
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new generation of ferrociphenols leads to a great diversity of reactive metabolites, and exhibits remarkable antiproliferative properties†
Yong
Wang
 ab,
Patrick M.
Dansette
c,
Pascal
Pigeon
ab,
Siden
Top
*b,
Michael J.
McGlinchey
d,
Daniel
Mansuy
*c and
Gérard
Jaouen
*ab
ab,
Patrick M.
Dansette
c,
Pascal
Pigeon
ab,
Siden
Top
*b,
Michael J.
McGlinchey
d,
Daniel
Mansuy
*c and
Gérard
Jaouen
*ab
aPSL, Chimie ParisTech, 11 rue Pierre et Marie Curie, F-75005 Paris, France
bSorbonne Universités, UPMC Univ Paris 6, UMR 8232 CNRS, IPCM, Place Jussieu, F-75005 Paris, France. E-mail: siden.top@upmc.fr; gerard.jaouen@upmc.fr
cLaboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, UMR 8601 CNRS, Université Paris Descartes, PRES Paris Cité Sorbonne, 45 rue des Saints Pères, 75270 Paris Cedex 06, France. E-mail: Daniel.Mansuy@parisdescartes.fr
dUCD School of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland
First published on 16th November 2017
Abstract
Organometallic compounds bearing the redox motif [ferrocenyl-ene-phenol] have very promising antiproliferative properties which have been further improved by incorporating pertinent substituents able to engender new mechanisms. Here we show that novel ferrociphenols bearing a hydroxypropyl chain exhibit strong antiproliferative effects, in most cases much better than those of cisplatin, tamoxifen, or of previously described ferrociphenols devoid of this terminal OH. This is illustrated, in the case of one of these compounds, by its IC50 values of 110 nM for MDA-MB-231 triple negative breast cancer cells and of 300 nM for cisplatin-resistant A2780cisR human ovarian cancer cells, and by its GI50 values lower than 100 nM towards a series of melanoma and renal cancer cell lines of the NCI-60 panel. Interestingly, oxidative metabolism of these hydroxypropyl-ferrociphenols yields two kinds of quinone methides (QMs) that readily react with various nucleophiles, such as glutathione, to give 1,6- and 1,8-adducts. Protonation of these quinone methides generates numerous reactive metabolites leading eventually to many rearrangement and cleavage products. This unprecedented and fully characterized metabolic profile involving a wide range of electrophilic metabolites that should react with cell macromolecules may be linked to the remarkable profile of antiproliferative activities of this new series. Indeed, the great diversity of unexpected reactive metabolites found upon oxidation will allow them to adapt to various situations present in the cancer cell. These data initiate a novel strategy for the rational design of anticancer molecules, thus opening the way to new organometallic potent anticancer drug candidates for the treatment of chemoresistant cancers.
Introduction
Despite recent advances in the treatment of certain cancers (for example, controlled targeting of the tumour, nanocapsule formulations, new biopharmaceutical agents with specific activity) the number of fatalities linked to cancers whose outcomes remain poor (e.g. melanoma, pancreatic and ovarian cancer, triple-negative breast cancer, glioma etc.), and which do not respond well to proapoptotic stimuli, continues to rise.1–4 This has encouraged exploration of the potential role of inorganic compounds in this area, following the success of the coordination complexes of Pt, often in oxidation state +II, for which DNA is a primary but not the only target, and which are now in regular use.5–8 Owing to a number of known issues with these complexes (relatively narrow therapeutic window, serious resistance and toxicity problems), recent research has turned towards organometallic complexes, principally those of gold and the Fe, Ru, Os triad, a novel approach that is now undergoing exponential growth.9–19 These compounds often target the mitochondrial system or inhibit kinases or redox proteins overexpressed in cancer cells. Thus, they provide a new channel that offers a complementary solution to that of the platinum derivatives, bringing different targets and mechanisms of action into play.Bioactive ferrocenes have provided a number of examples over the last few years to illustrate this approach, using a variety of synthetic strategies.20–31 The ferrociphenols stand out from the crowd of current candidates owing to the advanced state of biological studies in the area, and also because of the rich variety of their mechanisms of action.32–36 This richness is due to their ability to generate the redox motif {ferrocenyl-ene-phenol} in the cancer cell. The motif's activity can be modulated by various substitutions, thus permitting a diversity of possible targets, an effect that is known to delay or prevent phenomena of drug resistance to a given biomolecule.
In particular we have been able to show that hydroxytamoxifen 1, known for its antagonist effect on MCF-7 hormone-dependent (ERα+) breast cancer cells and for its lack of activity on hormone-independent (ERα−) MDA-MB-231 breast cancer cells, becomes strongly antiproliferative on both ERα+ and ERα− cells when modified to become the ferrociphenols 2a (Fig. 1).37 The IC50 of 2a on MDA-MB-231 (ERα−) cells is 0.5 μM, and a cytostatic effect occurs via senescence.38,39 This mechanism represents an alternative strategy to apoptosis in halting cell proliferation, and bypasses the problem of the resistance of cancers to proapoptotic stimuli. In addition, in those cancer cells for which apoptosis is possible, the apoptotic pathway is still operative. Moreover, compound 2a interacts with a redox target overexpressed in certain cancers such as thioredoxine reductase.40
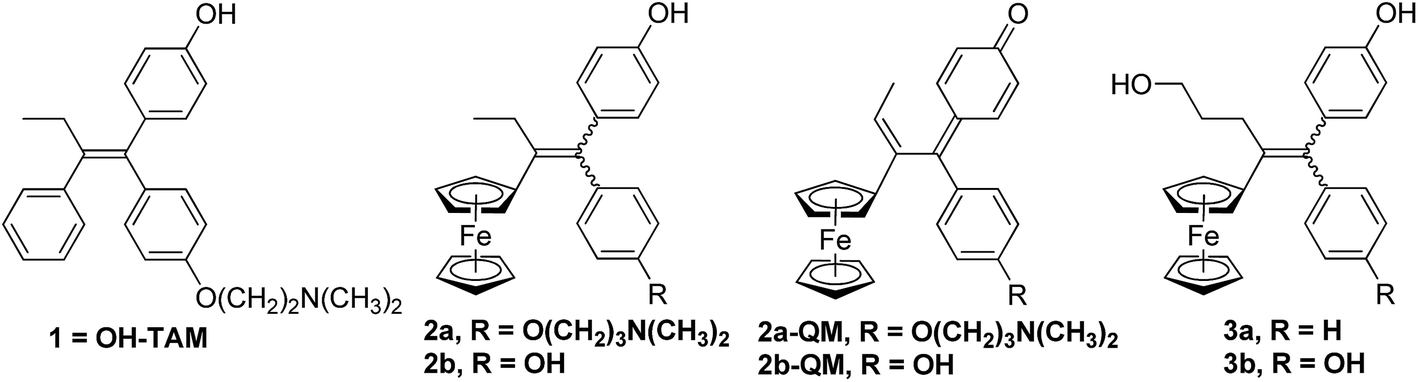 | ||
| Fig. 1 Hydroxytamoxifen 1, ferrociphenols 2 and their derived quinone methides 2-QM, hydroxypropyl-ferrociphenols 3. | ||
The basic chain in 2a can be removed to give the ferrociphenol, 2b, whose cytostatic effect on MDA-MB-231 cells is comparable to that of 2a. Subsequently, we were able to show that quinone methides 2-QM, arising from the initial oxidation of ferrocene FeII to the corresponding ferricinium FeIII species, are active metabolites of compounds 2 (Fig. 1).41–43 These compounds are active on cancer cells but not on healthy cells (astrocytes, melanocytes) where the redox effect is not observed.44,45 The effectiveness of these two compounds, 2a and 2b, delivered by lipid nanocapsules (LNCs), has been studied on experimental subcutaneous models of triple-negative breast cancer and malignant glioma, respectively. In both cases, a significant slowing of tumor progression was observed, confirming the antiproliferative activity of these compounds.46–48
In our search for a new series of molecules surpassing the antiproliferative effect of the above products we have recently obtained promising results by introducing functional groups at the end of the alkyl chain.49 In particular, the hydroxypropyl derivative 3b (Fig. 1) exhibited exceptional antiproliferative activity against inter alia liver hepatocellular carcinoma cells (HepG2) and triple negative breast cancer cells (MDA-MB-231) with IC50 values of 0.07 and 0.11 μM, respectively.49 Chemical oxidation of 3a/3b yielded an unprecedented tetrahydrofuran-substituted quinone methide (QM), 4a/4b, via an internal cyclization of the hydroxyl-alkyl chain (Fig. 2). The ferrocenyl group in 3 plays a key role not only as an intramolecular reversible redox “antenna” but also as a stabilized carbenium ion “modulator”.49 Moreover, the presence of the oxygen heterocycle in 4 enhances its stability and leads to a unique chemical evolution profile, leading to products 5, 6, 7, and 8 (Fig. 2) that were characterized spectroscopically (NMR, HRMS) or by X-ray crystallography.49
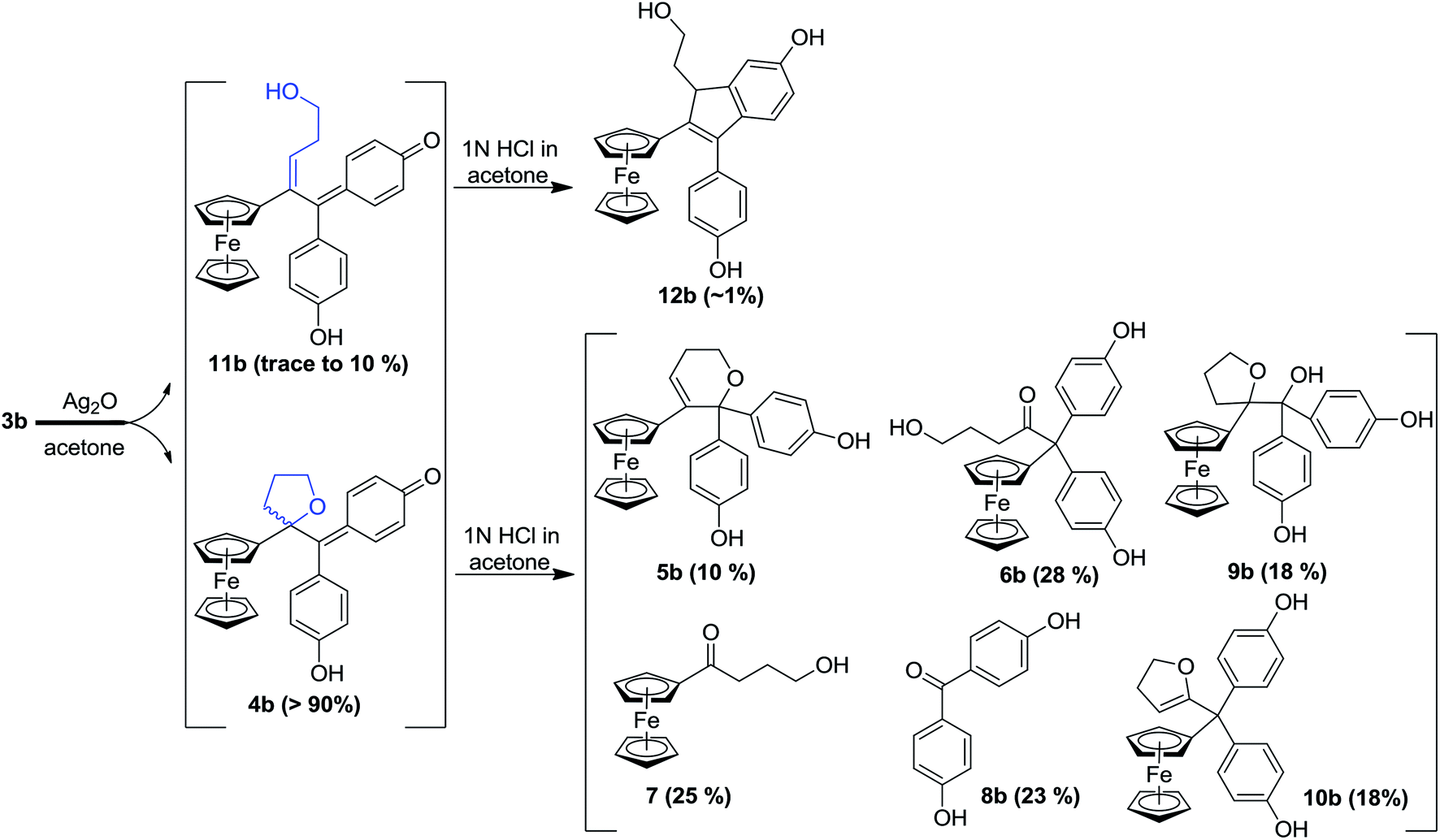 | ||
| Fig. 2 Quinone methides 4b and 11b from the oxidation of hydroxypropyl-ferrociphenols 3b and subsequent protonation (similar results were obtained in the case of 3a). Yields are based on starting compound 3b, but note that cleavage of the C–C bond of 9b yields both 7 and 8. | ||
To further explore the promising antiproliferative properties of compounds 3a and 3b and their unique evolution profile upon oxidation, we have studied the fate of these compounds upon chemical oxidation or after metabolism by liver microsomes, in the presence of nucleophiles such as thiols that are present in proteins and nucleic acids. The antiproliferative properties of compounds 3 and their metabolites towards breast cancer cells were evaluated. Moreover, we have compared the activities of 3b, 2b and cisplatin towards ovarian cancer cell lines sensitive or resistant to cisplatin. The antiproliferative effects of 3b were also evaluated by the National Cancer Institute on the NCI-60 human tumor cell line panel.50 The obtained data showed a quite remarkable profile of antiproliferative activities of 3b, and the formation of a surprisingly great diversity of reactive metabolites upon metabolic oxidation of 3b.
Results
Chemical oxidation of the hydroxypropyl-ferrociphenols, 3a and 3b
To compare the behaviour of the acyclic QMs, such as 2b-QM, and the tetrahydrofuran-substituted QMs 4a and 4b, we have explored the effects of several oxidants (including Ag2O, MnO2, H2O2) on their precursors 3a and 3b. As shown in Fig. 2, generation of the QMs 4, followed by addition of 1 N HCl aqueous solution in acetone, not only led to the previously reported molecules 5–8, but also to two new products, 9, the 1,6-adduct of water to 4, and 10, the product of ferrocenyl migration in a pinacol-type rearrangement (Fig. S1†). In contrast, protonation of 4b with HCl in dry diethyl ether yielded only two major products, 5b (35%) and 6b (47%). Compounds 7 and 8 were major products only when the protonation was carried out in acetone, a well-known solvent for triplet state photosensitized processes.51The novel products 10, arising from the protonation of 4, can come from a 1,2-migration of the ferrocenyl moiety, thus generating a cation stabilized by the adjacent oxygen in the five-membered ring (Fig. S1†). These rearrangement products, in which the 2,3-dihydrofuran ring and the ferrocenyl moiety are attached to the same carbon center, were unequivocally characterized spectroscopically, and by X-ray crystallography in the case of 10b whose structure appears in Fig. 3.
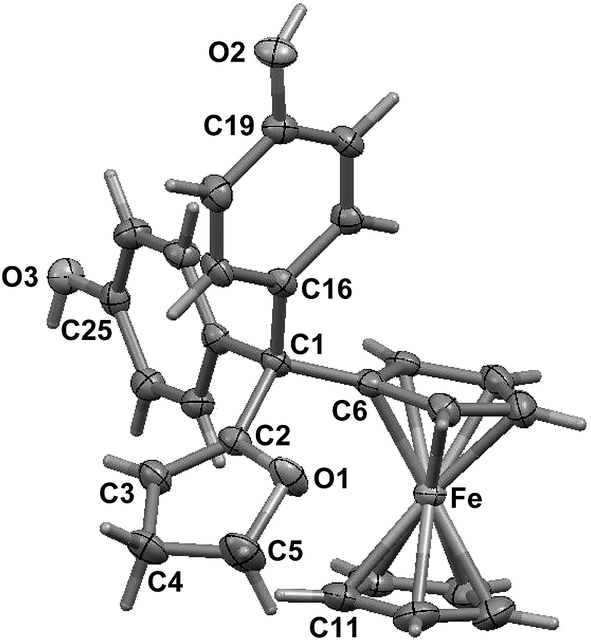 | ||
| Fig. 3 Molecular structure of 10b (thermal ellipsoids are shown at 50%). | ||
In addition to the products described above, the hydroxy-alkenyl quinone methides 11 (Fig. 2), analogous to the vinylic 2-QM, were also formed, and identified by HPLC and 1H NMR, even though their existence was somewhat transient. Thus, in the case of 11b for instance, the appearance of a multiplet and a triplet at 1.60 and 6.43 ppm, respectively, indicated the formation of a double bond within the starting hydroxypropyl group (Fig. S3†). The yield of 11b (varying from trace to 10%, relative to 4b, as monitored by 1H NMR) was dependent on the concentration of its precursor 3b (from 2 to 100 mM), and the quantity of oxidant (one to five equivalents of Ag2O). Indene product 12b resulting from an acid-catalyzed cyclisation of the hydroxy-alkenyl quinone methide 11b, as previously found in the case of vinylic quinone methides, 2-QM,52 (Fig. S2†) was also formed in low amounts upon oxidation of 3b. The identities of the new QM, 11b, and its corresponding indene, 12b, were also confirmed by their characteristic UV-VIS spectra (λmax ∼560 nm and ∼320 nm for vinyl QMs and indene derivatives, respectively53).
In this case, we found for the first time that chemical oxidation of the hydroxypropyl-ferrociphenol, 3, yields two kinds of quinone methides. Since the tetrahydrofuran-QMs 4, and the hydroxyalkenyl-QMs 11, arise from a common precursor, a ferrocenyl cation 13 (Fig. 4) initially generated by a two-electron oxidation of 3, the observed 4/11 ratio is the result of a competition between intramolecular cyclization with subsequent deprotonation to form 4, versus deprotonation to generate the conjugated carbon–carbon double bond in 11. This ratio and the proportions of the observed products 5–12 were greatly dependent on the nature of the used oxidant and acid, and on the amounts of oxidant relative to 3.
 | ||
| Fig. 4 1,6- and 1,8-adducts to quinone methides 4 and 11. | ||
Reactions of the tetrahydrofuran-substituted QMs 4a and 4b toward nucleophiles
The reactions of this new type of electrophilic intermediate with various nucleophiles, including thiols such as mercaptoethanol (ME), N-acetyl-L-cysteine methyl ester (NACM) and glutathione (GSH), were studied. Compounds 3a or 3b were first treated with silver oxide to generate 4a or 4b at room temperature and, after removal of excess Ag2O, the nucleophiles were added. In each case, progress of the reaction was monitored by TLC or HPLC.Methanol reacted via 1,6-addition to yield the pinacol-type methyl ethers, 14, in good yield (Fig. 4). By contrast, ME in acetone gave a poor yield of the 1,6-adducts, 15. However, both the rate of reaction and the yield of 15 were greatly enhanced by addition of NaOH to form the thiolate. Analogous reactions with NACM and glutathione in the presence of NaOH led to the corresponding 1,6-adducts, 16 and 17, respectively, in very good yields (Fig. 4). The adducts 14–16 were characterized by 1H NMR spectroscopy and mass spectrometry. Complete purification of 17 was not possible because of the high polarity of the GS function, and a crude sample was obtained.
When treated under more physiological conditions (50 mM phosphate buffer, pH 7.4, 37 °C), 4b led after 30 min to the aforementioned products 5b–10b in low to moderate yields. Under these conditions, the half-life of 0.15 mM 4b was approximately 25 min. However, incubation of 4b under these conditions in the presence of ME, NACM or GSH gave the corresponding 1,6-adducts, 15b, 16b or 17b, respectively, in yields greater than 80%. In the presence of these thiols, the half-life of 4b, as monitored by UV-VIS spectroscopy, was markedly reduced to 1.5, 3.2 and 2.3 min, respectively. It is noteworthy that all these 1,6-adducts were progressively transformed into 4-hydroxy-1-ferrocenylbutan-1-one 7 and diarylmethanone 8 (Fig. S4–S8†). However, they were stable for at least a week in the absence of air and light.
Oxidation of compounds 3a and 3b by horseradish peroxidase (HRP)
Incubation of 3a and 3b with HRP and H2O2 for 30 min (0.25 mM 3, 0.1% HRP and 4 equivalents of H2O2) led to products that have been observed upon chemical oxidation of 3a and 3b and that were found to derive from quinone methides 4 and 11. The proportions of these products varied as a function of the used conditions (3/HRP/H2O2 ratios, reaction time) (Tables S2 and S3†). However, compounds 7 and 8 were always the major oxidation products, and the products deriving from the hydroxyalkenyl-QM 11 were only formed in trace amounts. When low amounts of HRP and H2O2 were used (0.1 mM 3, 0.02% HRP and one equivalent of H2O2) their total yield increased to about 10%.Oxidative metabolism of compounds 3a and 3b by liver microsomes
Incubation of 3a or 3b with rat liver microsomes in the presence of NADPH, the reducing agent necessary for the activity of microsomal monooxygenases, led to the formation of all the products that were observed upon oxidation of 3a or 3b by chemical oxidants or HRP and H2O2 (Tables S2 and S3†). They were characterized by comparison of their HPLC retention times, and also their MS and MS-MS characteristics, with those of authentic samples obtained by chemical synthesis. None of these metabolites were observed under identical incubations without NADPH. Incubations of 3a and 3b with human liver microsomes under identical conditions led to results very similar to those obtained with rat liver microsomes. Ketones 7 and 8 were always the major products and represented more than 40% of all metabolites, whereas dihydropyran 5 was formed in lower amounts and compounds 6, 9 and 10 were formed in trace amounts. These data showed that microsomal oxidation of 3 mainly led to products deriving from QM 4. However, minor formation of metabolites 12 and 18, that derived from the hydroxyalkenyl quinone methide 11, showed that both QM 4 and 11 were formed as initial oxidation products. It is noteworthy that a decrease of the microsomal proteins concentration in the incubate led to an increase of the ratio of 18 relative to all other metabolites (from less than 5% to about 10% when decreasing the protein concentration by a factor 1.7). This result is to be compared to analogous data obtained in oxidation of 3 with chemical oxidants or HRP and H2O2, and indicates that the intermediate formation of QM 4 is the major pathway occurring in oxidation of 3, but that the relative importance of the formation of QM 11 is greater under milder oxidizing conditions (use of lower oxidant or oxidation catalyst amounts).Incubation of ferrociphenols 3a and 3b with rat liver microsomes in the presence of NADPH and various thiols, led to QM-thiol adducts, as well as the metabolites we have already observed under identical conditions in the absence of thiols (Tables S2 and S3†). All the 1,6- and 1,8-thiol adducts (15–17 and 19–21, respectively, Fig. 4) arising from their corresponding QM precursors were detected in trace to moderate amounts, and were characterized by comparison of their HPLC retention times and MS spectra with those of authentic samples. The 1,6-adducts are less polar and exhibit longer retention times than their 1,8-adduct counterparts; moreover, their mass spectral fragmentation patterns were also significantly different (Table S4†). In microsomal incubations of 3 in the presence of ME or NACM, ketones 7 and 8 were still the major products and represented more than 40% of all metabolites, whereas the proportions of 1,6-thiol adducts relative to all metabolites remained below 10% and only low amounts of 1,8-thiol adducts were observed under these conditions. Interestingly, incubation of 3 with liver microsomes, in the presence of NADPH and GSH, gave 17 as a major GSH adduct (40% of all metabolites) with a minor amount of the 1,8-GSH adduct 21. Upon increasing the protein concentration, the relative proportion of 7 and 8 increased and 17 was still formed in an about 5–10% yield relative to all metabolites whereas 21 was only formed in trace amounts (Tables S2 and S3†). Accordingly, incubation of authentic 1,6-thiol adducts 15–17 with liver microsomes and NADPH mainly led to 7 and 8. The stability of these 1,6-thiol adducts towards microsomal oxidation is as follows: 17 > 16 > 15, suggesting that the size and polarity of the thiol substituents adjacent to the tetrahydrofuran ring play an important role in their microsomal oxidation into 7 and 8.
Antiproliferative effects of 3a and 3b and their oxidation metabolites against TNBC MDA-MB-231 cells
The antiproliferative activities of compounds 3a and 3b, and of the products derived from their oxidation in the presence or absence of thiols, against hormone-refractory breast cancer MDA-MB-231 cells, are shown in Table 1. Compounds 3a and 3b were the most active, with IC50 values of 1.1 and 0.11 μM, respectively. However, the transient intermediates appearing during their oxidative metabolism, the quinone methides 4, their water 1,6-adducts 9, and their thiol 1,6-adducts, 15 and 16, were also active with IC50 values between 2 and 10 μM; these data are in sharp contrast with the very low activity of the final major metabolites 7 and 8 that exhibited IC50 values above 100 μM. These IC50 values of intermediates 4, 9, 15 and 16 are quite remarkable, especially if one takes into account their instability in the assay medium, and possible problems of cell penetration. This suggests that they should have potent antiproliferative effects when they are formed inside the cell, close to important cell targets, upon oxidative metabolism of 3.| Compound | IC50a (μM) | Compound | IC50a (μM) |
|---|---|---|---|
| a Measured after 5 days of culture (mean of two independent experiments ± SD). b Values taken from ref. 49. | |||
| 2b | 0.64 ± 0.06 | 5b | 2.03 ± 0.79 |
| 3a | 1.16 ± 0.02 | 6b | 4.14 ± 1.33 |
| 4a (QM)b | 1.89 ± 0.08 | 7 | ≈150 |
| 6a | 8.17 ± 1.56 | 8b | ≈295 |
| 9a (1,6-OH) | 6.12 ± 0.97 | 10b | 9.92 ± 0.78 |
| 15a (1,6-ME) | 7.51 ± 0.52 | 9b (1,6-OH) | 9.60 ± 1.29 |
| 16a (1,6-NACM) | 4.36 ± 0.26 | 14b (1,6-OMe) | 3.07 ± 0.01 |
| 3b | 0.11 ± 0.02 | 15b (1,6-ME) | 2.36 ± 0.14 |
| 4b (QM)b | 4.39 ± 1.47 | 16b (1,6-NACM) | 2.43 ± 0.4 |
Cytotoxicity studies of compounds 2b and 3b against cisplatin-sensitive and cisplatin-resistant human ovarian cancer cell lines
The cytotoxic activities of the vinyl derivative, 2b, and the hydroxypropyl complex, 3b, were further evaluated against two human ovarian cancer cell lines, cisplatin-sensitive A2780 and cisplatin-resistant A2780cisR cells (Table 2). Their activities toward a healthy human lung fibroblast cell line, MRC-5, were also measured. The A2780cisR variant is known to encompass all of the known major mechanisms of resistance to cisplatin:54 decreased drug transport, enhanced DNA repair/damage tolerance, elevated cellular thiol (GSH) level, and blockage of cell-death pathways. As shown in Table 2, 2b and 3b showed a potent antiproliferative activity not only against wildtype A2780, but also against resistant A2780cisR cells, with a resistance factor smaller than one, contrary to cisplatin (CDDP) that exhibits a resistance factor of about ten.55 It is noteworthy that 3b showed a 7-fold greater cytotoxicity than 2b against A2780 and A2780cisR cells, and a 38-fold greater cytotoxicity than cisplatin against A2780cisR cells. These observations suggest that compound 3b may be much less affected by high cellular GSH levels or other physiological adaptations occurring in resistant cancer cells. Moreover, 3b is approximately sixteen times less toxic towards normal human cells MRC-5 than towards A2780 cancer cells, whereas 2b and cisplatin have lower selectivity factors of approximately 2 and 8.5, respectively.| Compound | IC50a (μM) | RFb | IC50a (μM) MRC-5 | SFc | |
|---|---|---|---|---|---|
| A2780 | A2780cisR | ||||
| a Measured after 72 h of culture (mean of three independent experiments ± SD). b RF, resistance factor: IC50 toward A2780cisR/IC50 toward A2780 ratio. c SF, selectivity factor: IC50 toward MRC-5/IC50 for A2780 ratio. d Values taken from ref. 55. | |||||
| 2b | 3.5 ± 0.94 | 2.3 ± 0.7 | 0.66 | 7.28 ± 0.39 | 2.1 |
| 3b | 0.53 ± 0.01 | 0.30 ± 0.03 | 0.57 | 8.54 ± 0.32 | 16.1 |
| CDDPd | 1.2 ± 0.20 | 11.5 ± 0.3 | 9.6 | 16.2 ± 0.6 | 8.5 |
Antiproliferative effects of 2b and 3b on the NCI cell line panel
The antiproliferative effects of 3b were further evaluated by the National Cancer Institute on the NCI-60 human tumor cell line panel,50 which consists of approximately 60 cell lines within nine tumor type subpanels. They were compared to those found for compound 2b,56 tamoxifen (TAM) and cisplatin (CDDP) (Fig. 5, Tables S5 and S6†). The cells were treated for 48 h at five concentrations ranging from 0.01 to 100 μM. Three endpoints were calculated: GI50 (50% growth inhibition concentration), TGI (100% growth inhibition concentration), and LC50 (50% lethal concentration). Compounds 2b and 3b exhibited a broad spectrum of activity over a 3-log range of GI50, which means that these compounds are not indiscriminately cytotoxic against cellular growth. Compounds 2b and 3b (mean GI50 around 0.5 μM and 0.4 μM respectively) are approximately ten-fold and thirty-fold more potent (in terms of GI50 values) than tamoxifen and cisplatin, respectively (Table S6†). Compound 3b shows a high potency towards a wide range of cancer cell lines, with a particularly high activity against renal, melanoma and CNS cancer cell lines. Thus, its GI50 values for M14, SK-MEL-5, UACC-62 (3/8 melanoma sub-lines) and 786-0, A498, ACHN, CAKI-1 (4/8 renal cancer sub-lines) were found to be lower than 100 nM. Renal cancers are often chemoresistant because of the high expression of multi-drug resistant (MDR1) protein, and elevated level of P-glycoprotein in renal cancer cells.57,58 Melanoma is a highly aggressive neoplasm and has a poor response to conventional chemotherapy,59 meaning that intrinsic drug resistance is also a serious problem in melanoma treatment. The high potency of compound 3b towards the phenotypes of renal cancer and melanoma is promising for the use of such organometallic complexes for the treatment of such resistant cancers.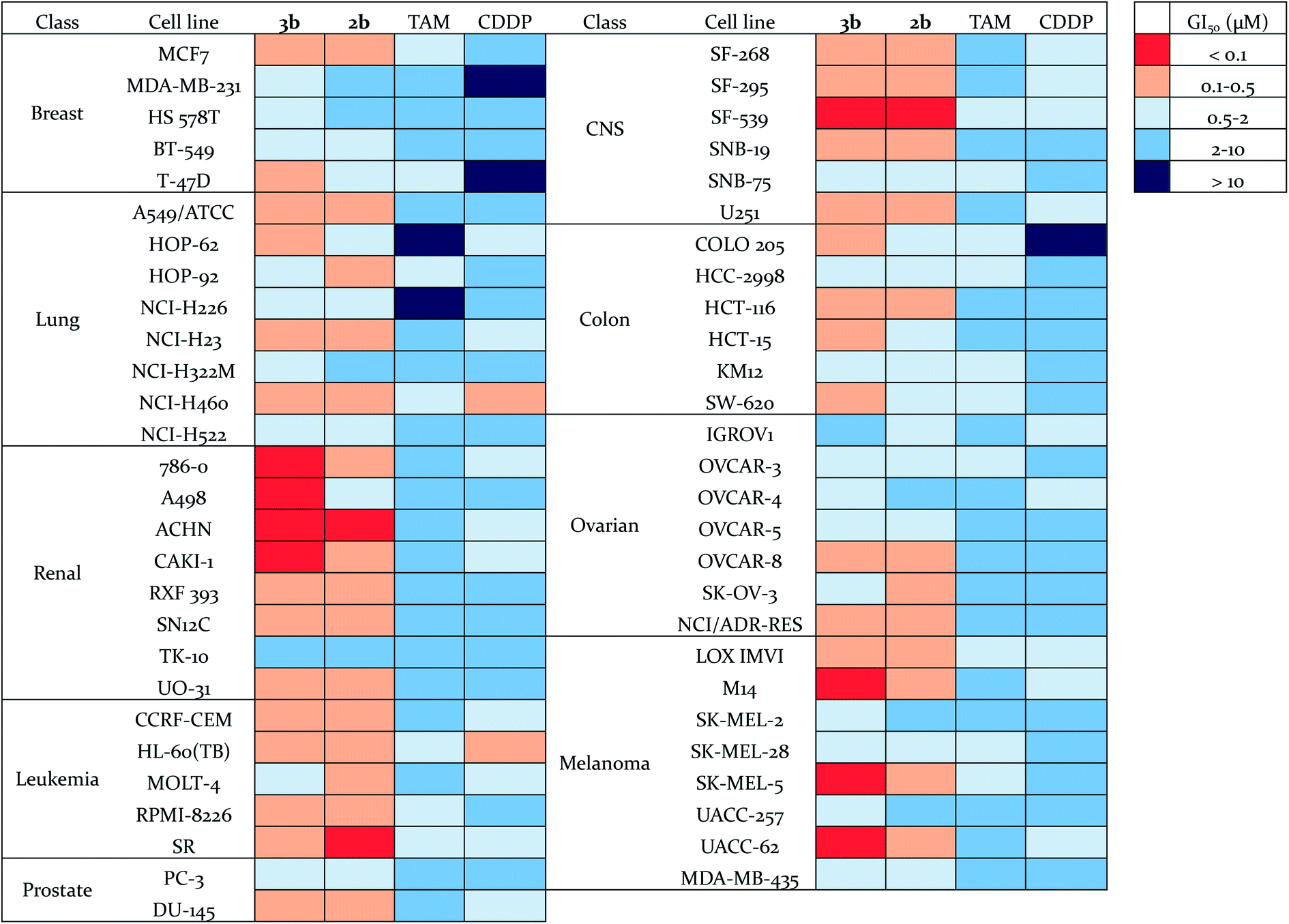 | ||
| Fig. 5 Heat map for GI50 values of 3b, 2b, TAM and CDDP. The deep red color indicates the highest activity, whereas the deep blue color represents the lowest activity. | ||
Discussion
The novel ferrociphenols 3 bearing a terminal OH group in their alkyl moiety exhibit strong antiproliferative properties, very frequently much better than those of ferrociphenols 2, cisplatin or tamoxifen. This is illustrated, in the case of 3b, by its IC50 values of 110 nM for MDA-MB-231 breast cancer cells (Table 1) and of 530 nM for A2780 human ovarian cancer cells (Table 2), and by its GI50 values lower than 100 nM towards a series of renal cancer and melanoma cells (Fig. 5). Interestingly, compound 3b is quite active towards cisplatin-resistant A2780cisR ovarian cancer cells (Table 2).Ferrociphenols 3 also exhibit a remarkable profile of metabolic oxidation, when compared to ferrociphenols 2. This is due to the formation, after their two-electron oxidation, of a new kind of QMs 4, involving an oxygen-containing heterocycle, in addition to the vinyl QMs 11 (Fig. 4), analogous to those formed upon oxidation of ferrociphenols 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 42 (Fig. 1 and S2†). To the best of our knowledge, this is the first case involving two such chemotypes of QM pathways from a single substrate, whether in organic or organometallic chemistry. We found that this occurred not only upon oxidation by chemical oxidants such as Ag2O, but also by enzymatic systems such as HRP in the presence of H2O2, or cytochrome P450-dependent monooxygenases that are present in liver microsomes. Evolution of QMs 4 in the incubation medium leads to a surprisingly great diversity of reactive intermediates and final metabolites. After protonation, they lead to rearrangement products such as 5, 6, and 10, and pinacol-type products 9 (Fig. 2). In incubations performed in the presence of an added nucleophile, such as thiols ME, NACM, or GSH, this nucleophile can compete with H2O for reaction with protonated 4 to give 1,6-thiol adducts 15–17 (Fig. 4). The cleavage of the central C–C bond of intermediates 9, 14, 15, 16, and 17 further leads to the final stable products 7 and 8. The mechanism of this C–C bond cleavage remains to be determined. However, one might tentatively propose two possible mechanisms. Firstly, photocleavage of the C–C bond leading to a radical pair has been already proposed (Fig. S1†).51 Another viable mechanism could be a one-electron oxidation of the phenol function, followed by beta-cleavage of the intermediate radical leading to a radical alpha to the ferrocene ring and a new quinone, the hydrolysis of which would lead to 8. Further oxidation of the latter radical would lead to 7 (Fig. 6). The particularly high yields of 7 and 8 observed upon oxidation of 3 by liver microsomes in the presence of NADPH would be in favour of the second mechanism, because the radicals formed by one-electron oxidation of the phenol function of compounds 9 and 15–17 by high-valent Fe
42 (Fig. 1 and S2†). To the best of our knowledge, this is the first case involving two such chemotypes of QM pathways from a single substrate, whether in organic or organometallic chemistry. We found that this occurred not only upon oxidation by chemical oxidants such as Ag2O, but also by enzymatic systems such as HRP in the presence of H2O2, or cytochrome P450-dependent monooxygenases that are present in liver microsomes. Evolution of QMs 4 in the incubation medium leads to a surprisingly great diversity of reactive intermediates and final metabolites. After protonation, they lead to rearrangement products such as 5, 6, and 10, and pinacol-type products 9 (Fig. 2). In incubations performed in the presence of an added nucleophile, such as thiols ME, NACM, or GSH, this nucleophile can compete with H2O for reaction with protonated 4 to give 1,6-thiol adducts 15–17 (Fig. 4). The cleavage of the central C–C bond of intermediates 9, 14, 15, 16, and 17 further leads to the final stable products 7 and 8. The mechanism of this C–C bond cleavage remains to be determined. However, one might tentatively propose two possible mechanisms. Firstly, photocleavage of the C–C bond leading to a radical pair has been already proposed (Fig. S1†).51 Another viable mechanism could be a one-electron oxidation of the phenol function, followed by beta-cleavage of the intermediate radical leading to a radical alpha to the ferrocene ring and a new quinone, the hydrolysis of which would lead to 8. Further oxidation of the latter radical would lead to 7 (Fig. 6). The particularly high yields of 7 and 8 observed upon oxidation of 3 by liver microsomes in the presence of NADPH would be in favour of the second mechanism, because the radicals formed by one-electron oxidation of the phenol function of compounds 9 and 15–17 by high-valent Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O cytochrome P450 species would be generated inside the enzyme active site, and be efficiently oxidized by the FeIV–OH intermediate to give 7 and 8.
O cytochrome P450 species would be generated inside the enzyme active site, and be efficiently oxidized by the FeIV–OH intermediate to give 7 and 8.
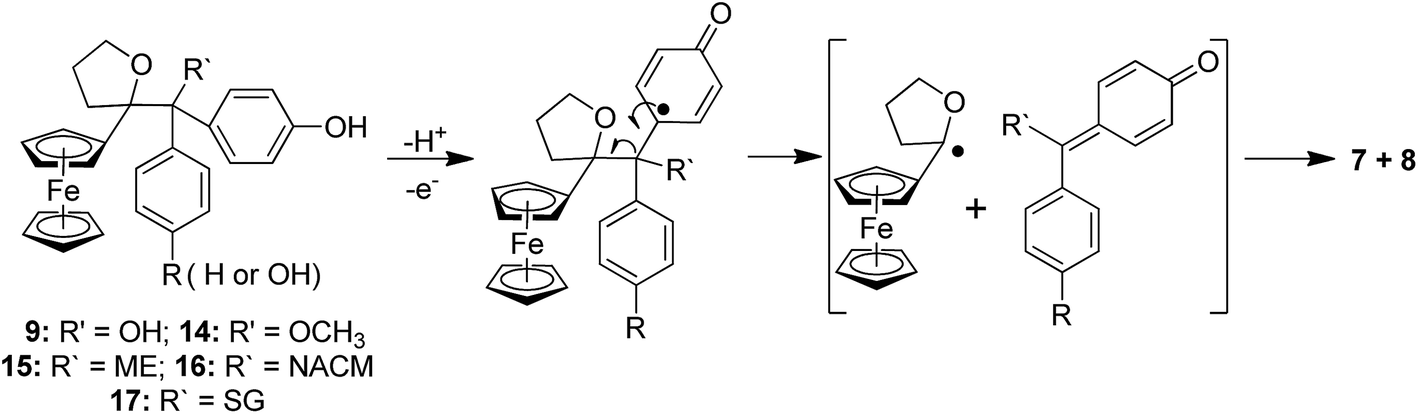 | ||
| Fig. 6 Possible mechanism for the oxidative cleavage of ferrocenyl pinacol-type compounds by liver microsomes in the presence of NADPH. | ||
Comparison of the oxidative metabolism of the hydroxypropyl-ferrociphenols 3 with that of ferrociphenols 2 reveals that a much greater number of reactive, electrophilic intermediates are formed in the former case. This includes the tetrahydrofuran-QMs, 4, and all the cationic intermediates involved in the formation of 5, 6 and 9. Upon protonation, QMs 4 were found to react with a variety of oxygen- and sulfur-containing nucleophiles, such as water, methanol, mercaptoethanol, N-acetyl-L-cysteine methyl ester and glutathione, to give the corresponding 1,6-adducts 9, 14, 15, 16, and 17. When formed inside the cell, these QMs should covalently react with protein and nucleic acid nucleophiles. The radical intermediates thought to be involved in the formation of 4-hydroxy-1-ferrocenylbutan-1-one, 7, and the diaryl ketones, 8, from 9 and 17 could also react with cell macromolecules. Moreover, compounds 5, 6, 9, and 15–17 themselves were also found to have significant antiproliferative properties against breast cancer cells (Table 1).
It is thus likely that the remarkable antiproliferative properties of ferrociphenols 3 are linked to the great diversity of reactive intermediates and metabolites formed during their oxidative metabolism. In that regard, the interesting activity of 3b towards human ovarian A2780cisR cisplatin resistant cancer cells (Table 2), whose resistance mechanism would refer to an elevated cellular thiol (GSH) level, could be related to a high level of GSH adduct, 17b, that should exhibit antiproliferative activity. In a more general manner, the great diversity of reactive metabolites formed upon oxidative metabolism of 3 should allow them to be more aggressive and cytotoxic for cancer cells that are known to have a higher level of oxidative stress than normal cells. This great diversity of reactive metabolites that could bind to various cell targets should allow them to adapt themselves to various situations existing in cancer cells (Fig. 7). It is well-known that some chemotherapy drugs exert their anticancer effects by forming a diversity of reactive metabolites; typically, 5-fluorouracil is widely used in the treatment of a range of solid tumors, and its mechanism involves the intracellular conversion to several active metabolites.60,61 The effective cure of acute promyelocytic leukemia by the inorganic drug arsenic trioxide (As2O3) is also believed to involve its in vivo conversion to yield a diversity of active metabolites.62
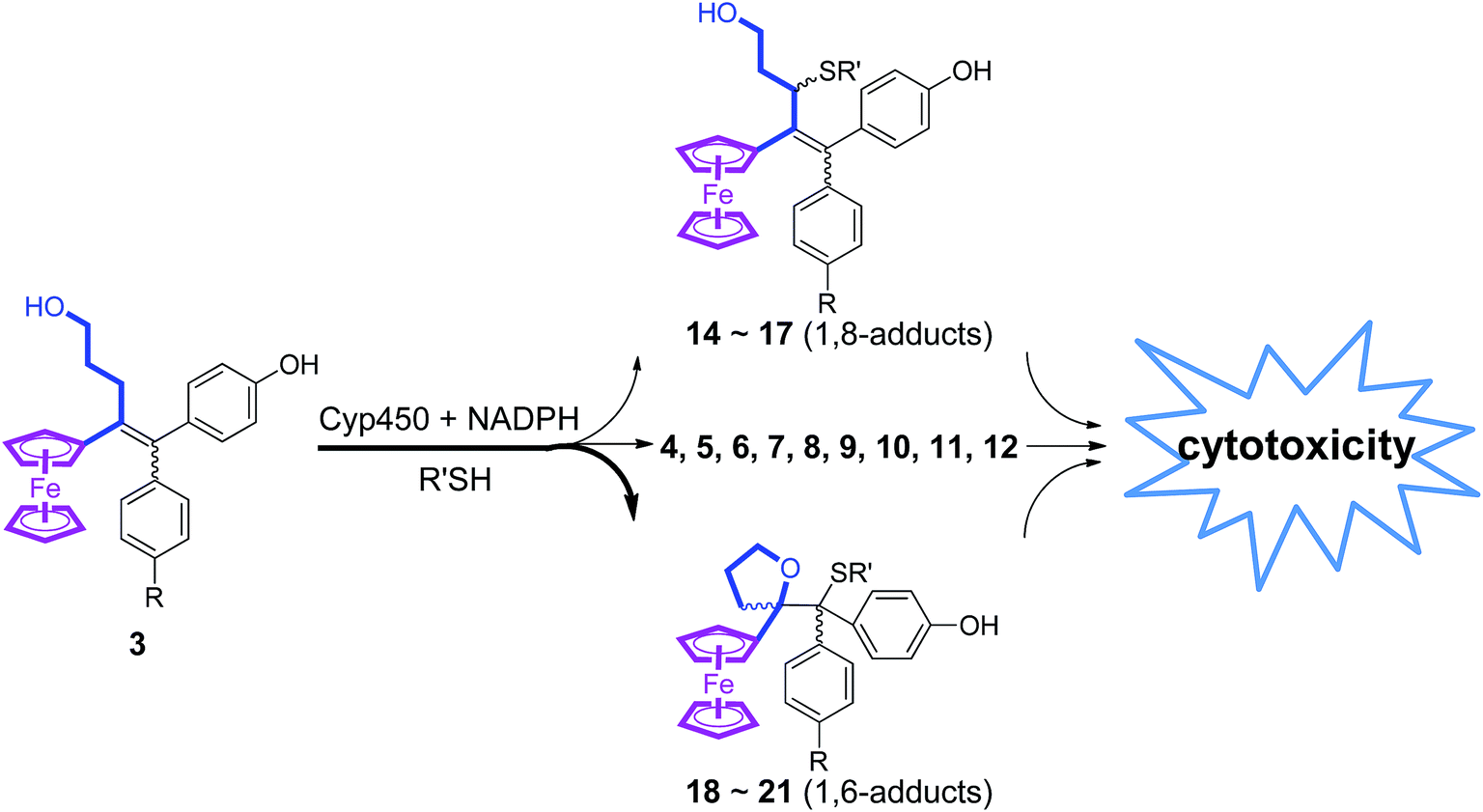 | ||
| Fig. 7 The diversity of reactive intermediates and metabolites involved in the oxidation of ferrociphenols 3. | ||
Further optimization concerning in vivo toxicity, mode of administration, pharmacokinetics and pharmacodynamics are currently in progress to maximize the full potential of the hydroxypropyl-ferrociphenols for clinical application. The hydroxypropyl-ferrociphenols 3 not only yield a great diversity of reactive species under metabolic oxidation, but should also be capable of finding the preferential metabolic pathway to accommodate the redox microenvironment of cells. This appears to be the first report of small molecules that are able to achieve a kind of “self-regulation” in terms of the virtual redox microenvironment. This unprecedented metabolic profile may initiate a new strategy for the rational design of anticancer molecules based on prodrugs, thus opening the way to new potent organometallic drug candidates for the treatment of chemoresistant cancers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. W. thanks the PGG foundation, PSL University and Feroscan for financial support, and we thank Geoffrey Gontard (IPCM, CNRS UMR 8232) for the X-ray structure determinations, Jérôme Bignon (Plateforme CIBI, ICSN, Gif) for cytotoxicity tests, the National Cancer Institute Developmental Therapeutics Program for in vitro testing, Barbara McGlinchey for linguistic assistance and Fatima Mechta-Grigoriou (Insitut Curie, Inserm U830) for helpful disscussions.References
- N. S. Gandhi, R. K. Tekade and M. B. Chougule, J. Controlled Release, 2014, 194, 238–256 CrossRef CAS PubMed.
- E. Hutchinson, Mol. Oncol., 2014, 8, 1–8 CrossRef.
- F. X. Gu, R. Karnik, A. Z. Wang, F. Alexis, E. Levy-Nissenbaum, S. Hong, R. S. Langer and O. C. Farokhzad, Nano Today, 2007, 2, 14–21 CrossRef.
- M. O. Palumbo, P. Kavan, W. H. Miller Jr, L. Panasci, S. Assouline, N. Johnson, V. Cohen, F. Patenaude, M. Pollak, R. T. Jagoe and G. Batist, Front. Pharmacol., 2013, 4, 57 Search PubMed.
- B. Rosenberg and L. Vancamp, Cancer Res., 1970, 30, 1799–1802 CAS.
- B. Lippert, Cisplatin: Chemistry and Biochemistry of a Leeding Anticancer Drug, John Wiley and Sons, New York, 1999 Search PubMed.
- S. Dhar and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22199–22204 CrossRef CAS PubMed.
- P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5–14 CrossRef CAS.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- A. A. Nazarov, C. G. Hartinger and P. J. Dyson, J. Organomet. Chem., 2014, 751, 251–260 CrossRef CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- V. Brabec, S. E. Howson, R. A. Kaner, R. M. Lord, J. Malina, R. M. Phillips, Q. M. A. Abdallah, P. C. McGowan, A. Rodger and P. Scott, Chem. Sci., 2013, 4, 4407–4416 RSC.
- M. Dörr and E. Meggers, Curr. Opin. Chem. Biol., 2014, 19, 76–81 CrossRef PubMed.
- L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284 RSC.
- M. A. Cinellu, I. Ott and A. Casini, in Bioorganometallic Chemistry, ed. G. Jaouen and M. Salmain, Wiley-VCH, 2015, ch. 4, pp. 117–140 Search PubMed.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed.
- Medicinal Organometallic Chemistry, ed. G. Jaouen and N. Metzler-Nolte, Springerlink, 2010, vol. 32 Search PubMed.
- S. S. Braga and A. M. S. Silva, Organometallics, 2013, 32, 5626–5639 CrossRef CAS.
- B. Balaji, B. Balakrishman, S. Perumalla, A. A. Karande and A. R. Chakravarty, Eur. J. Inorg. Chem., 2015, 8, 1398–1407 CrossRef.
- O. Payen, S. Top, A. Vessieres, E. Brule, M. A. Plamont, M. J. McGlinchey, H. Muller-Bunz and G. Jaouen, J. Med. Chem., 2008, 51, 1791–1799 CrossRef CAS PubMed.
- K. Kowalski, L. Szczupak, L. Oehninger, I. Ott, P. Hikisz, A. Koceva-Chyla and B. Therrien, J. Organomet. Chem., 2014, 772, 49–59 CrossRef.
- J. Amin, I. S. Chuckowree, M. Wang, G. J. Tizzard, S. J. Coles and J. Spencer, Organometallics, 2013, 32, 5818–5825 CrossRef CAS.
- P. James, J. Neudoerfl, M. Eissmann, P. Jesse, A. Prokop and H.-G. Schmalz, Org. Lett., 2006, 8, 2763–2766 CrossRef CAS PubMed.
- H. V. Nguyen, A. Sallustrau, J. Balzarini, M. R. Bedford, J. C. Eden, N. Georgousi, N. J. Hodges, J. Kedge, Y. Mehellou, C. Tselepis and J. H. R. Tucker, J. Med. Chem., 2014, 57, 5817–5822 CrossRef CAS PubMed.
- A. Mooney, R. Tiedt, T. Maghoub, N. O'Donovan, J. Crown, B. White and P. T. M. Kenny, J. Med. Chem., 2012, 55, 5455–5466 CrossRef CAS PubMed.
- S. B. Deepthi, R. Trivedi, L. Giribabu, P. Sujitha and C. G. Kumar, Dalton Trans., 2013, 42, 1180–1190 RSC.
- S. Daum, V. F. Chekhun, I. N. Todor, N. Y. Lukianova, Y. V. Shvets, L. Sellner, K. Putzker, J. Lewis, T. Zenz, I. A. M. de Graaf, G. M. M. Groothuis, A. Casini, O. Zozulia, F. Hampel and A. Mokhir, J. Med. Chem., 2015, 58, 2015–2024 CrossRef CAS PubMed.
- P. F. Salas, C. Herrmann, J. F. Cawthray, C. Nimphius, A. Kenkel, J. Chen, C. de Kock, P. J. Smith, B. O. Patrick, M. J. Adam and C. Orvig, J. Med. Chem., 2013, 56, 1596–1613 CrossRef CAS PubMed.
- R. Pulukkody, R. B. Chupik, S. K. Montalvo, S. Khan, N. Bhuvanesh, S.-M. Lim and M. Y. Darensbourg, Chem. Commun., 2017, 53, 1180–1183 RSC.
- P. Messina, E. Labbe, O. Buriez, E. A. Hillard, A. Vessieres, D. Hamels, S. Top, G. Jaouen, Y. M. Frapart, D. Mansuy and C. Amatore, Chem.–Eur. J., 2012, 18, 6581–6587 CrossRef CAS PubMed.
- G. Jaouen, A. Vessieres and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817 RSC.
- S. Top, J. Tang, A. Vessieres, D. Carrez, C. Provot and G. Jaouen, Chem. Commun., 1996, 955–956 RSC.
- G. Jaouen, S. Top, A. Vessieres, G. Leclercq and M. J. McGlinchey, Curr. Med. Chem., 2004, 11, 2505–2517 CrossRef CAS PubMed.
- S. Top, A. Vessieres, G. Leclercq, J. Quivy, J. Tang, J. Vaissermann, M. Huche and G. Jaouen, Chem.–Eur. J., 2003, 9, 5223–5236 CrossRef CAS PubMed.
- G. Jaouen and S. Top, in Advances in Organometallic Chemistry and Catalysis: The Silver/Gold Jubilee International Conference on Organometallic Chemistry Celebratory Book, ed. A. J. L. Pombeiro, Wiley, 2014, ch. 42, pp. 563–580 Search PubMed.
- A. Vessieres, C. Corbet, J. M. Heldt, N. Lories, N. Jouy, I. Laios, G. Leclercq, G. Jaouen and R. A. Toillon, J. Inorg. Biochem., 2010, 104, 503–511 CrossRef CAS PubMed.
- C. Bruyere, V. Mathieu, A. Vessières, P. Pigeon, S. Top, G. Jaouen and R. Kiss, J. Inorg. Biochem., 2014, 141, 144–151 CrossRef CAS PubMed.
- A. Citta, A. Folda, A. Bindoli, P. Pigeon, S. Top, A. Vessieres, M. Salmain, G. Jaouen and M. P. Rigobello, J. Med. Chem., 2014, 57, 8849–8859 CrossRef CAS PubMed.
- E. Hillard, A. Vessières, L. Thouin, G. Jaouen and C. Amatore, Angew. Chem., Int. Ed., 2006, 45, 285–290 CrossRef CAS PubMed.
- Y. Wang, M.-A. Richard, S. Top, P. M. Dansette, P. Pigeon, A. Vessières, D. Mansuy and G. Jaouen, Angew. Chem., Int. Ed., 2016, 55, 10431–10434 CrossRef CAS PubMed.
- D. Hamels, P. M. Dansette, E. A. Hillard, S. Top, A. Vessieres, P. Herson, G. Jaouen and D. Mansuy, Angew. Chem., Int. Ed., 2009, 48, 9124–9126 CrossRef CAS PubMed.
- Q. Michard, G. Jaouen, A. Vessieres and B. A. Bernard, J. Inorg. Biochem., 2008, 102, 1980–1985 CrossRef CAS PubMed.
- E. Allard, C. Passirani, E. Garcion, P. Pigeon, A. Vessières, G. Jaouen and J.-P. Benoit, J. Controlled Release, 2008, 130, 146–153 CrossRef CAS PubMed.
- E. Allard, D. Jarnet, A. Vessieres, S. Vinchon-Petit, G. Jaouen, J. P. Benoit and C. Passirani, Pharm. Res., 2011, 27, 56–64 CrossRef PubMed.
- A. L. Laine, E. Adriaenssens, A. Vessieres, G. Jaouen, C. Corbet, E. Desruelles, P. Pigeon, R. A. Toillon and C. Passirani, Biomaterials, 2013, 34, 6949–6956 CrossRef CAS PubMed.
- A. L. Laine, A. Clavreul, A. Rousseau, C. Tétaud, A. Vessieres, E. Garcion, G. Jaouen, R. A. Toillon and C. Passirani, Nanomed. Nanotechnol. Biol. Med., 2014, 10, 1667–1677 CrossRef CAS PubMed.
- Y. Wang, P. Pigeon, S. Top, M. J. McGlinchey and G. Jaouen, Angew. Chem., Int. Ed., 2015, 54, 10230–10233 CrossRef CAS PubMed.
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS PubMed.
- I. G. Gut, P. D. Wood and R. W. Redmond, J. Am. Chem. Soc., 1996, 118, 2366–2373 CrossRef CAS.
- M.-A. Richard, D. Hamels, P. Pigeon, S. Top, P. M. Dansette, H. Z. S. Lee, A. Vessières, D. Mansuy and G. Jaouen, ChemMedChem, 2015, 10, 981–990 CrossRef CAS PubMed.
- V. Scalcon, A. Citta, A. Folda, A. Bindoli, M. Salmain, I. Ciofini, S. Blanchard, J. D. Cazares-Marinero, Y. Wang, P. Pigeon, G. Jaouen, A. Vessieres and M. P. Rigobello, J. Inorg. Biochem., 2016, 165, 146–151 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- I. Romero-Canelon, L. Salassa and P. J. Sadler, J. Med. Chem., 2013, 56, 1291–1300 CrossRef CAS PubMed.
- M. Görmen, P. Pigeon, S. Top, E. A. Hillard, M. Huché, C. G. Hartinger, F. de Montigny, M.-A. Plamont, A. Vessières and G. Jaouen, ChemMedChem, 2010, 5, 2039–2050 CrossRef PubMed.
- H. T. Cohen and F. J. McGovern, N. Engl. J. Med., 2005, 353, 2477–2490 CrossRef CAS PubMed.
- W. T. Bellamy, Annu. Rev. Pharmacol. Toxicol., 1996, 36, 161–183 CrossRef CAS PubMed.
- D. Grossman and D. C. Altieri, Cancer Metastasis Rev., 2001, 20, 3–11 CrossRef CAS PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- P. Álvarez, J. A. Marchal, H. Boulaiz, E. Carrillo, C. Vélez, F. Rodríguez-Serrano, C. Melguizo, J. Prados, R. Madeddu and A. Aranega, Expert Opin. Ther. Pat., 2012, 22, 107–123 CrossRef PubMed.
- G.-Q. Chen, L. Zhou, M. Styblo, F. Walton, Y. Jing, R. Weinberg, Z. Chen and S. Waxman, Cancer Res., 2003, 63, 1853–1859 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for syntheses and biological evaluation, supplementary Fig. 1–8 and Tables 1–6, X-ray crystallographic data, cif file. CCDC 1527404. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04213b |
| This journal is © The Royal Society of Chemistry 2018 |