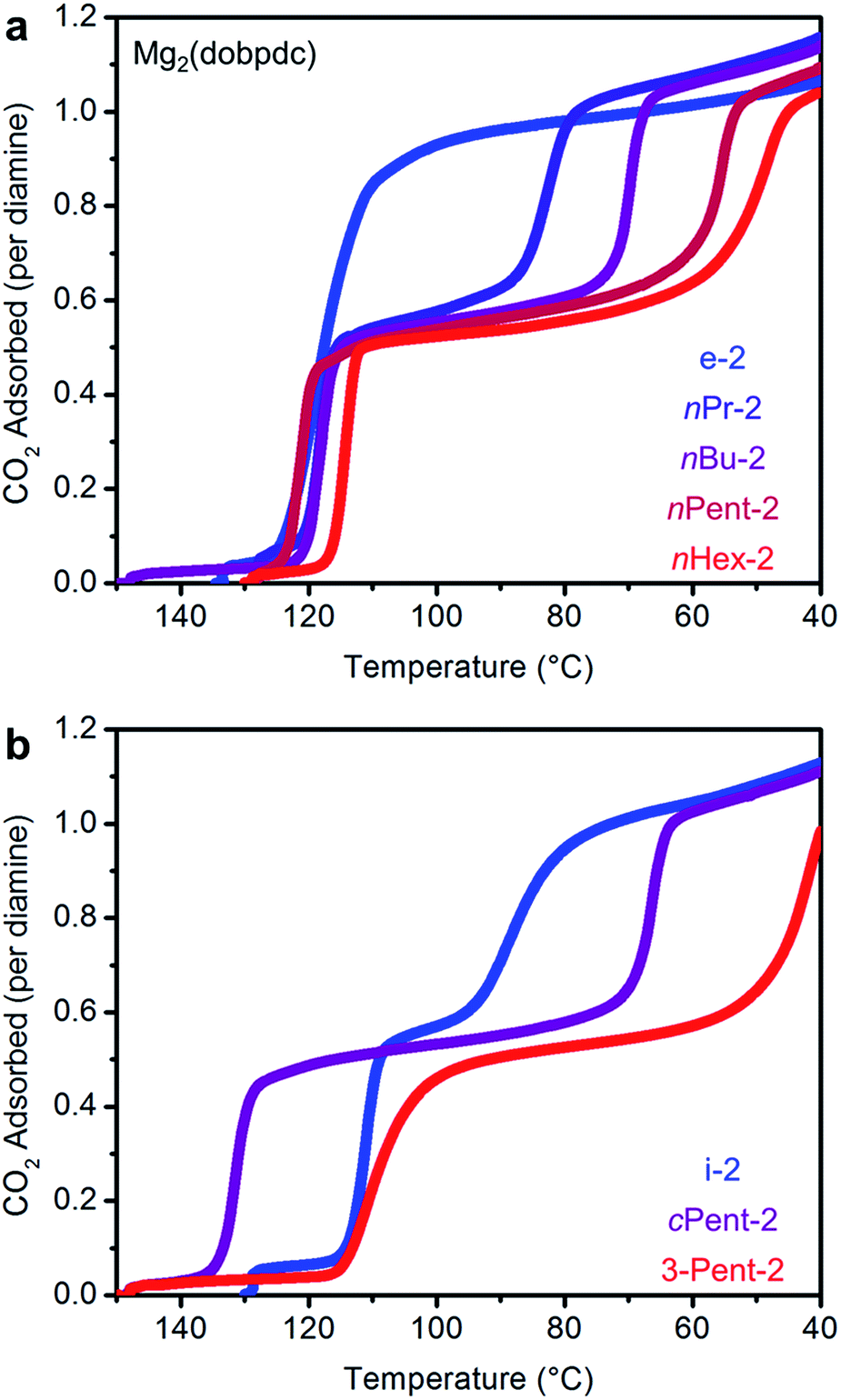 | ||
| Fig. 2 Pure CO2 adsorption isobars for a series of 1°,2°-alkylethylenediamine-appended variants of Mg2(dobpdc) bearing (a) linear and (b) branched alkyl groups. The lower temperature CO2 adsorption step moves to successively lower temperatures as the substituent size increases. In both plots, the gradient from blue to red reflects the increasing size of the substituent on the secondary amine. | ||
As previously reported, e-2–Mg2(dobpdc) displays a single CO2 adsorption step at a relatively high temperature (125 °C), corresponding to an adsorption step at ∼0.5 mbar in the 40 °C adsorption isotherm (Fig. S7†).14b Conversely, i-2–Mg2(dobpdc) exhibits a two-step CO2 adsorption profile, which we previously ascribed to a conformational shift during the formation of ammonium carbamate chains.14b This behavior is a hallmark of increasing the size of the alkyl group on the secondary amine in 1°,2°-alkylethylenediamines, as two sharp CO2 adsorption/desorption steps were also observed for Mg2(dobpdc) appended with nPr-2, nBu-2, nPent-2, nHex-2, cPent-2, and 3-Pent-2 (Fig. 2 and S6†). In every case, each step corresponds to approximately 50% of the expected CO2 capacity, assuming the adsorption of one CO2 per diamine (Fig. 2 and S6†).19 Increasing the molecular weight of the diamine also led to a gradual decrease in gravimetric capacity due to the higher molecular weight of the diamine (Fig. S6†). With the exception of e-2, two adsorption steps were also observed in the 40 °C CO2 isotherms of the adsorbents shown in Fig. 2, confirming that this behavior is not a kinetic effect (Fig. S7†). Notably, two-step CO2 adsorption/desorption profiles were also observed for Mg2(dobpdc) functionalized with 1°,3°-alkylethylenediamines (ESI Section 4†) and 2°,2°-alkylethylenediamines bearing N-substituents larger than methyl groups,14b suggesting that this behavior is common to variants of Mg2(dobpdc) appended with sterically encumbered alkylethylenediamines.
The occurrence of two distinct adsorption/desorption steps in these materials suggests that they undergo two cooperative transitions upon CO2 adsorption and desorption, as has been previously reported for certain flexible metal–organic frameworks.20 Such behavior is undesirable for carbon capture applications because it could lead to diminished working capacities if the second step occurs at a pressure that is too high for the target flue gas stream. For example, cooling 3-Pent-2–Mg2(dobpdc) under a stream of dry simulated coal flue gas (15% CO2 in N2) revealed that the second adsorption step was inoperative at 40 °C (Fig. S9†), leading to half the expected CO2 capacity.
A comparison of the adsorption and desorption step temperatures for the family of 1°,2°-alkylethylenediamines provides insight into the effect of the alkyl group size on the positions of the two steps (Fig. 2). For example, the higher temperature adsorption steps occur at similar temperatures for the series of diamines with linear alkyl groups (inflection points: e-2 = 119 °C, nPr-2 = 117 °C, nBu-2 = 119 °C, nPent-2 = 121 °C, nHex = 114 °C) (Fig. 2a). Consistent with this finding, the first CO2 adsorption steps in the 40 °C CO2 isotherms of these adsorbents all occur at ∼0.5 mbar (Fig. S7†). Likewise, the higher temperature CO2 desorption steps are also at similar temperatures (inflection points: e-2 = 131 °C, nPr-2 = 126 °C, nBu-2 = 127 °C, nPent-2 = 133 °C, nHex-2 = 131 °C) (Fig. S6†). In contrast, the second CO2 adsorption step temperatures steadily decrease as the size of the alkyl group increases (inflection points: nPr-2 = 81 °C, nBu-2 = 70 °C, nPent-2 = 56 °C, nHex-2 = 48 °C) (Fig. 2a), as do the CO2 desorption step temperatures (inflection points: nPr-2 = 88 °C, nBu-2 = 78 °C, nPent-2 = 65 °C, nHex-2 = 65 °C) (Fig. S6†). Therefore, the steric encumbrance of the 2° amine does not significantly affect the thermodynamics of CO2 adsorption for the higher temperature step, but does significantly influence the lower temperature adsorption step.
A related trend can be observed in the corresponding series of branched 1°,2°-alkylethylenediamines (Fig. 2b). The inflection point of the first adsorption step occurs at a similar temperature for i-2–Mg2(dobpdc) (114 °C) and 3-Pent-2–Mg2(dobpdc) (111 °C), whereas the second CO2 adsorption step occurs at a lower temperature for the bulkier 3-Pent-2 (42 °C) than for i-2 (91 °C). Notably, cPent-2–Mg2(dobpdc) possesses a considerably higher adsorption step temperature (inflection point: 129 °C) than the other 1°,2°-alkylethylenediamines, reflecting more thermodynamically favorable adsorption of CO2 in this material (Fig. 2b). This favorable adsorption behavior likely stems from more efficient packing of cyclopentyl groups in the ammonium carbamate chains compared to other alkyl substituents. Nonetheless, the inflection point of the second CO2 adsorption step for cPent-2–Mg2(dobpdc) occurs at 66 °C, which is between that of the smaller i-2–Mg2(dobpdc) (91 °C) and larger 3-Pent-2–Mg2(dobpdc) (42 °C). Thus, these three diamines also follow the trend of decreasing temperatures for the second CO2 adsorption step with increasing steric bulk of the diamine alkyl substituent.
Increased water co-adsorption for adsorbents displaying two CO2 adsorption steps
Notwithstanding the undesirable two-step CO2 adsorption/desorption profiles of bulky diamine-appended variants of Mg2(dobpdc), their high thermal stabilities (Fig. S4†) led us to evaluate their applicability for CO2 capture under humid conditions. As previously reported,14a the co-adsorption of water upon cooperative CO2 adsorption in diamine-appended metal–organic frameworks can be rapidly assessed using humid TGA isobaric experiments, wherein the incident gas stream is bubbled through water before reaching the adsorbent. One shortcoming of this method is that the identity of the adsorbed species cannot be definitively established. Nonetheless, direct comparison of the wet and dry N2 and CO2 adsorption isobars can still provide insight into the ability of adsorbents to capture CO2 under humid conditions. The results of these studies are summarized in Fig. 3 and 4.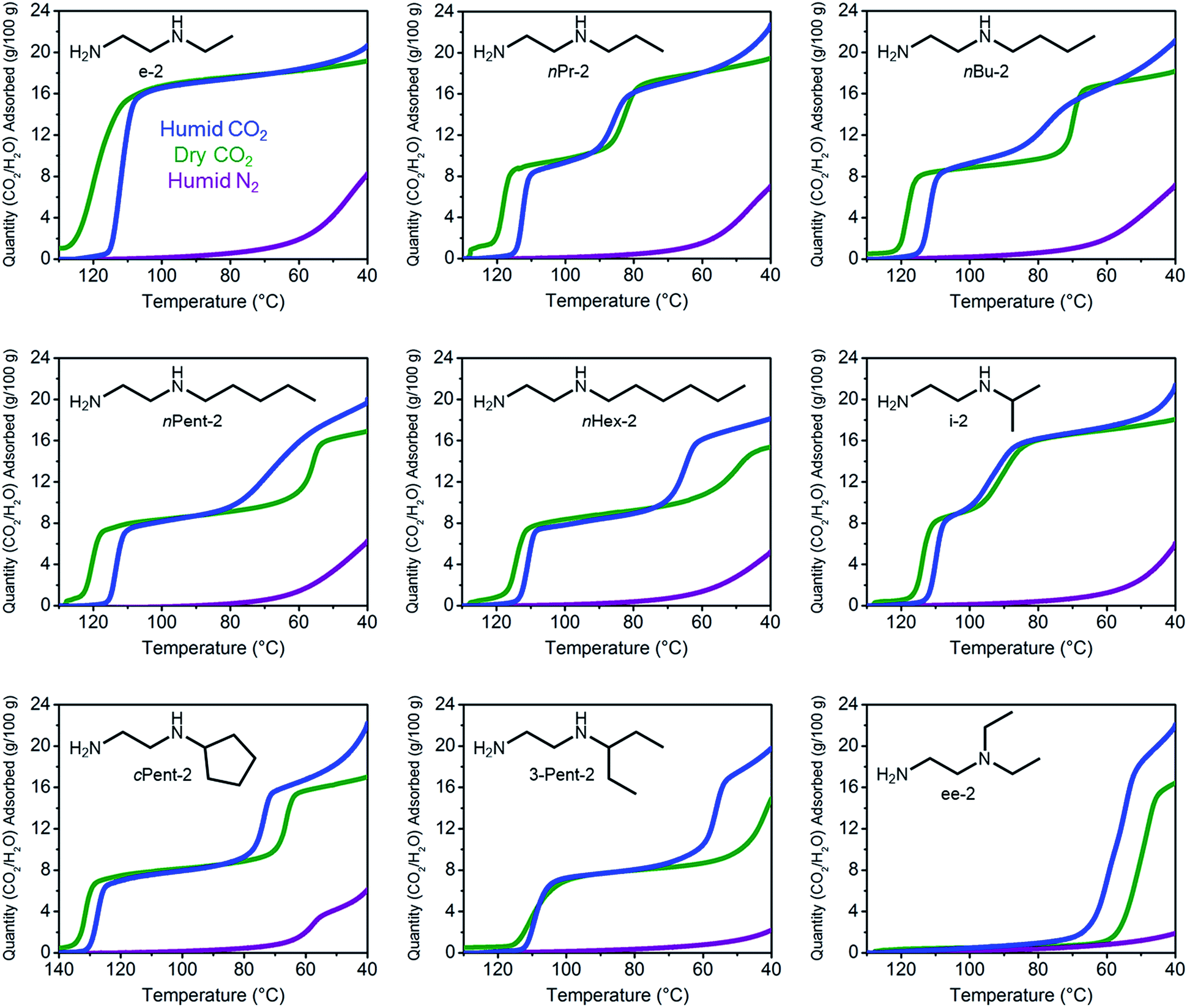 | ||
| Fig. 3 Dry CO2 (green), humid CO2 (blue), and humid N2 (purple) adsorption isobars for a series of 1°,2°-alkylethylenediamine-appended variants of Mg2(dobpdc), as well as ee-2–Mg2(dobpdc) for comparison. | ||
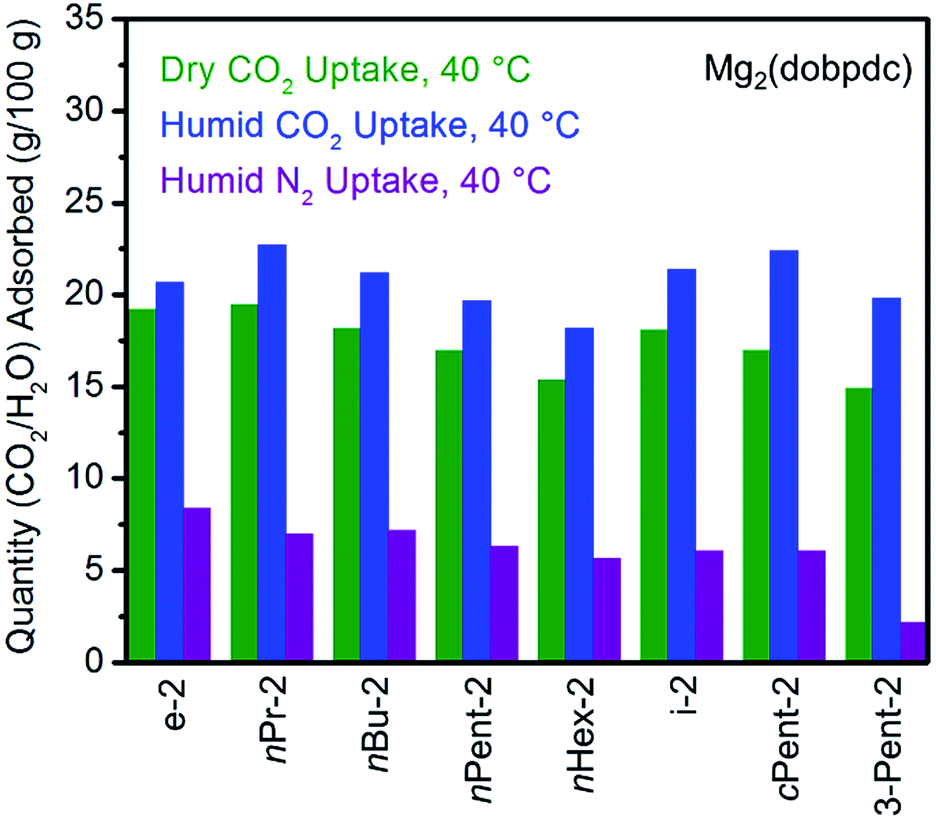 | ||
| Fig. 4 Summary of the adsorption capacities from Fig. 3 at 40 °C for a series of 1°,2°-alkylethylenediamine-appended variants of Mg2(dobpdc) under dry CO2 (green), humid CO2 (blue), and humid N2 (purple). | ||
All the 1°,2°-alkylethylenediamine-appended Mg2(dobpdc) variants exhibited minimal diamine loss upon activation under humid N2 at high temperatures (130–150 °C) (Table S1†).22 Subsequent cooling of the adsorbents under a humid N2 stream (purple curves, Fig. 3) allowed for an estimation of the amount of water adsorbed due to the minimal N2 uptake of these materials.14a,b,d,15b,c Water adsorption decreased as the size of the alkyl group increased, from ∼8.3 g/100 g for e-2–Mg2(dobpdc) to ∼2.2 g/100 g for 3-Pent-2–Mg2(dobpdc) (Fig. 4). This trend is likely due to the decreased ability of the unbound amine to hydrogen bond with water as the hydrophobic substituent becomes larger. We previously observed a similar trend in the single-component water adsorption isotherms of e-2–Mg2(dobpdc) and i-2–Mg2(dobpdc).14b
Most importantly for carbon capture applications, all of the 1°,2°-alkylethylenediamine-appended Mg2(dobpdc) variants maintained step-shaped adsorption of CO2 in the presence of water (blue curves, Fig. 3). In all cases, the higher temperature step was similar in height and occurred at a similar or slightly lower (<10 °C difference) temperature under humid CO2 compared to dry CO2. In contrast, for materials exhibiting two-step CO2 adsorption profiles, the lower temperature step generally shifted to higher temperatures under humid conditions (Fig. 3). This phenomenon is likely due to the stabilizing influence of water on the ammonium carbamate chains formed during the second step,8,14a which are likely less thermodynamically stable than those originating from the higher temperature CO2 adsorption step.14b Consistent with this hypothesis, ee-2–Mg2(dobpdc) (Fig. 3) and pyrr-2–Mg2(dobpdc) (Fig. S16†) also display higher CO2 adsorption step temperatures under humid conditions compared to dry conditions as a result of their less thermodynamically stable ammonium carbamate chains.14b
Differences between the total mass uptake under dry (green) and humid (blue) CO2 streams can be attributed primarily to water co-adsorption (Fig. 4). Although we anticipated that increasing the bulk of the diamine would decrease water co-adsorption, the smallest diamine in this series (e-2) actually exhibits the least co-adsorption of water, ∼1.5 g/100 g or 0.2 molecules of water per diamine (Fig. 4). In contrast, all of the bulkier 1°,2°-alkylethylenediamines that display two-step CO2 adsorption profiles co-adsorb more water per diamine at 40 °C under humid conditions (Fig. 4 and Table S2†). For example, this increased degree of water co-adsorption led to a higher gravimetric discrepancy between the wet and dry CO2 isobars for n-Hex-2–Mg2(dobpdc) (2.8 g/100 g or 0.5 molecules of water per diamine). Notably, 1°,3°-alkylethylenediamine-substituted variants of Mg2(dobpdc) exhibit even more dramatic water co-adsorption (>4 g/100 g assuming all water uptake) when cooled under humid CO2, confirming that less stable ammonium carbamate chains display an increased proclivity towards favorably interacting with water (Fig. 3 and S16†). The increased water co-adsorption by bulky 1°,2°-alkylethylenediamine-appended variants of Mg2(dobpdc) is likely a direct result of the presence of secondary, less stable ammonium carbamate chains (corresponding to the lower temperature CO2 adsorption steps). In contrast, e-2–Mg2(dobpdc) forms stable, tightly packed chains with minimal accessible surface for hydrogen-bonding or ion-dipole interactions with water. Nonetheless, minimal water co-adsorption was observed for all of these materials at temperatures above 60 °C, suggesting that water co-adsorption can, in general, be minimized by adsorbing CO2 from humid streams at higher temperatures.
Adsorption/desorption cycling under humid conditions
In order to assess whether increasing the size of the alkyl group on the diamine leads to improved stability to diamine loss in a temperature swing adsorption process, we performed adsorption/desorption cycling experiments on 1°,2°-alkylethylenediamine-appended variants of Mg2(dopbdc) under humid conditions (Fig. 5). Heating to 140 °C was found to be necessary to fully desorb both CO2 and water from these materials. While e-2–Mg2(dobpdc) displays a sharp CO2-adsorption step and minimal water co-adsorption, its propensity towards diamine loss during adsorption/desorption cycling precludes it from being useful for carbon capture applications (Fig. 5). For example, cycling this material from adsorption under a simulated coal flue gas stream (humid 15% CO2 in N2, 40 °C, 5 min)21 to desorption under humid pure CO2 (140 °C, 5 min) led to approximately 13% diamine loss after 60 cycles (∼0.2% loss per cycle). With the exception of nPr-2 (23% loss over 60 cycles), diamine loss upon cycling was less dramatic for Mg2(dobpdc) functionalized with the larger congeners of e-2, namely nBu-2 (11% diamine loss over 60 cycles), nPent-2 (3% loss), and nHex-2 (1% loss). A similar trend was observed with branched alkyl groups (i-2: 24%; cPent-2: 8%; 3-Pent-2: 5% diamine loss over 60 cycles) (Fig. S10†). Notably, the CO2/H2O cycling capacities of nPent-2–Mg2(dobpdc) and nHex-2–Mg2(dobpdc) remained stable over 60 adsorption/desorption cycles, and thus these adsorbents are the most suitable for long-term application in a carbon capture process.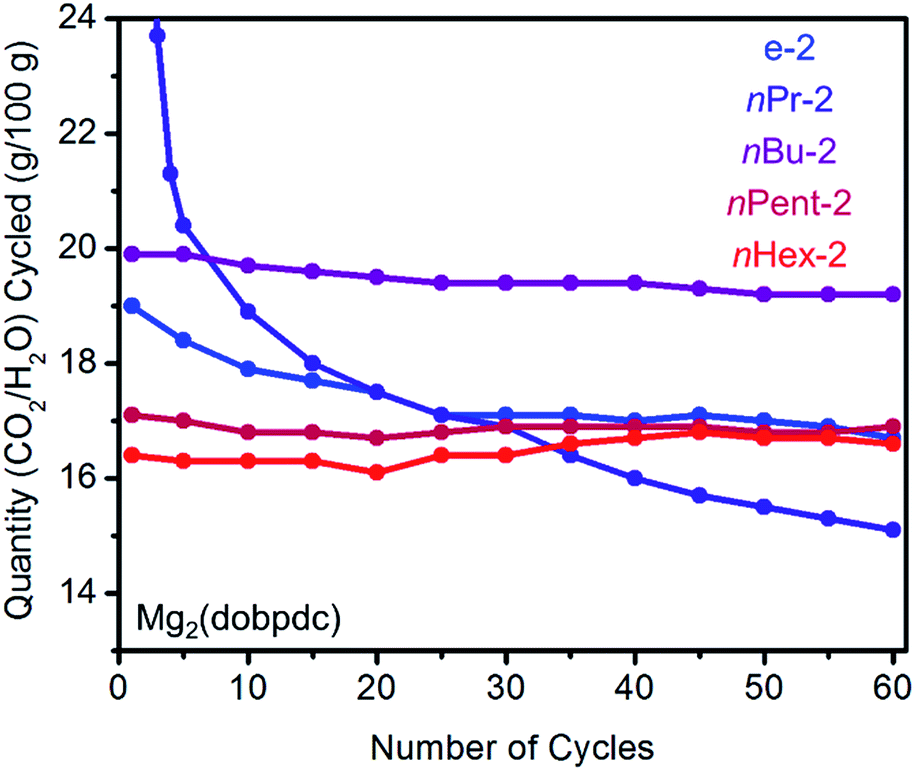 | ||
| Fig. 5 Cycling stability of 1°,2°-alkylethylenediamine-appended variants of Mg2(dobpdc) in a simulated temperature swing adsorption process. Adsorption conditions: humid 15% CO2 in N2, 40 °C, 5 min; desorption conditions: humid CO2, 140 °C, 5 min. Over 60 cycles, the diamine loadings decreased by 13%, 23%, 11%, 3%, and 1% for e-2, nPr-2, nBu-2, nPent-2, and nHex-2, respectively. | ||
Origin of the two CO2 adsorption steps
Examination of the single crystal X-ray diffraction structure of Zn2(dobpdc), which is isostructural to Mg2(dobpdc), provides a likely explanation for the origin of the two-step adsorption behavior exhibited by bulky alkylethylenediamine-append variants of Mg2(dobpdc) (Fig. 6).14b The hexagonal channels of Zn2(dobpdc) are not uniform; instead, they possess pairs of more closely associated Zn2+ centers in the a–b plane. As a consequence, for the diamine-appended frameworks, increasing the size of the alkyl group on the secondary amine leads to increasingly disfavorable steric interactions between adjacent diamines in the a–b plane. These unfavorable interactions would be exacerbated upon CO2 insertion into the M–N bond, which would bring the alkyl-substituted amines into steric conflict with one another. Therefore, we hypothesize that for adsorbents demonstrating two CO2 adsorption steps, the higher temperature step corresponds to initial formation of ammonium carbamate chains at half of the diamine sites, as illustrated in Fig. 6, with the formation of these ammonium carbamate chains blocking CO2 insertion at the paired sites in the a–b plane. An increase in the driving force for adsorption (as achieved, for example, by decreasing the temperature in isobaric measurements or increasing the pressure in isothermal measurements) would be required to facilitate formation of the second, more hindered set of ammonium carbamate chains. Notably, the second set of ammonium carbamate chains would be less thermodynamically stable, and is thus the most likely binding site for water co-adsorption (Fig. 3 and 4).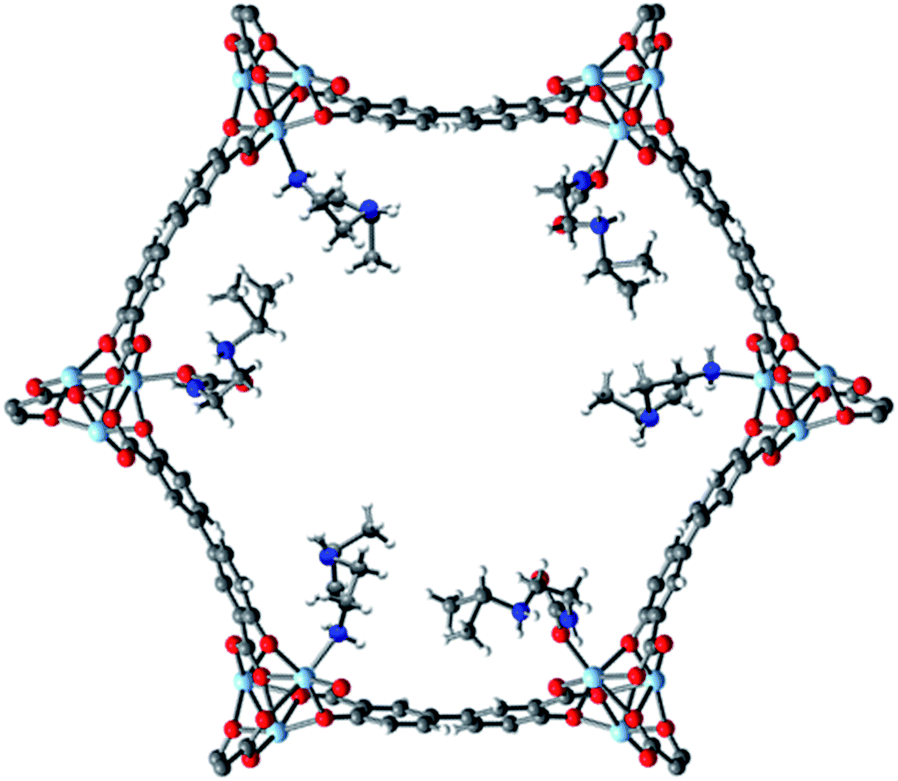 | ||
| Fig. 6 Proposed structure of the intermediate formed after the first adsorption step (50% capacity) of i-2–Zn2(dobpdc), which is isostructural to i-2–Mg2(dobpdc). As shown here, CO2 inserts into the Zn–N bonds to form ammonium carbamate chains along alternating rows of metal sites down the pore axis (c-axis). This mode of insertion occurs due to unfavorable interactions between paired diamine sites in the a–b plane. The second adsorption step leading to 100% capacity proceeds via insertion at the remaining diamine sites. Light blue, grey, red, dark blue, and white spheres correspond to Zn, C, O, N, and H, respectively. This model structure was prepared by overlaying the previously-reported single-crystal structures of Zn2(dobpdc)(i-2)1.94 and Zn2(dobpdc)(i-2–CO2)(i-2),14b both of which contain a single metal site in the asymmetric unit. | ||
There are two competing trends in the applicability of 1°,2°-alkylethylenediamine-appended variants of Mg2(dobpdc) for carbon capture: increasing the size of the alkyl group on the 2° amine leads to improved stability to diamine loss upon adsorption/desorption cycling, but also leads to two CO2 adsorption steps due to steric interactions within the a–b plane. These destabilizing interactions diminish the potential adsorption capacities of the materials under flue gas conditions and lead to increased water co-adsorption due to the formation of less stable ammonium carbamate chains. Based on these results, we hypothesized that reducing the steric interactions between adjacent diamines should mitigate the two-step CO2 adsorption/desorption profiles, thus leading to materials that combine the best features of the adsorbents described above, namely, stability to adsorption/desorption cycling, one sharp CO2 adsorption step, and minimal water co-adsorption.
Synthesis of the expanded framework Mg2(dotpdc) and grafting with 1°,2°-alkylethylenediamines
One strategy for minimizing unfavorable interactions between adjacent diamines is to change the base framework to an isoreticular structure with a larger spacing between the metal sites in the a–b plane. Specifically, changing the linker from dobpdc4− to the longer terphenyl ligand 4,4′′-dioxido-[1,1′:4′,1′′-terphenyl]-3,3′′-dicarboxylate (dotpdc4−) should better separate the ammonium carbamate chains from one another, as the opposing metal centers in the a–b plane would be approximately 5 Å farther apart (Fig. 7).18 Importantly, the separation between Mg2+ centers along the c-axis should remain at essentially the same distance in Mg2(dotpdc) as in Mg2(dobpdc), as previously demonstrated for the corresponding iron framework,18 a requirement for facilitating cooperative ammonium carbamate chain formation.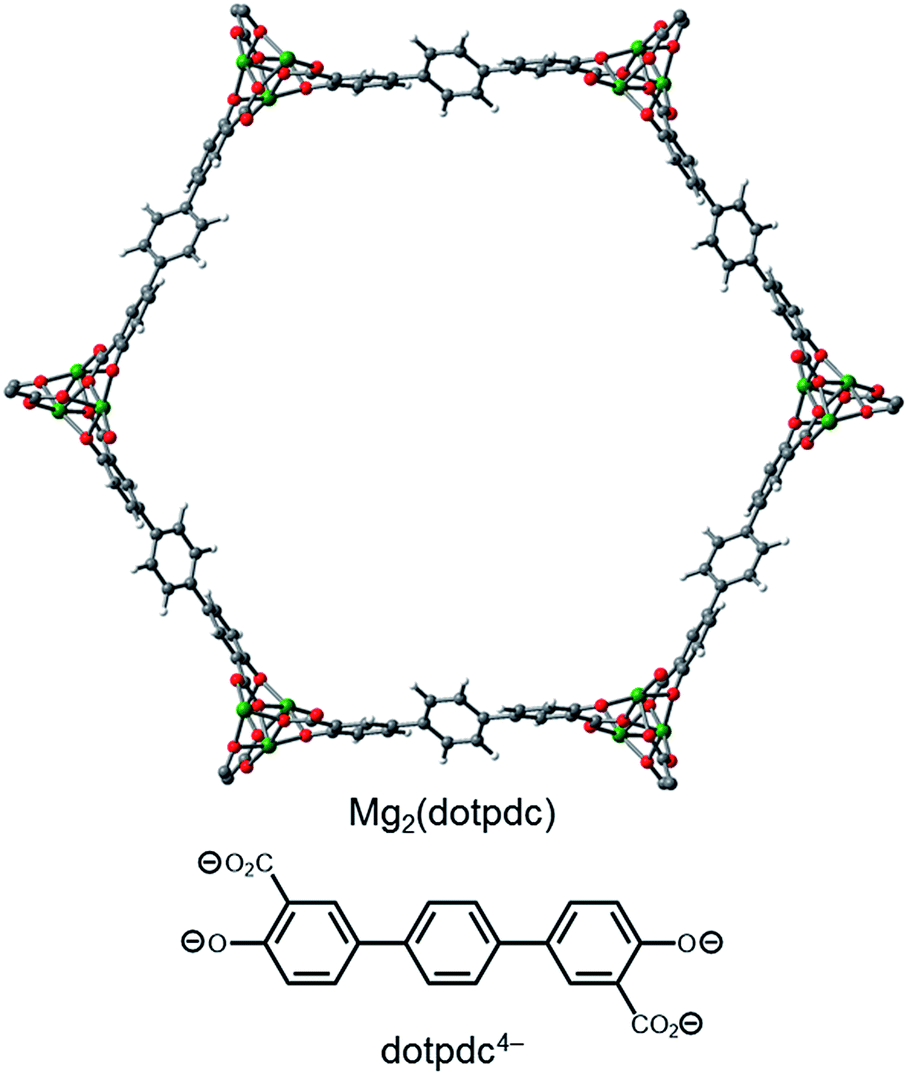 | ||
| Fig. 7 Structure of the organic linker dotpdc4− = 4,4′′-oxido-[1,1′:4′,1′′-terphenyl]-3,3′′-dicarboxylate and a model of the hexagonal pore of Mg2(dotpdc). Green, grey, red, and white spheres correspond to Mg, C, O, and H, respectively. | ||
Although Mg2(dotpdc) had not been synthesized prior to this work, related frameworks incorporating functionalized terphenyl ligands have been reported.12d,16,18,23 Gratifyingly, the same solvothermal conditions used to prepare Mg2(dobpdc) from H4dobpdc and Mg(NO3)2·6H2O14c afforded Mg2(dotpdc) in high yield as a pale yellow crystalline solid. The powder X-ray diffraction pattern of this material is consistent with a framework isoreticular in structure to Mg2(dobpdc) with a larger unit cell (Fig. S17†). In contrast to many metal–organic frameworks with large pore diameters, Mg2(dotpdc) does not show evidence of pore collapse after solvent evacuation10c,24 and does not form an interpenetrated structure,16 leading to a high 77 K N2 Brunauer–Emmett–Teller (BET) surface area of 3100 m2 g−1 (Langmuir surface area: 5840 m2 g−1) (Fig. S20†). The presence of accessible Mg2+ sites in Mg2(dotpdc) was confirmed by the sharp uptake of CO2 at low pressures in the adsorption isotherms collected at 25, 35, and 45 °C (Fig. S22†). At low loadings, the differential heat of CO2 adsorption is −40 kJ mol−1 (Fig. S23†), as determined from the Clausius–Clapeyron relationship (eqn (2)). This Δhads value is similar to those previously reported for related metal–organic frameworks with open Mg2+ sites, such as Mg2(dobpdc) (−43 kJ mol−1)14d and Mg2(dobdc) (dobdc4− = 2,5-dioxido-1,4-benzenedicarboxylate) (−42 kJ mol−1).25
The bulky 1°,2°-alkylethylenediamines that display two CO2 adsorption steps in Mg2(dobpdc) were grafted to Mg2(dotpdc). The standard grafting procedure led to >100% diamine loading in most cases, due to the presence of excess diamine in the large pores of Mg2(dotpdc) (Table S4†). Therefore, the activation temperatures of diamine-appended variants of Mg2(dotpdc) were carefully optimized based on N2 decomposition curves to facilitate complete removal of the excess diamine from the pores without loss of the metal-bound diamines (Fig. S26†). Using this strategy, Mg2(dotpdc) variants functionalized with the majority of the bulky 1°,2°-alkylethylenediamines shown in Fig. 2, including nHept-2, could be prepared with high loadings (>90%) remaining after activation (Table S4†). However, to date we have been unable to reproducibly prepare high quality samples of e-2–Mg2(dotpdc), possibly due to degradation of the framework upon exposure to this diamine.
CO2 adsorption, water co-adsorption, and adsorption/desorption cycling in 1°,2°-alkylethylenediamine-appended variants of Mg2(dotpdc)
Consistent with our hypothesis regarding the origin of the two-step CO2 adsorption/desorption behavior in Mg2(dobpdc), all of the 1°,2°-alkylethylenediamine-appended variants of Mg2(dotpdc) exhibit a single CO2 adsorption step in isobaric measurements (see Fig. S28† for individual CO2 adsorption/desorption isobars). In addition, the CO2 adsorption isotherm at 40 °C of Mg2(dotpdc) appended with nHept-2, the largest diamine studied, shows a single adsorption step (Fig. S29†). In many cases, the CO2 adsorption steps were shorter than expected assuming the adsorption of 1 CO2 per diamine, which is likely due to poor framework crystallinity and/or the presence of defects impeding the complete formation of ammonium carbamate chains.The CO2 adsorption steps in Mg2(dotpdc) appended with 1°,2°-alkylethylenediamines bearing linear alkyl groups occur at nearly the same temperature (inflection points: nPr-2 = 123 °C, nBu = 126 °C, nPent = 126 °C, nHex = 127 °C, nHept = 127 °C) (Fig. 8a). Notably, the CO2 adsorption step temperatures for the Mg2(dotpdc) frameworks are approximately 10 °C higher than for the corresponding Mg2(dobpdc) variants. Given the chemical similarity of the Mg2+ centers in Mg2(dobpdc) and Mg2(dotpdc), the slight increase in thermodynamic favorability of CO2 adsorption in Mg2(dotpdc) compared to Mg2(dobpdc) may result from the elimination of disfavorable steric interactions in the expanded framework. In contrast, branching on the alkyl group has a more substantial effect on the CO2 adsorption step temperatures (inflection points: i-2 = 111 °C, cPent-2 = 124 °C, 3-Pent-2 = 98 °C) (Fig. 8b).
 | ||
| Fig. 8 Pure CO2 adsorption isobars for a series of 1°,2°-alkylethylenediamine-appended variants of Mg2(dotpdc) bearing (a) linear and (b) branched alkyl groups. In both plots, the gradient from blue to red reflects the increasing size of the substituent on the secondary amine. | ||
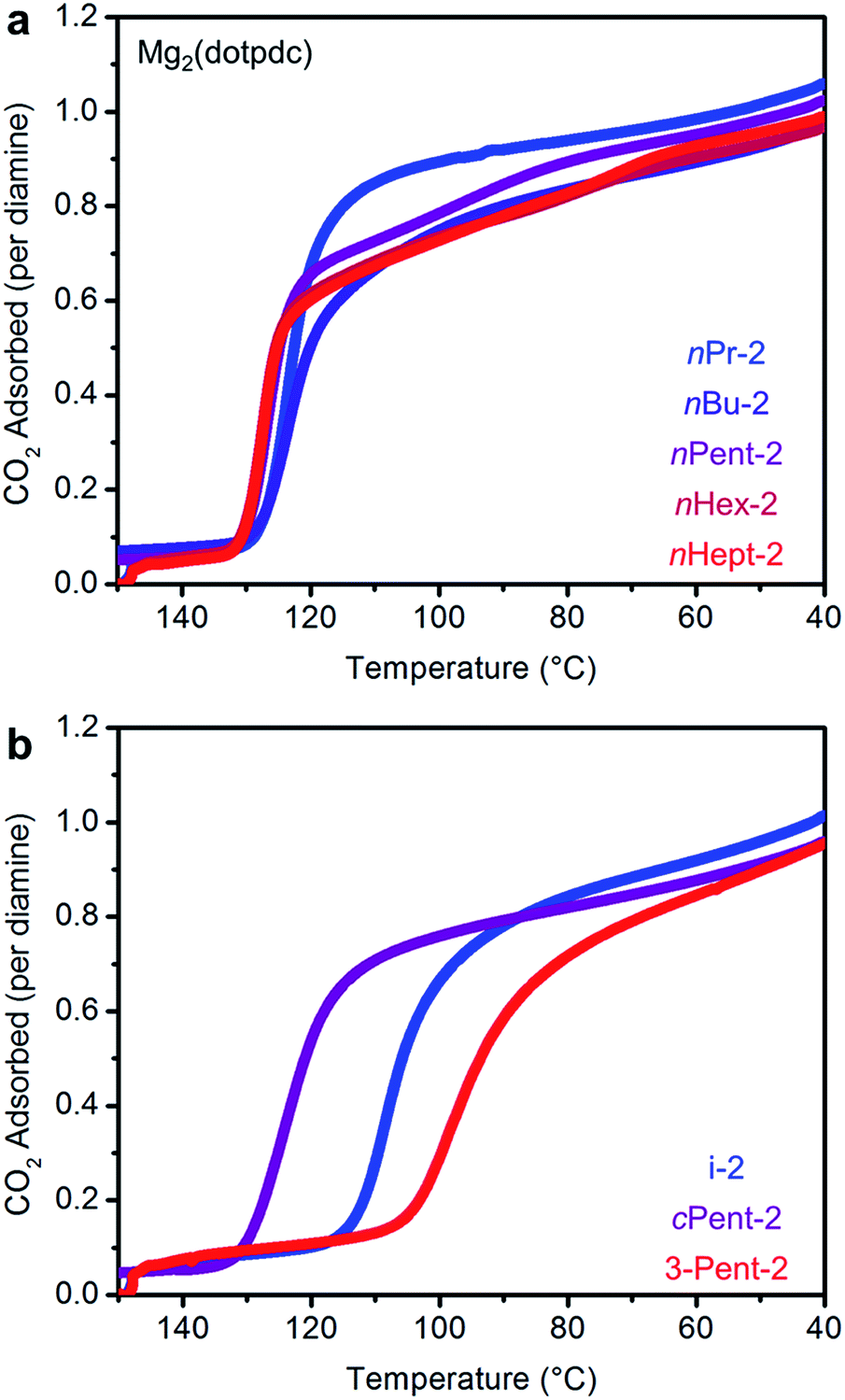
Having successfully eliminated the problematic two-step adsorption profiles of frameworks appended with bulky 1°,2°-alkylethylenediamines, we further evaluated CO2 adsorption under humid conditions in these materials (Fig. 9; see Fig. S30† for individual adsorption isobars). Given their single CO2 adsorption steps, 1°,2°-alkylethylenediamine-appended variants of Mg2(dotpdc) should form a single type of strongly bound ammonium carbamate chains. Therefore, diamines functionalized with hydrophobic alkyl groups should further display minimal water co-adsorption under humid conditions. Gratifyingly, all of the diamine-appended variants of Mg2(dotpdc) exhibit step-shaped CO2 adsorption with minimal excess mass uptake under humid conditions at 40 °C (as reflected in the differences between green and blue bars in Fig. 9). For example, nHept-2–Mg2(dotpdc) exhibited co-adsorption of only ∼0.8 g H2O/100 g, corresponding to 0.2 molecules per diamine (see Table S5†). Interestingly, this is the same amount of water co-adsorbed per diamine in e-2–Mg2(dobpdc), which also displays a single CO2 adsorption step. We note that the wet N2 isobars of these materials (purple bars, Fig. 9) confirm that water adsorption occurs readily in the absence of CO2, likely facilitated by hydrogen-bonding to the secondary amine. Therefore, the minimal water co-adsorption after CO2 adsorption is probably due to the formation of highly-stabilized ammonium carbamate chains lined with hydrophobic alkyl groups that exclude water. Importantly, the minimal amount of water co-adsorption observed with 1°,2°-alkylethylenediamine-appended variants of Mg2(dotpdc) should diminish the parasitic energy costs associated with water desorption upon regeneration of the adsorbent.
 | ||
| Fig. 9 Summary of the adsorption capacities at 40 °C for a series of 1°,2°-alkylethylenediamine-appended variants of Mg2(dotpdc) under dry CO2 (green), humid CO2 (blue), and humid N2 (purple). | ||
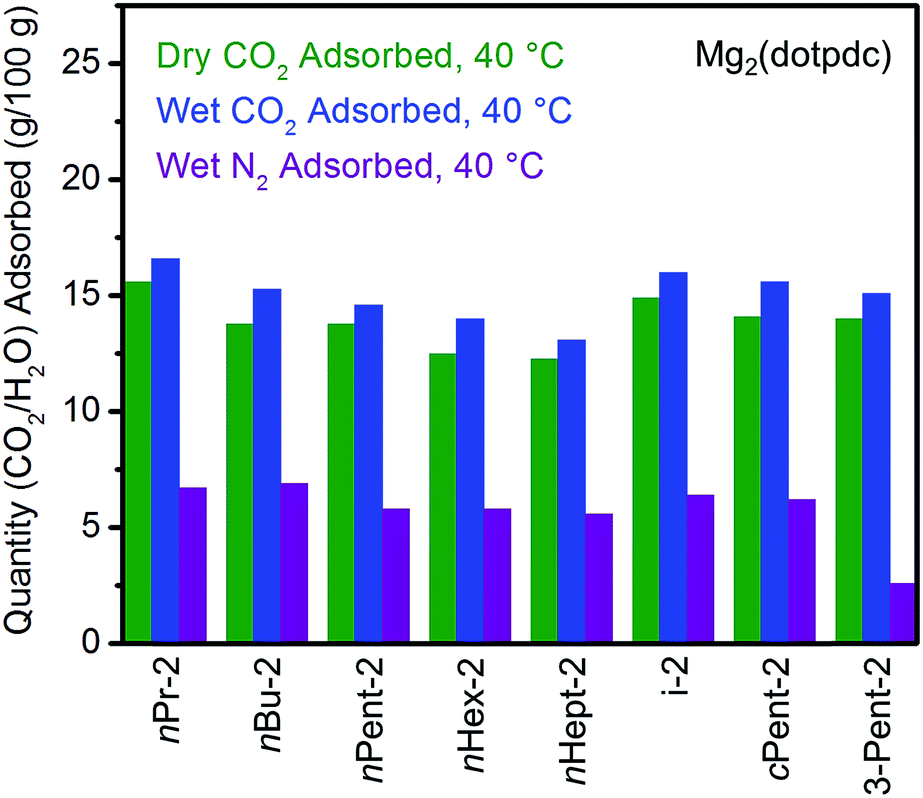
The cycling stabilities of several diamine-appended variants of Mg2(dotpdc) were also assessed (Fig. 10). As with Mg2(dobpdc) (Fig. 5), Mg2(dotpdc) variants functionalized with higher molecular weight diamines display greater thermal stability to diamine loss (Fig. S27†). Consistently, n-Pent-2–Mg2(dotpdc) and n-Hept-2–Mg2(dotpdc) exhibit stable adsorption/desorption cycling with only 3% diamine loss over 60 cycles. Given the low degree of water co-adsorption in these materials, the cycling capacities are almost entirely due to CO2 adsorption/desorption, and thus these materials demonstrate reasonably high CO2 working capacities (>9 g/100 g or >2.0 mmol g−1) for cycling under humid conditions.
 | ||
| Fig. 10 Cycling stability of 1°,2°-alkylethylenediamine-appended variants of Mg2(dotpdc) in a simulated temperature swing adsorption process. Adsorption conditions: humid 15% CO2 in N2, 40 °C, 5 min; desorption conditions: humid CO2, 140 °C, 5 min. Over 60 cycles, the diamine loadings decreased by 10%, 3%, and 3% for cPent-2, nPent-2, and nHept-2, respectively. | ||
Synthesis of Mg2(pc-dobpdc) and crystal structure of Zn2(pc-dobpdc)
Although changing the parent framework from Mg2(dobpdc) to Mg2(dotpdc) eliminated the two-step CO2 adsorption profiles observed with bulky 1°,2°-alkylethylenediamines, the latter framework presents several drawbacks. For instance, diamine-appended variants of Mg2(dotpdc) exhibit ∼20% lower gravimetric capacities due to the higher molecular weight of the organic linker and ∼40% lower volumetric capacities due to the ∼30% lower crystallographic density of Mg2(dotpdc).26,27 Therefore, we sought a framework with a crystallographic density similar to that of Mg2(dobpdc) capable of minimizing the unfavorable steric interactions between adjacent ammonium carbamate chains. As discussed above, the two-step adsorption profiles of diamine-appended variants of Mg2(dobpdc) originate from the distorted hexagonal pores, as illustrated with N,N-dimethylacetamide (DMA) solvent molecules (Fig. 11, top). In contrast, a framework with uniform hexagonal channels would exhibit a longer distance between each diamine and its nearest neighbor in the a–b plane and therefore might not exhibit a thermodynamic preference for forming alternating ammonium carbamate chains.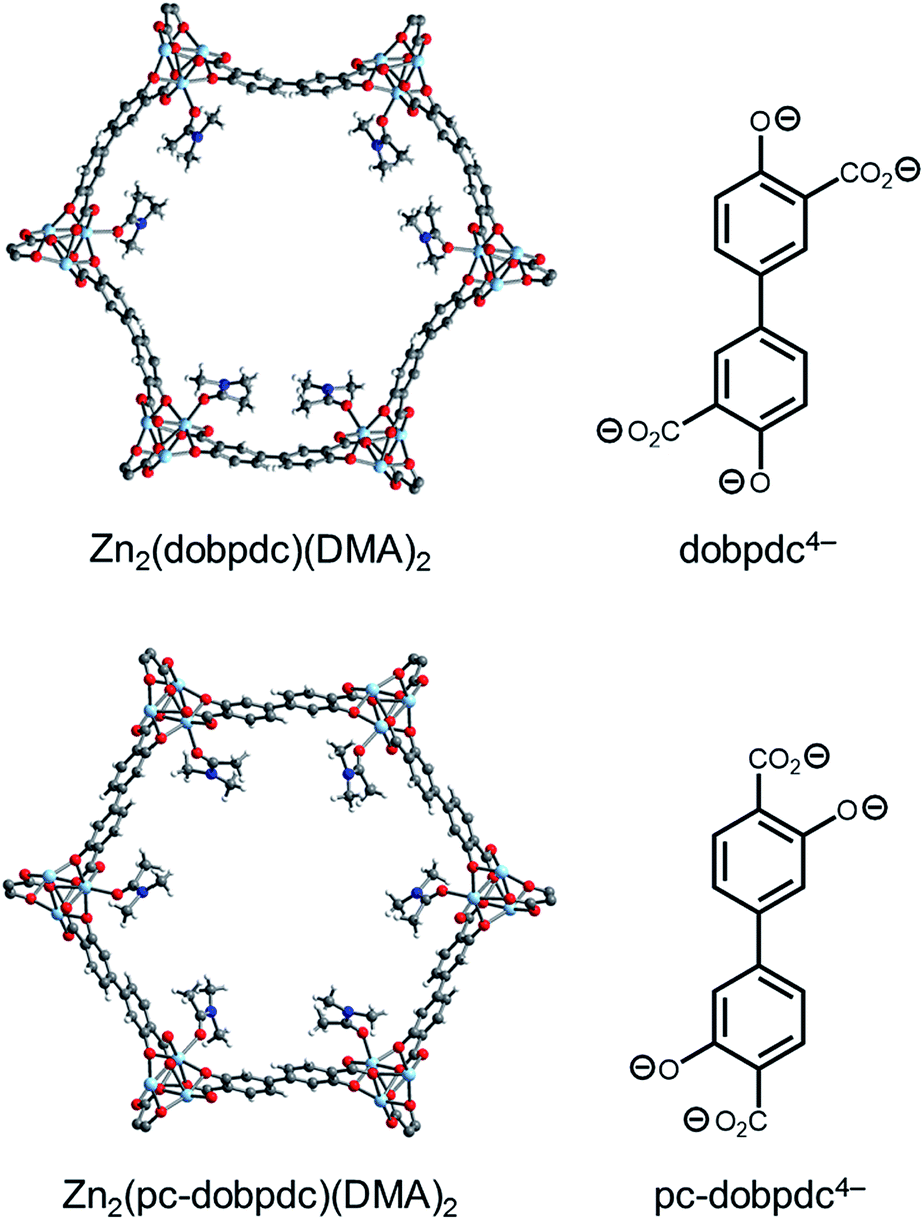 | ||
| Fig. 11 Single-crystal X-ray diffraction structures of Zn2(dobpdc)(DMA)2 (top) and Zn2(pc-dobpdc)(DMA)2 (bottom) collected at 100 K.14b The structures of the ligands are included for comparison. Light blue, grey, red, dark blue, and white spheres correspond to Zn, C, O, N, and H, respectively. DMA = N,N-dimethylacetamide. | ||
We have previously reported that frameworks incorporating the isomeric ligands 2,5-dioxido-1,4-benzenedicarboxylate and 2,4-dioxido-1,5-benzenedicarboxylate exhibit slightly different pore architectures that lead to differences in the gas adsorption properties.28 Inspired by this work, we sought a suitable structural isomer of Mg2(dobpdc) with a more symmetric pore structure. As such, we produced single crystals of Zn-IRMOF-74-II, which we refer to here as Zn2(pc-dobpdc) (pc-dobpdc4− = 3,3′-dioxidobiphenyl-4,4′-dicarboxylate, pc = para-carboxylate), a recently reported framework prepared with a linker isomeric in structure to dobpdc4−.16 Gratifyingly, the single-crystal X-ray diffraction structure of Zn2(pc-dobpdc)(DMA)2 possesses uniformly hexagonal channels (Fig. 11, bottom). This subtle change in framework structure is accompanied by a change from the inversion-twinned crystals in the enantiomorphic space groups P3221/P3121 for Zn2(dobpdc)(DMA)2 to achiral crystals in the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) for Zn2(pc-dobpdc)(DMA)2, which leads to the coplanarity of the two aromatic rings in Zn2(pc-dobpdc)(DMA)2 compared to the 38.3(2)° twist in Zn2(dobpdc)(DMA)2. Apart from these differences, the structures of Zn2(pc-dobpdc)(DMA)2 and Zn2(dobpdc)(DMA)2 are quite similar, with nearly identical unit cell lengths along the c-axis (6.7186(2) Å and 6.6937(4) Å, respectively). Notably, the crystallographic density of Zn2(pc-dobpdc)(DMA)2 (1.103 g cm−3) is approximately the same as that of Zn2(dobpdc)(DMA)2 (1.066 g cm−3), such that the volumetric CO2 uptake capacities of the diamine-appended variants of these frameworks should be similar.
for Zn2(pc-dobpdc)(DMA)2, which leads to the coplanarity of the two aromatic rings in Zn2(pc-dobpdc)(DMA)2 compared to the 38.3(2)° twist in Zn2(dobpdc)(DMA)2. Apart from these differences, the structures of Zn2(pc-dobpdc)(DMA)2 and Zn2(dobpdc)(DMA)2 are quite similar, with nearly identical unit cell lengths along the c-axis (6.7186(2) Å and 6.6937(4) Å, respectively). Notably, the crystallographic density of Zn2(pc-dobpdc)(DMA)2 (1.103 g cm−3) is approximately the same as that of Zn2(dobpdc)(DMA)2 (1.066 g cm−3), such that the volumetric CO2 uptake capacities of the diamine-appended variants of these frameworks should be similar.
Based on the single-crystal X-ray diffraction structure of Zn2(pc-dobpdc)(DMA)2, we surmised that diamine-appended variants of Mg2(pc-dobpdc) should possess more regularly spaced diamines in the a–b plane than the corresponding Mg2(dobpdc) analogues. Although the Mg analogue of Zn2(pc-dobpdc) has been prepared previously,16 the reported BET surface area of 2510 m2 g−1 is significantly lower than that of Mg2(dobpdc) (3330 m2 g−1).14c This is unexpected given the similar single-crystal X-ray diffraction structures of their Zn-analogues (Fig. 11). Following the published procedure but employing more thorough washing with N,N-dimethylformamide and methanol enabled the synthesis of Mg2(pc-dobpdc) with an increased 77 K N2 BET surface area of 3000 m2 g−1 (Fig. S38†).29,30 Using this higher surface area material, we determined that the Δhads for CO2 at low loadings in this framework is similar (−38 kJ mol−1) to that of other adsorbents bearing open Mg2+ sites (Fig. S39 and S40†).14d,25 Therefore, the major structural difference between Mg2(dobpdc) and Mg2(pc-dobpdc) is the disposition of the Mg2+ sites within the hexagonal channels.
CO2 adsorption, water co-adsorption, and adsorption/desorption cycling in 1°,2°-alkylethylenediamine-appended variants of Mg2(pc-dobpdc)
The standard procedure of exchanging the bound methanol on the Mg2+ sites of Mg2(pc-dobpdc) with 1°,2°-alkylethylenediamines was employed to prepare the corresponding diamine-appended frameworks. In most cases, high diamine loadings of ≥90% could be obtained, and even with the large diamine nHept-2, a reasonably high diamine loading of 79% was reliably obtained (Table S7†). Remarkably, all of the linear 1°,2°-alkylethylenediamine-appended variants of Mg2(pc-dobpdc), even nHept-2–Mg2(pc-dobpdc), display a single sharp CO2 adsorption step (Fig. 12a) at nearly the same temperature (inflection points: e-2 = 117 °C, nPr-2 = 116 °C, nBu-2 = 117 °C, nPent-2 = 116 °C, nHex-2 = 112 °C, nHept-2 = 112 °C) (see Fig. S45† for individual CO2 adsorption/desorption isobars). Additionally, the CO2 adsorption isotherm at 40 °C of Mg2(pc-dobpdc) appended with the largest diamine, nHept-2, confirmed the presence of a single CO2 adsorption step at 0.7 mbar (Fig. S46†). Similarly, Mg2(pc-dobpdc) variants appended with branched 1°,2°-alkylethylenediamines (Fig. 12b) and bulky 1°,3°-alkylethylenediamines (Fig. S51†) display a single CO2 adsorption step, with adsorption temperatures comparable to those observed in the corresponding Mg2(dotpdc) variants. Thus, the subtle change in the orientation of the metal sites in Mg2(pc-dobpdc) prevents the two-step CO2 adsorption/desorption profiles observed with these diamines in Mg2(dobpdc). Notably, due to the minimal hysteresis observed upon CO2 desorption (Fig. S45†), these adsorbents could be regenerated under pure CO2 at temperatures below 140 °C, with the exception of cPent-2–Mg2(pc-dobpdc). Because of their low CO2 step pressures and higher gravimetric and volumetric uptake capacities compared to the corresponding Mg2(dotpdc)-based adsorbents, these 1°,2°-alkylethylenediamine-appended variants of Mg2(pc-dobpdc) are more promising for carbon capture applications.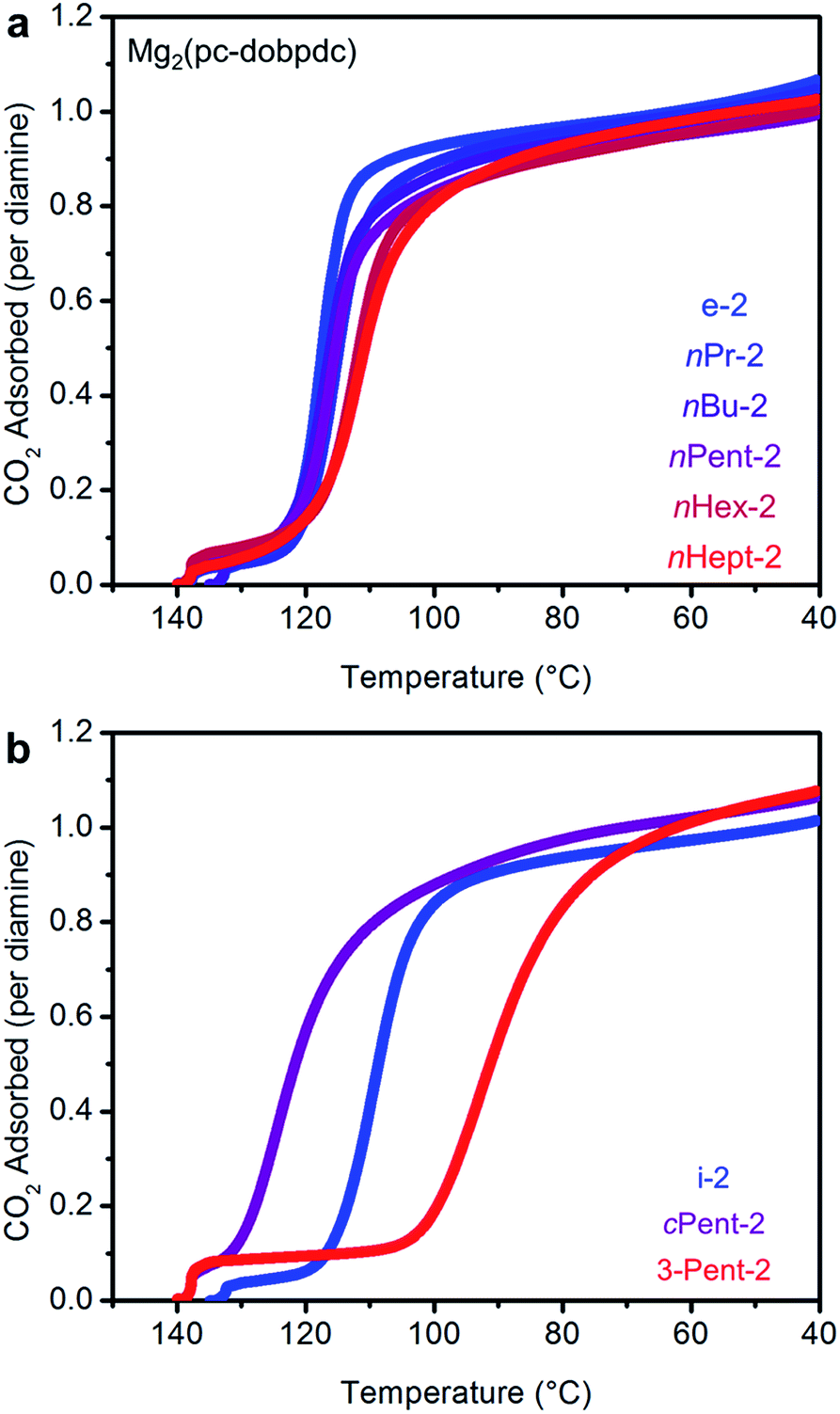 | ||
| Fig. 12 Pure CO2 adsorption isobars for a series of 1°,2°-alkylethylenediamine-appended variants of Mg2(pc-dobpdc) bearing (a) linear and (b) branched alkyl groups. In both plots, the gradient from blue to red reflects the increasing size of the substituent on the secondary amine. | ||
In order to further evaluate the applicability of diamine-appended variants of Mg2(pc-dobpdc) for CO2 capture, their performance under humid conditions was assessed by TGA (Fig. 13, see Fig. S47† for individual adsorption isobars). As observed with Mg2(dobpdc) and Mg2(dotpdc), diamine-appended variants of Mg2(pc-dobpdc) exhibit step-shaped adsorption of CO2 under humid conditions at temperatures similar to those under dry CO2 (Fig. S47†). In contrast to the results shown in Fig. 3 and 4, the variant functionalized with the smallest diamine, e-2–Mg2(pc-dobpdc), exhibited the most co-adsorption of water under humid conditions in this series, on both a gravimetric (∼5.6 g H2O/100 g) and molar (∼0.8 molecules of water per diamine) basis (Fig. 13). In addition, steadily decreasing amounts of water co-adsorption are apparent for the Mg2(pc-dobpdc) materials as the alkyl group on the diamine becomes larger, leading to only ∼1.7 g H2O/100 g (∼0.3 molecules of water per diamine) of water co-adsorption in nHept-2–Mg2(pc-dobpdc) (Table S8†). This steady decrease in the degree of water co-adsorption contrasts with the results observed with both the Mg2(dobpdc) and Mg2(dotpdc) families. Indeed, for Mg2(dobpdc), more water co-adsorption was observed with larger diamines due to the presence of the second CO2 adsorption step (Fig. 4), whereas for Mg2(dotpdc), all diamines exhibited approximately the same molar amount of water co-adsorption (0.2–0.3 molecules of water per diamine) (Table S5†), potentially due to the increased hydrophobicity of the terphenyl framework.
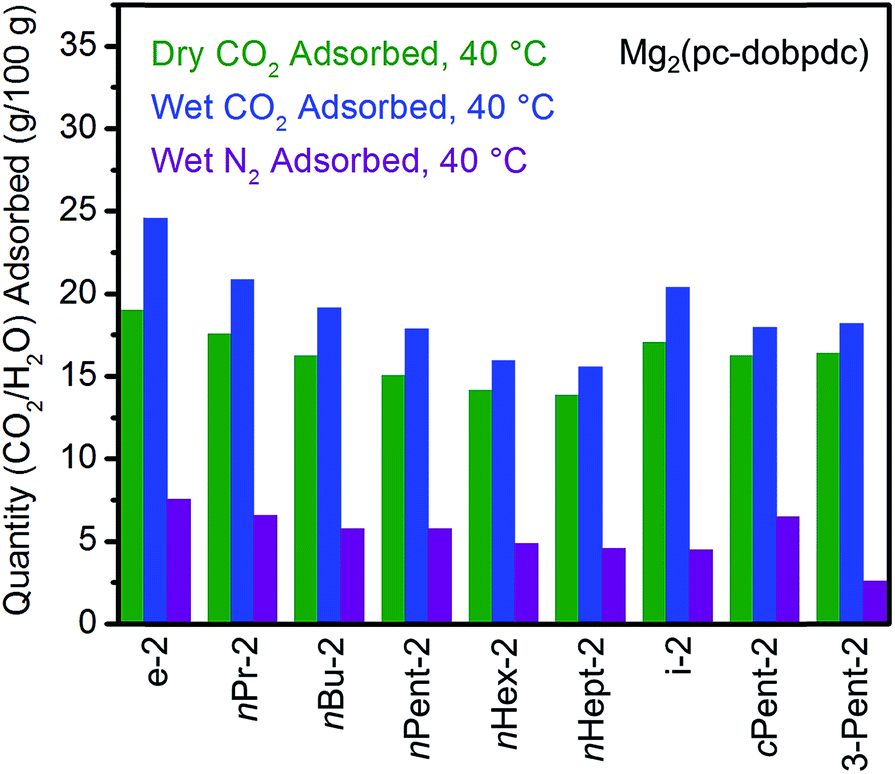 | ||
| Fig. 13 Summary of the adsorption capacities at 40 °C for a series of 1°,2°-alkylethylenediamine-appended variants of Mg2(pc-dobpdc) under dry CO2 (green), humid CO2 (blue), and humid N2 (purple). | ||
As discussed above, diamines with large alkyl groups exhibit improved cycling stabilities (Fig. 5 and 10) compared to their lower molecular weight analogues. Consistent with this trend, nHept-2–Mg2(pc-dobpdc) displays excellent thermal stability with negligible diamine loss (<1%) over 60 cycles, whereas nPent-2–Mg2(pc-dobpdc) shows gradual diamine loss (∼5%) upon cycling (Fig. 14). In addition, nHept-2–Mg2(pc-dobpdc) exhibits a high cycling capacity of ∼13 g/100 g, which should correspond primarily to CO2 (∼11.3 g/100 g = 2.6 mmol g−1) given the minimal amount of water co-adsorbed in humid isobaric measurements (Fig. 13). As expected, this cycling capacity is higher than that observed for nHept-2–Mg2(dotpdc) (∼8.1 g/100 g, Fig. 10), due to the higher molecular weight of the framework in the latter case. Therefore, nHept-2–Mg2(pc-dobpdc) warrants further study for carbon capture applications due to its sharp CO2 adsorption/desorption steps, minimal water co-adsorption under humid conditions, high thermal stability, and low CO2 adsorption step pressure.
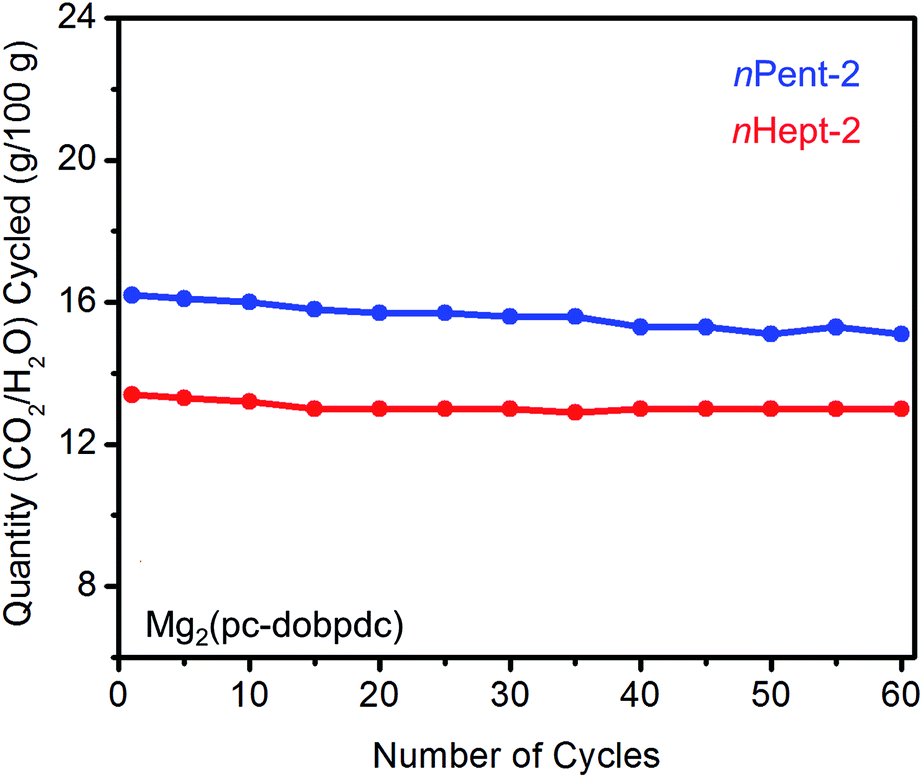 | ||
| Fig. 14 Cycling stability of 1°,2°-alkylethylenediamine-appended variants of Mg2(pc-dobpdc) in a simulated CCS process. Adsorption conditions: humid 15% CO2 in N2, 40 °C, 5 min; desorption conditions: humid CO2, 140 °C, 5 min. Over 60 cycles, the diamine loadings decreased by 5% for nPent-2 and <1% for nHept-2. | ||
Conclusions
The applicability of 1°,2°-alkylethylenediamine-appended metal–organic frameworks for carbon capture has been assessed by evaluating their CO2 capture performance under humid conditions as well as their stability to adsorption/desorption cycling in a simulated temperature swing adsorption process. We have identified several important trends that are relevant to their application for carbon capture.First, diamines bearing large alkyl groups impede uniform ammonium carbamate chain formation in Mg2(dobpdc), likely due to unfavorable steric interactions in the a–b plane of the framework. The destabilization of adjacent ammonium carbamate chains leads to two-step CO2 adsorption/desorption profiles, as well as increased water co-adsorption under humid conditions. The steric interactions between adjacent diamines can be minimized by judiciously positioning the metal sites farther apart from one another, using frameworks with either a longer organic linker (Mg2(dotpdc)) or a more uniformly hexagonal pore structure (Mg2(pc-dobpdc)). Notably, these findings represent the first examples of cooperative CO2 chemisorption in metal–organic frameworks other than the M2(dobpdc) (M = Mg, Mn, Fe, Co, Zn) series.14,15
Second, increasing the molecular weight of the diamine provides a general strategy for improving the thermal stability of the resulting diamine-appended metal–organic framework towards diamine loss under humid conditions. However, increasing the size of the diamine also leads to decreased surface areas and gravimetric CO2 uptake capacities. Therefore, only the largest diamine necessary to achieve stable adsorption/desorption cycling should be employed for a carbon capture process.
Third, when developing new adsorbents for carbon capture from a humid gas stream, it is important to target adsorbents that not only competitively capture CO2 under humid conditions, but also do so with minimal water co-adsorption. Importantly, for 1°,2°-alkylethylenediamine-appended variants of Mg2(pc-dobpdc), increasing the size of the alkyl group leads to steadily decreasing molar amounts of water co-adsorption, indicating that the addition of large alkyl groups to the pore environment is a strategy for potentially minimizing water co-adsorption. In addition, our findings with diamine-appended variants of Mg2(dotpdc) suggest that the framework architecture also has an effect on the overall hydrophobicity of the pores. Therefore, synergistic optimization of both the diamine and framework structure can be used to minimize water co-adsorption in diamine-appended metal–organic frameworks. Notably, the majority of these adsorbents display minimal water co-adsorption at temperatures above 60 °C, indicating that parasitic costs can also be reduced by carrying out carbon capture at elevated temperatures.
These extensive studies indicated the particular promise of nHept-2–Mg2(dotpdc) and nHept-2–Mg2(pc-dobpdc) for carbon capture, due to their sharp CO2 adsorption steps at low pressures (≤0.7 mbar at 40 °C), minimal water co-adsorption under humid conditions, and high thermal stability to diamine loss upon adsorption/desorption cycling in a simulated temperature swing adsorption process. Given its higher gravimetric and volumetric CO2 adsorption capacities, nHept-2–Mg2(pc-dobpdc) is the more promising of the two adsorbents, and future studies will focus on developing a scalable synthesis of Mg2(pc-dobpdc) and further assessing the application of nHept-2–Mg2(pc-dobpdc) for carbon capture from humid gas streams.
Conflicts of interest
The authors declare the following competing financial interests: J. R. L. has a financial interest in Mosaic Materials, Inc., a start-up company working to commercialize metal–organic frameworks for gas separations, including CO2 capture applications. The University of California, Berkeley and ExxonMobil Research and Engineering Company have applied for a joint patent on some of the materials discussed herein, on which J. R. L., S. C. W., P. J. M., J. D. M., and R. L. S. are listed as inventors.Acknowledgements
We gratefully acknowledge ExxonMobil Research and Engineering Company for financial support of this work. The initial synthesis and characterization of m-2–Mg2(dobpdc), e-2–Mg2(dobpdc), i-2–Mg2(dobpdc), and ee-2–Mg2(dobpdc) was funded by the Advanced Research Projects Agency – Energy (ARPA-E), U.S. Department of Energy, under award number DE-AR00040. The crystallographic studies of Zn2(pc-dobpdc) were supported through the Center for Gas Separations Relevant to Clean Energy Technologies, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under Award DE-SC0001015, which used the resources of the Advanced Light Source at Lawrence Berkeley National Laboratory, a user facility supported by the Director, Office of Science, Office of Basic Energy Sciences, of the DOE under Contract No. DE-AC02-05CH11231. We thank the National Institute of General Medical Sciences of the National Institutes of Health for a postdoctoral fellowship for P. J. M. (F32GM120799). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We further thank the Miller Institute for Basic Research in Science for a post-doctoral fellowship for J. D. M., Miguel Gonzalez (UC Berkeley) for experimental assistance and helpful discussions, Dr Joseph Falkowski (ExxonMobil Researching and Engineering Company) for helpful discussions, and Dr Katie Meihaus (UC Berkeley) for editorial assistance with this manuscript.Notes and references
- R. K. Pachauri and L. A. Meyer, Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, International Government Panel on Climate Change, Geneva, Switzerland, 2014 Search PubMed.
- (a) S. Chu, Science, 2009, 325, 1599 CrossRef CAS PubMed; (b) R. S. Haszeldine, Science, 2009, 325, 1647 CrossRef CAS PubMed.
- (a) M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. MacDowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallet, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130 RSC; (b) A. S. Bhown and B. C. Freeman, Environ. Sci. Technol., 2011, 45, 8624 CrossRef CAS PubMed; (c) G. T. Rochelle, Science, 2009, 325, 1652 CrossRef CAS PubMed.
- (a) S. B. Fredriksen and K.-J. Jens, Energy Procedia, 2013, 37, 1770 CrossRef CAS; (b) C. Gouedard, D. Picq, F. Launay and P.-L. Carrette, Int. J. Greenhouse Gas Control, 2012, 10, 244 CrossRef CAS.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815 RSC.
- For selected reviews, see: (a) S.-Y. Lee and S. J. Park, Ind. Eng. Chem., 2015, 23, 1 CrossRef CAS; (b) A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438 CrossRef CAS; (c) S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796 CrossRef CAS PubMed.
- (a) W. R. Woerner, A. M. Plonka, X. Chen, D. Banerjee, P. K. Thallapally and J. B. Parise, J. Phys. Chem. C, 2016, 120, 360 CrossRef CAS; (b) C. Kim, H. S. Cho, S. Chang, S. J. Cho and M. Choi, Energy Environ. Sci., 2016, 9, 1803 RSC; (c) J. A. Mason, T. M. McDonald, T.-H. Bae, J. E. Bachman, K. Sumida, J. J. Dutton, S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2015, 137, 4787 CrossRef CAS PubMed; (d) Y. Wang and M. D. LeVan, J. Chem. Eng. Data, 2010, 55, 3189 CrossRef CAS.
- (a) E. E. Ünveren, B. O. Monkul, S. Sarioğlan, N. Karademir and E. Alper, Petroleum, 2017, 3, 37 CrossRef; (b) S. A. Didas, M. A. Sakwa-Novak, G. S. Foo, C. Sievers and C. W. Jones, J. Phys. Chem. Lett., 2014, 5, 4194 CrossRef CAS PubMed; (c) Z. Bacsik, N. Ahlsten, A. Ziadi, G. Zhao, A. E. Garcia-Bennett, B. Martín-Matute and N. Hedin, Langmuir, 2011, 27, 11118 CrossRef CAS PubMed; (d) A. Sayari and Y. Belmabkhout, J. Am. Chem. Soc., 2010, 132, 6312 CrossRef CAS PubMed; (e) R. Serna-Guerrero, E. Da'na and A. Sayari, Ind. Eng. Chem. Res., 2008, 47, 9406 CrossRef CAS.
- (a) R. Veneman, W. Zhao, Z. Li, N. Cai and D. W. F. Brilman, Energy Procedia, 2014, 63, 2336 CrossRef CAS; (b) X. Xu, C. Song, B. G. Miller and A. W. Scaroni, Ind. Eng. Chem. Res., 2005, 44, 8113 CrossRef CAS; (c) R. S. Franchi, P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2005, 44, 8007 CrossRef CAS.
- (a) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed; (b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 123044 CrossRef PubMed; (c) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674 CrossRef CAS PubMed; (b) R. Sabouni, H. Kazemian and S. Rohani, Environ. Sci. Pollut. Res., 2014, 21, 5427 CrossRef CAS PubMed; (c) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed; (d) Y. Liu, Z. U. Wang and H.-C. Zhou, Greenhouse Gases: Sci. Technol., 2012, 2, 239 CrossRef CAS; (e) J.-R. Li, Y. Ma, M. C. McCarthy, J. Sculley, J. Yu, H.-K. Jeong, P. B. Balbuena and H.-C. Zhou, Coord. Chem. Rev., 2011, 255, 1791 CrossRef CAS; (f) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef PubMed.
- For selected examples and reviews, see: (a) Y. Lin, C. Kong and L. Chen, RSC Adv., 2016, 6, 32598 RSC; (b) Z. Qiao, N. Wang, J. Jiang and J. Zhou, Chem. Commun., 2016, 52, 974 RSC; (c) P.-Q. Liao, X.-W. Chen, S.-Y. Liu, X.-Y. Li, Y.-T. Xu, M. Tang, Z. Rui, H. Ji, J. P. Zhang and X.-M. Chen, Chem. Sci., 2016, 7, 6528 RSC; (d) A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863 CrossRef CAS PubMed; (e) Y. Cao, F. Song, Y. Zhao and Q. Zhong, J. Environ. Sci., 2013, 25, 2081 CrossRef CASC. Montoro, E. García, S. Calero, M. A. Pérez-Fernández, A. L. López, E. Barea and J. A. R. Navarro, J. Mater. Chem., 2012, 22, 10155 RSC; (f) J. Liu, P. K. Thallapally, B. P. MGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308 RSC.
- For selected examples and reviews, see: (a) P. M. Bhatt, Y. Belmabkhout, A. Cadiau, K. Adil, O. Shekhah, A. Shkurenko, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2016, 138, 9301 CrossRef CAS PubMed; (b) C. A. Fernandez, S. K. Nune, H. V. Annapureddy, L. X. Dang, B. P. McGrail, F. Zheng, E. Polikarpov, D. L. King, C. Freeman and K. P. Brooks, Dalton Trans., 2015, 44, 13490 RSC; (c) S. K. Elsaidi, M. H. Mohamed, H. T. Schaef, A. Kumar, M. Lusi, T. Pham, K. A. Forrest, B. Space, W. Xu, G. J. Halder, J. Liu, M. Zaworotko and P. K. Thallapally, Chem. Commun., 2015, 51, 15530 RSC; (d) J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594 RSC; (e) P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80 CrossRef CAS PubMed; (f) T.-H. Chen, I. Popov, O. Zenasni, O. Daugulis and O. S. Milanić, Chem. Commun., 2013, 49, 6846 RSC; (g) S. D. Burd, S. Ma, J. A. Perman, B. J. Sikora, R. Q. Snurr, P. K. Thallapally, J. Tian, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 3663 CrossRef CAS PubMed; (h) C. Yang, U. Kaipa, Q. Z. Mather, X. Wang, V. Nesterov, A. F. Venero and M. A. Omary, J. Am. Chem. Soc., 2011, 133, 18094 CrossRef CAS PubMed; (i) J. G. Nguyen and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 4560 CrossRef CAS PubMed; (j) K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2010, 40, 498 RSC.
- (a) P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runčevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541 CrossRef CAS PubMed; (b) R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhown and J. R. Long, J. Am. Chem. Soc., 2017, 139, 10526 CrossRef CAS PubMed; (c) T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303 CrossRef CAS PubMed; (d) W. S. Drisdell, R. Poloni, T. M. McDonald, T. A. Pascal, L. F. Wan, C. D. Pemmaraju, B. Vlaisavlievich, S. O. Odoh, J. B. Neaton, J. R. Long, D. Prendergast and J. B. Kortright, Phys. Chem. Chem. Phys., 2015, 17, 2144 RSC; (e) T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056 CrossRef CAS PubMed.
- (a) H. Jo, W. R. Lee, N. W. Kim, H. Jung, K. S. Lim, J. E. Kim, D. W. Kang, H. Lee, V. Hiremath, J. G. Seo, H. Jin, D. Moon, S. S. Han and C. S. Hong, ChemSusChem, 2017, 10, 541 CrossRef CAS PubMed; (b) W. R. Lee, H. Jo, L.-M. Yang, H. Lee, D. W. Ryu, K. S. Lim, J. H. Song, D. Y. Min, S. S. Han, J. G. Seo, Y. K. Park, D. Moon and C. S. Hong, Chem. Sci., 2015, 6, 3697 RSC; (c) W. R. Lee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744 RSC.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018 CrossRef CAS PubMed.
- J.-W. Lee and R. Klajn, Chem. Commun., 2015, 51, 2036 RSC.
- D. J. Xiao, J. Oktawiec, P. J. Milner and J. R. Long, J. Am. Chem. Soc., 2016, 138, 14371 CrossRef CAS PubMed.
- Careful analysis of the pure CO2 desorption isobar of e-2–Mg2(dobpdc) (Fig. S8†) revealed the possible presence of two subtle CO2 desorption steps. Consistently, we have previously observed that the single-crystal X-ray diffraction structure of CO2-inserted e-2–Zn2(dobpdc) possesses two distinct ammonium carbamate chain conformations (ref. 14b).
- For examples with CO2, see: (a) M. Ichikawa, A. Kondo, H. Noguchi, N. Kojima, T. Ohba, H. Kajiro, Y. Hattori and H. Kanoh, Langmuir, 2016, 32, 9722 CrossRef CAS PubMed; (b) W.-P. Wu, Z.-S. Li, B. Liu, P. Liu, Z.-P. Xi and Y.-Y. Wang, Dalton Trans., 2015, 44, 10141 RSC; (c) S. Sanda, S. Parshamoni and S. Konar, Inorg. Chem., 2013, 52, 12866 CrossRef CAS PubMed; (d) H. J. Park and M. P. Suh, Chem. Commun., 2010, 46, 610 RSC; (e) S. Bourrelly, P. L. Llewellyn, C. Serre, F. Millange, T. Loiseau and G. Férey, J. Am. Chem. Soc., 2005, 127, 13519 CrossRef CAS PubMed; for examples with N2, see: (f) M. K. Taylor, T. Runčevski, J. Oktawiec, M. I. Gonzalez, R. L. Siegelman, J. A. Mason, J. Ye, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2016, 138, 15019 CrossRef CAS PubMed; (g) F. Salles, G. Maurin, C. Serre, P. L. Llewellyn, C. Knöfel, H. J. Choi, Y. Filinchuk, L. Oliviero, A. Vimont, J. R. Long and G. Férey, J. Am. Chem. Soc., 2010, 132, 13782 CrossRef CAS PubMed; (h) A. Kondo, H. Noguchi, L. Carlucci, D. M. Proserpio, G. Ciani, H. Kajiro, T. Ohba, H. Kanoh and K. Kaneko, J. Am. Chem. Soc., 2007, 129, 12362 CrossRef CAS PubMed.
- E. J. Granite and H. W. Pennline, Ind. Eng. Chem. Res., 2002, 41, 5470 CrossRef CAS.
- Activation of Mg2(dobpdc) variants appended with 2°,2° diamines such as N,N′-dimethylethylenediamine and N,N′-diethylethylenediamine under flowing humid N2 at high temperatures (130–150 °C) for extended periods of time led to substantial diamine volatilization from the Mg2+ sites. This is likely due to the weaker M–N bonds in these adsorbents compared to those with 1° amines bound to the metal sites.
- (a) K. S. Lim, W. R. Lee, H. G. Lee, D. W. Kang, J. H. Song, J. Hilgar, J. D. Rinehart, D. Moon and C. S. Hong, Inorg. Chem., 2017, 56, 7443 CrossRef CAS PubMed; (b) A. M. Fracaroli, P. Siman, D. A. Nagib, M. Suzuki, H. Furukawa, F. D. Toste and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 8352 CrossRef CAS PubMed.
- (a) Y.-S. Bae, D. Dubbeldam, A. Nelson, K. S. Walton, J. T. Hupp and R. Q. Snurr, Chem. Mater., 2009, 21, 4768 CrossRef CAS; (b) A. P. Nelson, O. K. Farha, K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 458 CrossRef CAS PubMed.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030 CAS.
- D. Gygi, E. D. Bloch, J. A. Mason, M. R. Hudson, M. I. Gonzalez, R. L. Siegelman, T. A. Darwish, W. L. Queen, C. M. Brown and J. R. Long, Chem. Mater., 2016, 28, 1128 CrossRef CAS.
- Determining the crystallographic density of Mg2(dotpdc) has proven difficult due to its modest crystallinity. The ratio of crystallographic densities of Mg2(dotpdc) and Mg2(dobpdc) was estimated from that of the isostructural iron frameworks. The crystallographic density of activated Fe2(dotpdc) was determined by powder X-ray diffraction to be approximately 0.462 g cm−3 (ref. 18). The crystallographic density of activated Fe2(dobpdc) was previously determined to be 0.6750 g cm−3 by powder X-ray diffraction (ref. 26). Therefore, the crystallographic density of Fe2(dotpdc) is 31% lower than that of Fe2(dobpdc). The 17% lower gravimetric capacity of Fe2(dotpdc) (1 CO2 per Fe2+ site = 4.52 mmol g−1) compared to Fe2(dobpdc) (1 CO2 per Fe2+ site = 5.46 mmol g−1) leads to an approximately 43% lower volumetric capacity in Fe2(dotpdc) (1 CO2 per Fe2+ site = 2.09 mmol cm−3) compared to that of Fe2(dobpdc) (1 CO2 per Fe2+ site = 3.69 mmol cm−3). Because the adsorption of CO2 in diamine-appended metal–organic frameworks occurs in a ratio of one CO2 per metal site, and the functionalization of the framework has a minimal effect on the unit cell, these ratios should roughly translate to diamine-appended variants.
- M. T. Kapelewski, S. J. Geier, M. R. Hudson, D. Stück, J. A. Mason, J. N. Nelson, D. J. Xiao, Z. Hulvey, E. Gilmour, S. A. FitzGerald, M. Head-Gordon, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2014, 136, 12119 CrossRef CAS PubMed.
- The expected BET surface area of Mg2(pc-dobpdc) was simulated to be 2299 m2 g−1 using previously reported computational methods (ref. 30). This value is lower than the measured surface area (3000 m2 g−1), but is also lower than the simulated surface area of Mg2(dobpdc) (3040 m2 g−1). Therefore, the lower surface area of Mg2(pc-dobpdc) compared to Mg2(dobpdc) is likely due to their different pore structures and not due to incomplete activation of Mg2(pc-dobpdc).
- H. Frost, T. Düren and R. Q. Snurr, J. Phys. Chem. B, 2006, 110, 9565 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, and full characterization (powder X-ray diffraction, infrared spectra, diamine loadings, dry N2 decomposition profiles, and CO2 adsorption data) for all new adsorbents. CCDC 1577354. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04266c |
| This journal is © The Royal Society of Chemistry 2018 |