DOI:
10.1039/C7SC04200K
(Minireview)
Chem. Sci., 2018,
9, 15-21
Mechanochemistry of the mechanical bond
Received
26th September 2017
, Accepted 27th November 2017
First published on 4th December 2017
Abstract
Strong and stable under tension? In this review we present the recent efforts investigating the mechanochemical properties of interlocked structures by atomic force microscopy and polymer mechanochemistry.
 Guillaume De Bo | Guillaume De Bo was born in Brussels (Belgium) and obtained his PhD in 2009 at the University of Louvain in Belgium under the guidance of Prof. István E. Markó, working on the synthesis of angular triquinanes. In 2009 he took a post-doctoral position in the laboratory of Prof. Jean-François Gohy and Prof. Charles-André Fustin (UCL, Belgium) to work on the assembly of mechanically linked block copolymers. He then moved to the UK to join the group of Prof. David A. Leigh, to work on the development of molecular machines. Since January 2016 he is a Royal Society University Research Fellow at the University of Manchester. His research interests include the mechanical activation of covalent and mechanical bonds for applications in materials, synthesis, and catalysis. |
Introduction
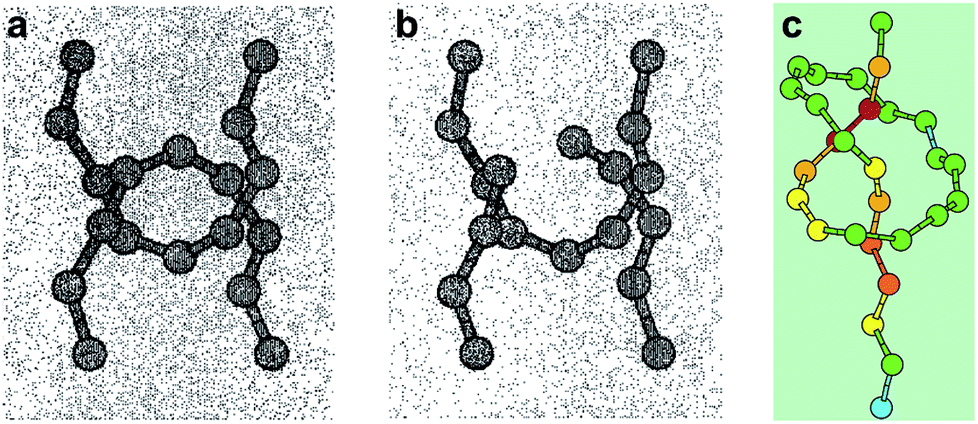
Mechanical bonds have always fascinated chemists because of their intriguing nature and an undeniable aesthetic appeal.1 Since the first synthesis of a catenane in 1960,2 mechanical bonds have been used in a variety of contexts and their dynamic properties have been exploited to build molecular machines3 and new materials.4 However, a fundamental question remains: what is the strength of a mechanical bond? Formally a mechanical bond is as strong as a covalent bond since one often needs to break the latter to open the former. Nevertheless, the very interlocked nature of this bond has the potential to weaken covalent bonds within the topological construct. For example it has been shown that the entanglement of polymer chains under tension can lead to the rupture of covalent bonds5 (Fig. 1a and b) and that the presence of a knot in a polymer weakens the chain at the entrance of the knotted structure (Fig. 1c).6,7 Hence, beyond the fundamental aspects of chemical bonding that this question could answer, a better understanding of the mechanical bond under tension is also of significant importance for the development of new materials. In this review we cover the early examples of the mechanochemical investigation of interlocked structures by atomic force microscopy8 (AFM) and polymer mechanochemistry.9,10
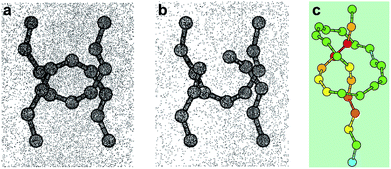 |
| | Fig. 1 Snapshots from molecular dynamics simulation of the carbon backbones of two entangled alkanes (a) before and (b) after the break of a C–C bond and (c) a knotted polymer strand showing the strain energy distribution (from blue (low) to red (high)). Reproduced from ref. 5 and 6 respectively. | |
Chemical bonds under tension
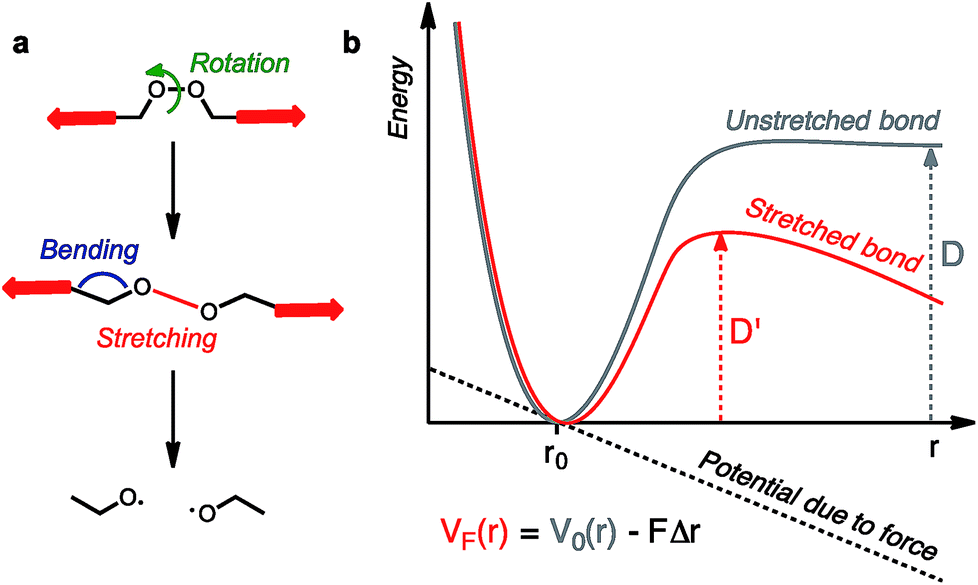
Molecules respond to external force via bond rotation, angle bending, and bond stretching until, at sufficiently high force, a bond scission occurs (Fig. 2a). Rupture of the weakest covalent bond usually proceeds in a homolytic fashion and this process can be described by a Morse potential (V0(r)) modified by the application of a constant force F over a distance Δr (Fig. 2b).11 The resulting deformed Morse potential (VF(r)) displays a lower bond dissociation energy (D′) than in the force-free potential (D, Fig. 2b).
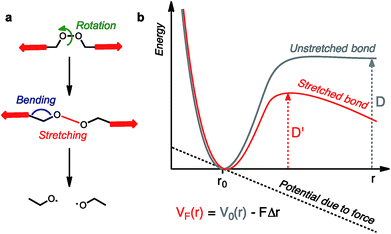 |
| | Fig. 2 Molecules under tension can undergo a series of deformations before the rupture of the weakest bond (a). The potential energy surface of a bond under tension can be described by a force-modified Morse potential (b). | |
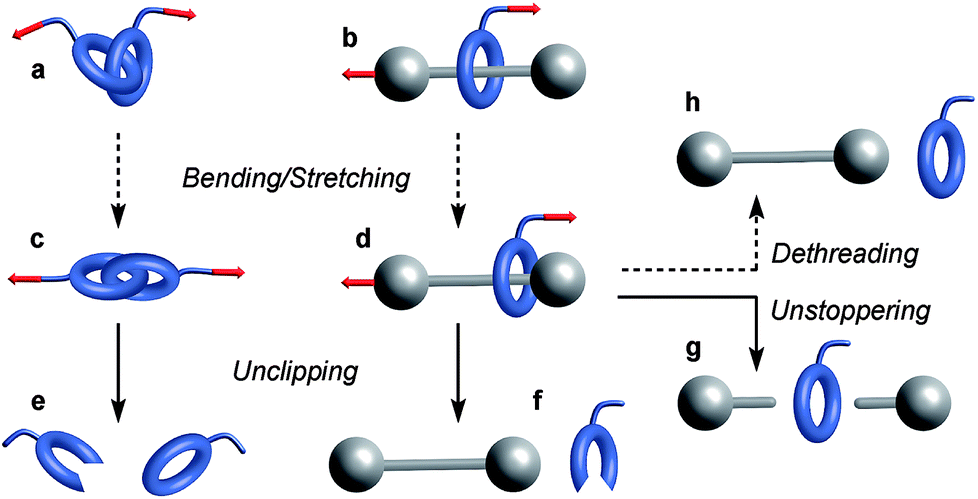
Similarly, bending and stretching is observed when a macrocycle is pulled out of its equilibrium position in a mechanical bond under tension (Fig. 3c and d). At higher force, the mechanical bond eventually breaks trough the rupture of a covalent bond in the macrocycle (Fig. 3e and f) or the thread (Fig. 3g). An additional dissociation pathway exists in rotaxanes whereby the dethreading of the macrocycle is facilitated by the deformation of the molecular backbone of the macrocycle and/or the stopper (Fig. 3h).
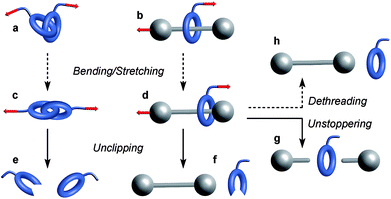 |
| | Fig. 3 Examples of mechanochemical behaviors observed in a catenane (a) and a rotaxane (b). Elongation of these mechanical bonds (c and d) can lead to their disassembly via the rupture of a covalent bond in the macrocycle (e and f) or the thread (g). Rotaxanes can also dissociate by dethreading (h). Plain and dashed arrows denote events involving the rupture or not of a covalent bond respectively. | |
Single molecule study by AFM
Initial AFM studies investigating the behaviour of mechanical bonds under tension have focused on dethreading rotaxanes and probing the strength of intercomponent interactions.12–18 In a typical experiment, one subcomponent of the mechanical bond is anchored to a surface while the other is tethered to the AFM tip. The tip is then raised at a defined speed to stretch the mechanical bond along the pulling axis and the resulting force is recorded as a function of the tip-surface distance.
Crossing barriers
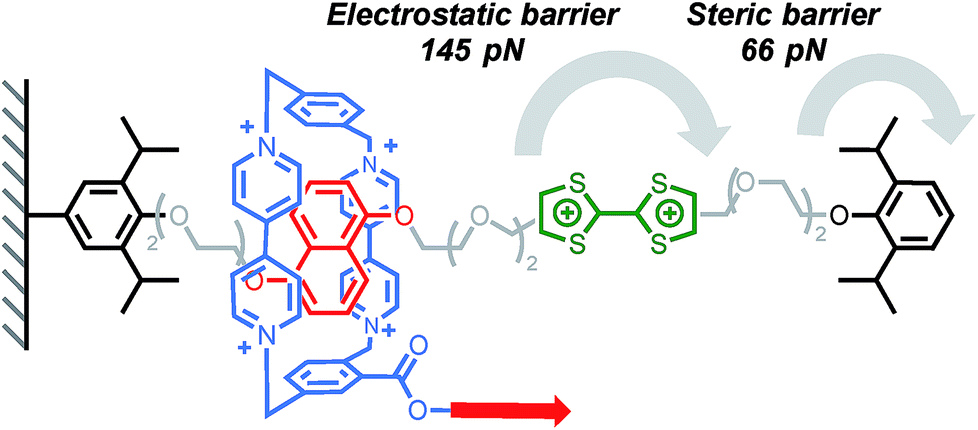
The ability of a macrocycle to cross or not a steric barrier (‘stopper’) is what distinguishes a rotaxane from a pseudorotaxane19,20 and, although the use of a bulky stopper21 is usually preferred to assemble rotaxanes, the fine tuning of the stoppering ability of a steric group22 is desirable for the assembly of complex molecular machines23–25 and motors.26,27 The ability of a bulky group to stop a macrocycle from dethreading has been the object of several experimental19,20,22,28–32 and computational19,22,33 studies, and it is no surprise that it has also been the subject of the first mechanochemical investigations. An early report by Ho, Stoddart, Houk and co-workers explored the electrostatic barrier and the steric barrier that a tetracationic macrocycle has to overcome upon deslipping from a rotaxane thread.12 Pulling experiments were performed on a redox switchable rotaxane (Scheme 1),34,35 where the cyclobis(paraquat-p-phenylene) (CBPQT4+) ring, tethered to the AFM tip, was pulled at a rate of 1.05 μm s−1 (the tips used had an average spring constant of 6 pN nm−1 leading to a loading rate of ∼6 nN s−1) from the neutral tetrathiafulvalene (TTF) station or the 1,5-dioxynaphthalene (DNP) station until deslippage occurred. In the first case, rupture occurs at 66 pN, a force orders of magnitude below that required to break a covalent bond (typically in the nN range);36 this is interpreted as the rupture of the interlocked architecture with the macrocycle crossing the steric barrier of the stopper unit. In the second case a force of 145 pN is required for the tetracationic CBPQT4+ to cross the electrostatic barrier of the TTF2+ unit. By combining AFM and computational data, the authors were able to determine the steric and electrostatic barrier energies (113 kJ mol−1 and 192 kJ mol−1 respectively). The energy required to cross the electrostatic barrier is significantly higher than the value measured in stress-free conditions on similar systems,37,38 which could be explained by the fact that in a pulling experiment the subcomponents can be bent out of shape and forced into high energy conformations and that mechanical bonds require greater elongation than covalent bonds and hence lower force (see equation in Fig. 2). Taking into account the energy required to break the CBPQT4+/TTF interaction (79 kJ mol−1), the authors calculated that the oxidation of TTF to TTF2+ in the rotaxane delivers a repulsive actuation energy of 272 kJ mol−1.
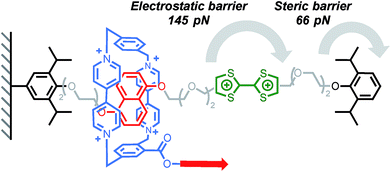 |
| | Scheme 1 AFM experiments probing the ability of a tetracationic macrocycle to cross electrostatic (green) and steric (black) barriers in Stoddart's bistable rotaxane (counterions are omitted for clarity). Red arrow indicates the direction of the force. | |
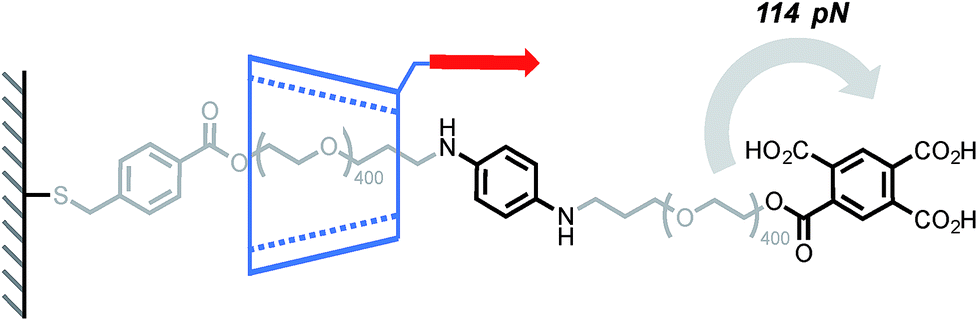
Cyclodextrins (CD) are well known to form polyrotaxanes by threading on polyethyleneglycol (PEG) chains,39 and these systems form the basis of Ito's slide-ring gels, which are particularly effective at dissipating mechanical energy.4 In this context, Round's study shed light on the sliding/deslipping behaviour of an α-CD on a PEG track.14 Several (poly)rotaxanes of similar structure but with different axel lengths were investigated by AFM (Scheme 2). The force-extension profile obtained after pulling a cyclodextrin at a rate of 500 nm s−1 (∼7–12 nN s−1) showed a peak at 114 pN and, for each rotaxane, the peak rupture length was consistent with the length of the PEG track plus the length of the tether linking the α-CD to the tip. A control experiment has shown that the detachment of the polymer from the tip occurs at higher force (∼271 pN). Taken together, these results suggest that the polymer breaks when the α-CD slips over the bulky tricarboxylic acid benzoyl stopper. Interestingly the relatively high force required to break these interlocked structures can be partially attributed to the affinity of the tricarboxylic unit for the α-CD since a force of 56 pN is recorded to separate the non-interlocked host–guest complex.
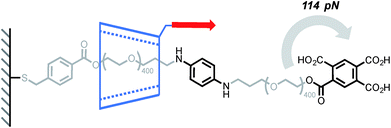 |
| | Scheme 2 AFM experiment probing the ability of α-cyclodextrin to slip over a tricarboxylic acid benzoyl stopper. Red arrow indicates the direction of the force. | |
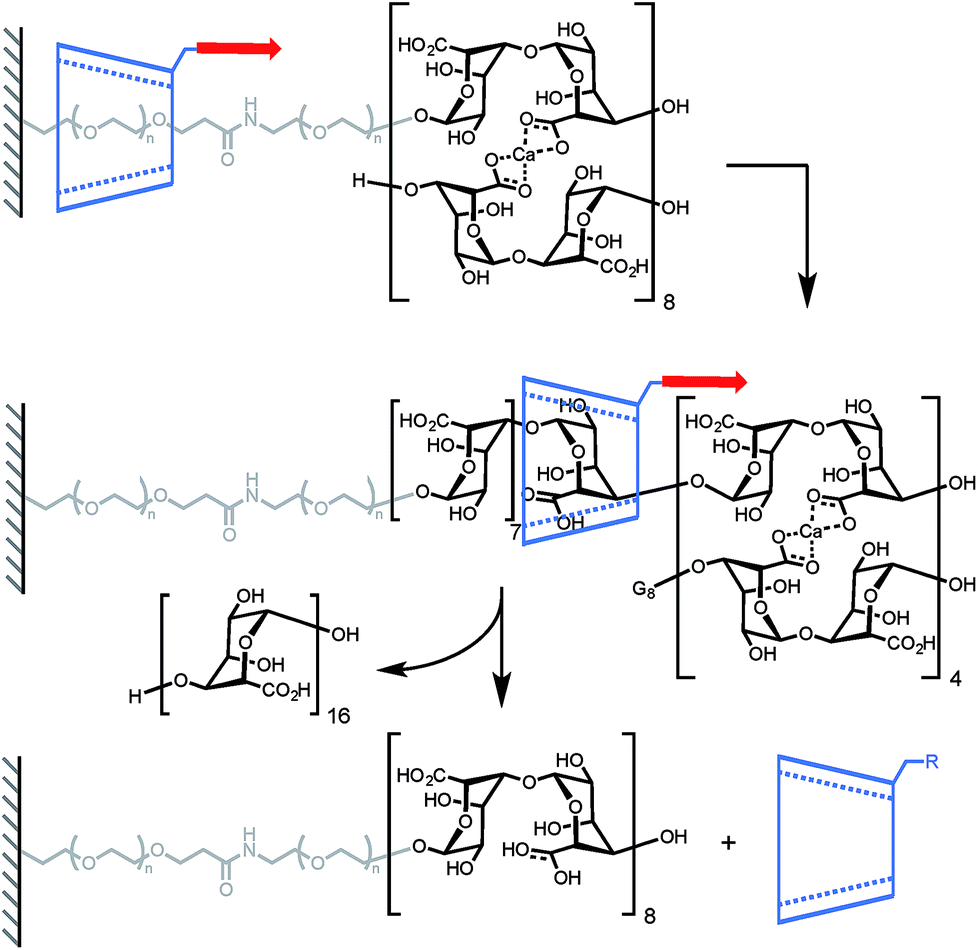
A mechanical bond can also prove to be very useful to probe the stability of a non-covalent complex. In another elegant experiment, Round and co-workers have used a CD-based pseudorotaxane to measure the strength of a calcium-mediated eggbox junction (a cross-link between sequences of oligoguluronic acids (oligoGs), Scheme 3).18 Because of their long equilibration time these complexes are difficult to characterize by AFM. With this design the system is able to fully equilibrate before the AFM tip is lowered to pick an α-CD in order to unzip the eggbox junction (α-CD can only accommodate one oligoG inside its cavity). The authors first determined how much force is required to slide along a single strand of oligoGs. Slipping over one guluronic acid requires ∼47 pN, amounting to a steric barrier of 48 kJ mol−1, while unzipping requires 58 pN, 112 pN and 141 pN (at 500 nm s−1, ∼6–20 nN s−1) for oligoGs containing 2, 4 and 8 Ca2+ respectively (corresponding to 31 kJ per mol per Ca2+ for the two longer oligoGs). The strength of the first cross-link actually depends on the number of subsequent cross-links and inspection of the force-extension curves of the different oligomers shows that the α-CD doesn't break one complex at a time but rather disrupts a sequence of 4 Ca2+ in a single burst.
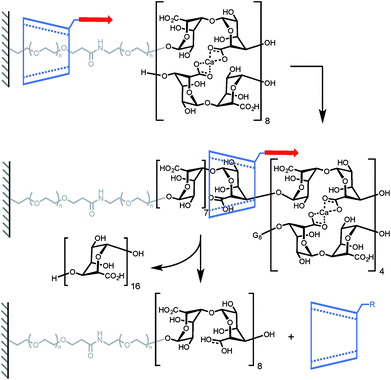 |
| | Scheme 3 AFM experiment probing the ability of α-cyclodextrin to dethread by unzipping a doubly-stranded oligoguluronic acid/Ca2+ complex. Red arrows indicate the direction of the force. | |
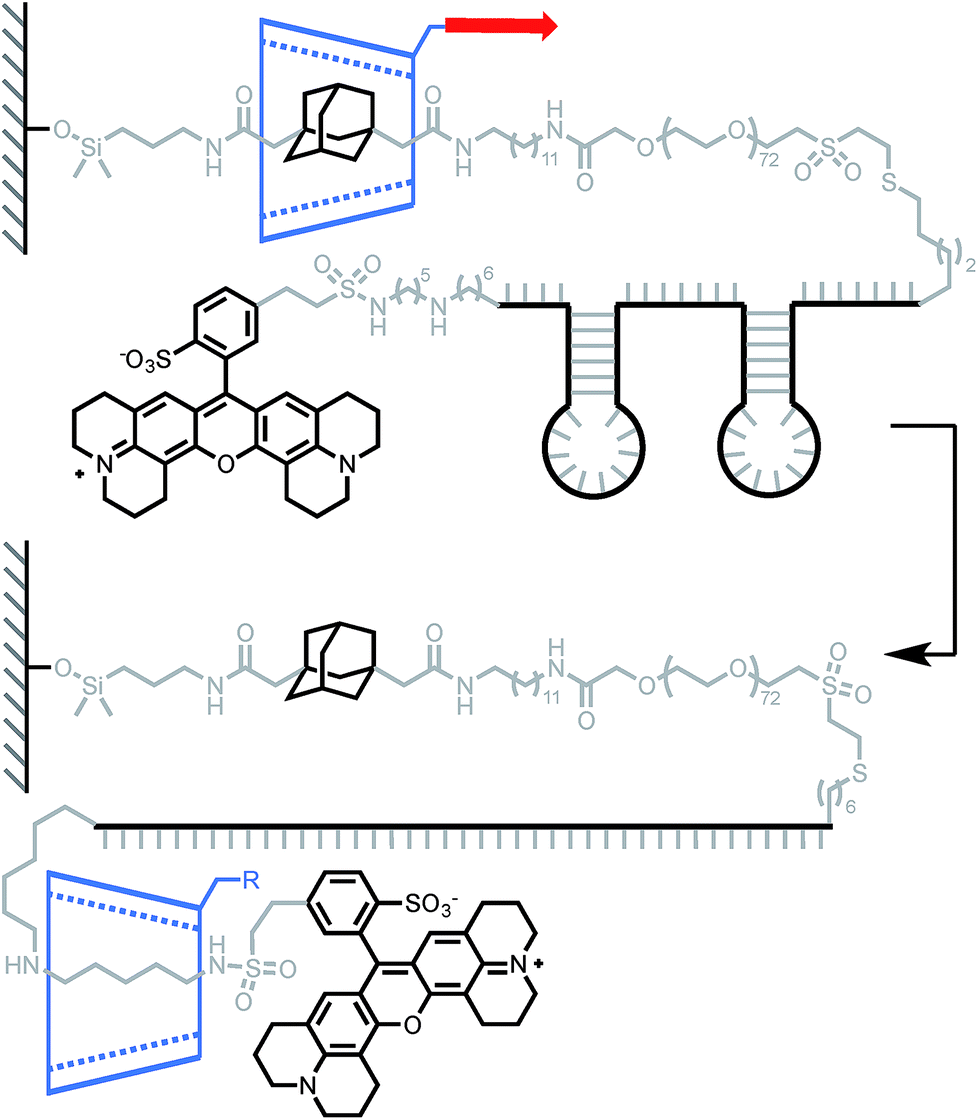
Lindsay and his team used a similar approach to measure the force required to unfold DNA secondary structures, with the original goal of applying this approach to the sequencing of biopolymers.13 Indeed, steered molecular dynamics (SMD) simulations have shown that small differences in the force required to slip a β-CD over a purine or pyrimidine base could be observed at high loading rate.40 Two rotaxanes were built with either a 49-base or an 81-base oligonucleotide, each containing two hairpins (Scheme 4). Pulling the β-CD along the PEG–DNA conjugate at high loading rates (∼10–100 nN s−1) didn't display any composition-dependent profile and only two rupture events could be observed for each rotaxane. They were attributed to the successive unfolding of the two hairpins, the first occurring around 376 pN in both oligonucleotides, and the second around 248 pN and 371 pN in the 49-base and 81-base oligonucleotides respectively. Hairpin opening requires a much higher force in the rotaxane than when it is pulled from both ends (∼15 pN).41 The authors attribute this to the fact that hairpins open at lower strain with β-CD, but it could also be indicative of a change in pulling geometry from unzipping to shearing. Another interesting feature of this system is that the average diameter of single-stranded DNA (1–2 nm) is rather large compared to the internal diameter of a β-CD (0.7 nm), so it cannot pass through easily. In this case, the applied force probably keeps the β-CD in a constant deformed state, which modifies the geometry of the internal cavity to better accommodate the passage of the DNA strand. The same effect might come into play in the rotaxane depicted in Scheme 1 and explain why a macrocycle can slip over a stopper that seems too large at first glance (instead of breaking apart).
 |
| | Scheme 4 AFM experiment probing the ability of β-cyclodextrin to dethread by unzipping a pair of hairpin DNA. Red arrow indicates the direction of the force. | |
Probing intercomponent interactions
The ability to control the dynamic properties of interlocked structures through the modulation of their intercomponent interactions is at the heart of their use as molecular machine elements.42 The possibility of using AFM to probe non-covalent interactions at- and out-of-equilibrium has been recognised43 and demonstrated in multiple systems.44
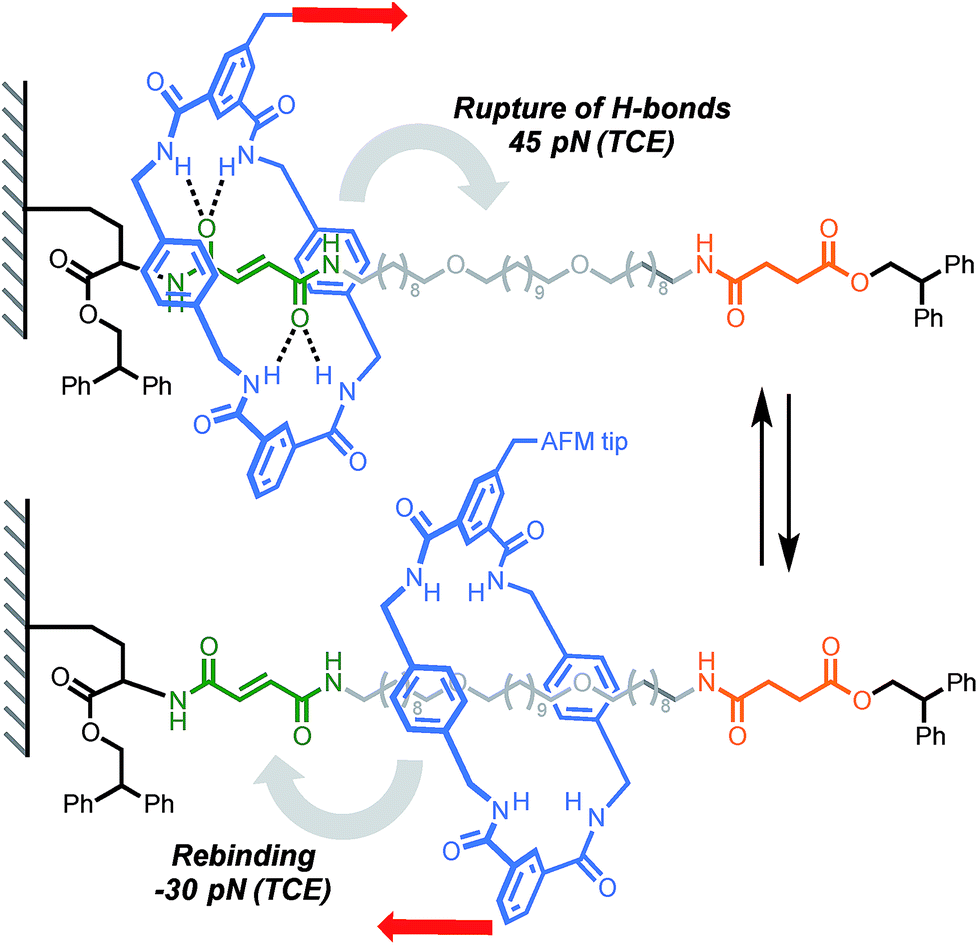
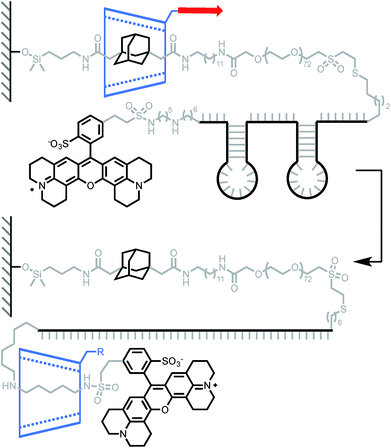
A report by Duwez, Leigh, Fustin and co-workers demonstrated the force-induced switching of a bistable rotaxane where a tetralactam macrocycle, connected to an AFM tip via a polyethyleneoxide (PEO) tether, is pulled away from a strongly binding fumaramide station towards a weakly binding succinic amide-ester station (Scheme 5).16 In DMF, a force of 27 pN was required to disrupt the hydrogen bond network connecting the ring and the fumaramide station (0.5 nN s−1). Higher forces were recorded in less polar solvents such as tetrachloroethane (TCE, 45 pN). Interestingly, an increase in tension was observed upon reduction of the tip-surface distance. This indicates that the macrocycle is able to generate a force (30 pN in TCE) against the external load exerted by the cantilever while rebinding to the fumaramide station, amounting to a work of ∼25 kJ mol−1, which is of similar magnitude to the force developed by biological machines (∼3–60 pN).45
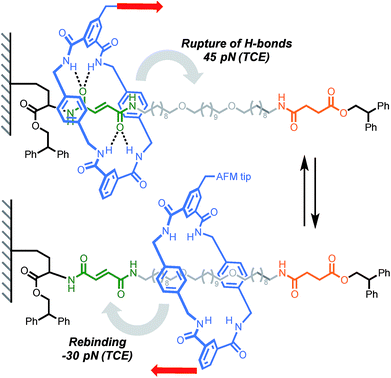 |
| | Scheme 5 AFM experiment probing the ability of a tetralactam macrocycle to escape a complementary station in Leigh's bistable rotaxane. Red arrows indicate the direction of the force. | |
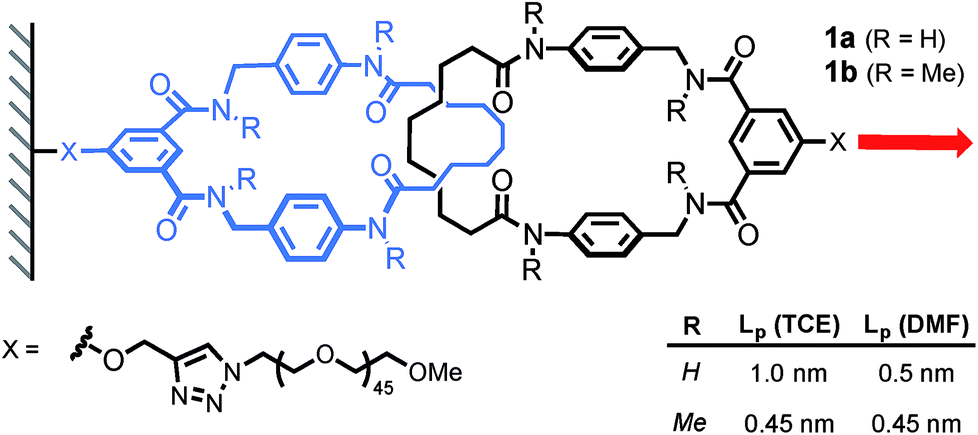
The same authors then probed the mobility of a tetralactam [2]catenane under tension.17 Using AFM, they recorded the force-extension curve of catenane 1a and its methylated counterpart 1b, in DMF and TCE (Scheme 6). The persistence length of each polymer (Lp) was extracted by fitting the curves with a worm-like chain model. Catenane 1b, which is unable to form any internal hydrogen bonds, displays the same persistence length (Lp) in both solvents (0.45 nm). On the other hand, catenane 1a shows a dramatic increase in Lp from DMF (0.5 nm) to TCE (1 nm), due to strong hydrogen bond interactions between the two rings that transform the catenane in a long rigid segment. This result nicely demonstrates the impact that a single mechanical bond can have on a polymer's properties.
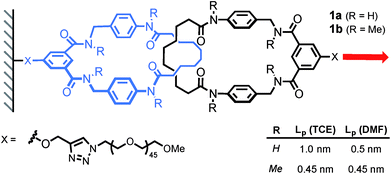 |
| | Scheme 6 AFM experiment probing the mobility of a [2]catenane under tension (1 nN s−1). Red arrow indicates the direction of the force. | |
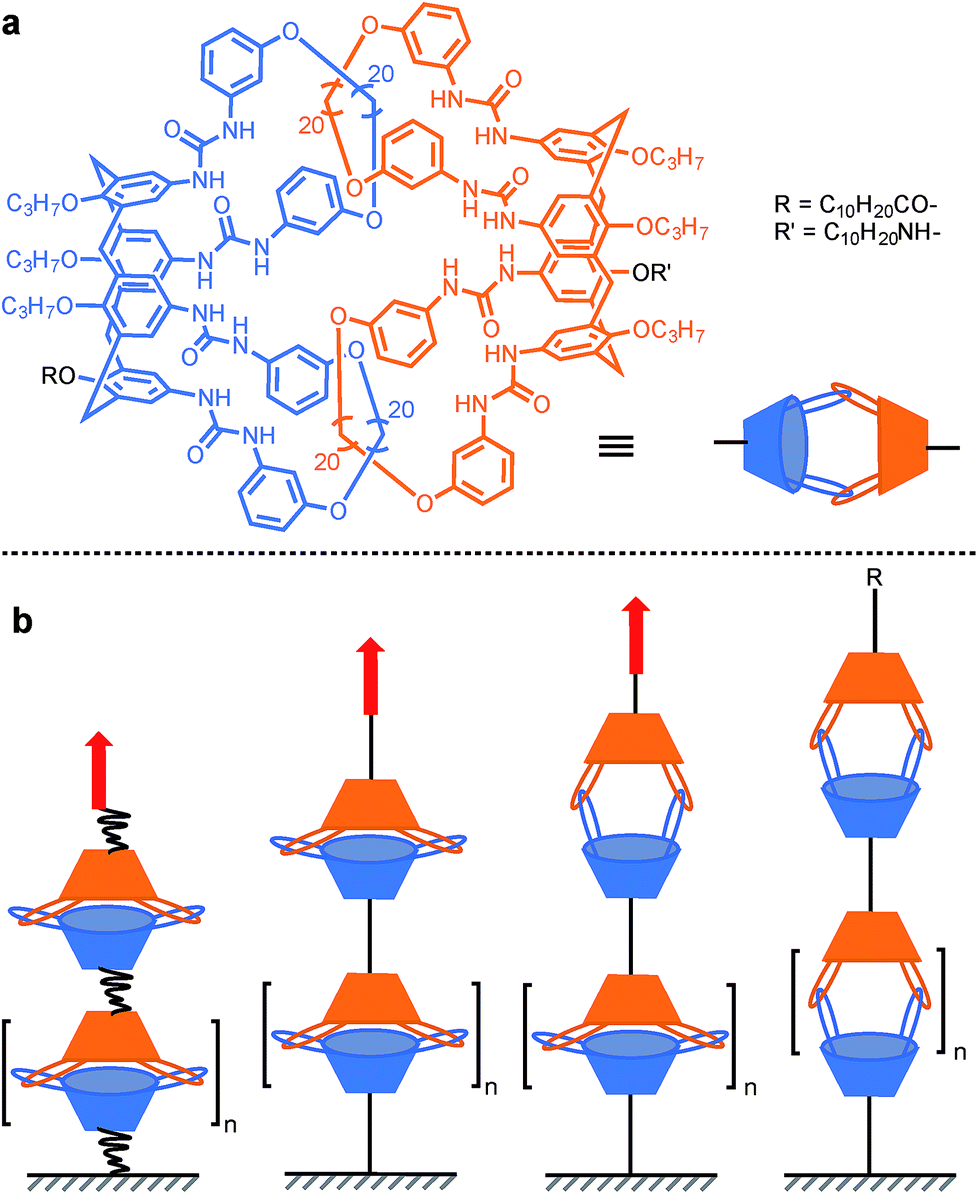
In order to probe the strength of supramolecular interactions within both quasi-equilibrium and out-of-equilibrium regimes, where the pulling rate is respectively close to or much higher than the natural bond dissociation rate, Janshoff and co-workers devised a system that prevents the bond rupture from being irreversible, based on interlocked calix[4]arenes.15 Dimeric capsules of calix[4]arenes are held together by their interdigitated urea's 16 hydrogen bonds, and concatenated by two pairs of interlocked hoops protruding from the larger rim of the cavitand (Scheme 7a). Short oligomers were obtained by tethering several of these capsules together. Pulling the oligomer along its axis reveals a sawtooth pattern in the force-extension curves that is indicative of individual non-cooperative dissociation of calixarene dimers. The dynamic strength of the hydrogen bond network was found to amount to 40–60 pN (mesitylene) for a quasi-equilibrium regime (60 pN s−1). This corresponds to ∼3–4 pN per hydrogen bond (for 8 strong and 8 weak interactions/dimer) and is in good agreement with the value observed for a single RNA base pair (∼4–5 pN per H-bond).46 Additional measurements further away from equilibrium (0.3–30 nN s−1) and molecular dynamics simulations uncovered two successive increase in length (pulling from R/R′, Scheme 7a), of ∼1.1 nm and ∼0.8 nm attributed to the rupture of the H-bonds and the opening of a sterically locked conformation respectively.
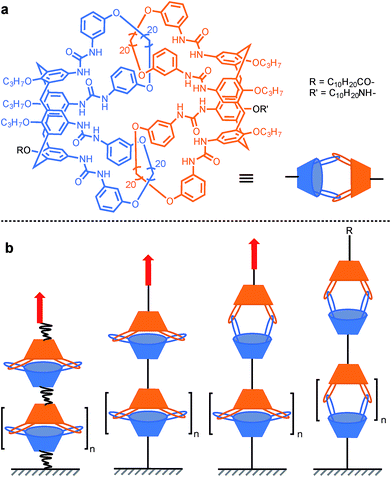 |
| | Scheme 7 AFM experiment probing the 16 hydrogen bonds uniting interlocked calix[4]arenes dimeric capsules. Red arrows indicate the direction of the force. | |
Mechanical activation in solution with ultrasound
Ultrasound-induced cavitation has the ability to stretch macromolecules at a much higher strain rate than AFM (typically in the order of N s−1).9,10 In this technique, acoustic waves provoke the nucleation, growth and collapse of microbubbles. As a result, a high-gradient elongational flow is created in the surroundings of collapsing bubbles. If a macromolecule is caught within this gradient, it is stretched and deformed until an eventual bond scission takes place in the central region of the molecular chain. This usually occurs in a homolytic fashion but more complex reaction pathways can be accessed when mechanically-active molecules (mechanophores) are introduced in the middle of a polymer.47 This approach has become the method of choice for studying the mechanochemistry of polymers in solution.9,10 The first mechanical bond activated in such a way was reported by Stoddart and his team.48 Sonication of a rotaxane-centred poly(methyl acrylate) led to a reduction of the molecular weight to approaching half of its initial value (Scheme 8). Control experiments confirmed the mechanochemical nature of the degradation. The disassembly of the mechanical bond was inferred from the diminution of the charge-transfer UV-vis absorption, associated with the interaction between the ring and the electron-rich DNP unit, but it was not possible to determine if it occurred through dethreading or via the cleavage of a covalent bond (on the axle or the macrocycle). Based on the force obtained by AFM (Scheme 1), deslipping of the macrocycle seems to be the most likely degradation pathway. However, since bond strength depends greatly on how fast the force is applied (i.e. the loading rate),49 alternative degradation pathways involving the rupture of a covalent bond cannot be ruled out in these high-strain conditions (Fig. 2).
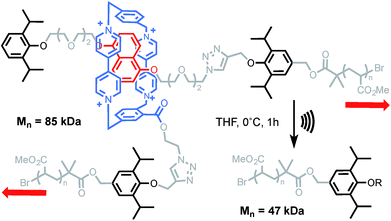 |
| | Scheme 8 Sonication of Stoddart's rotaxane incorporated into a PMA backbone. Counterions not shown for clarity. Red arrows indicate the direction of the force. | |
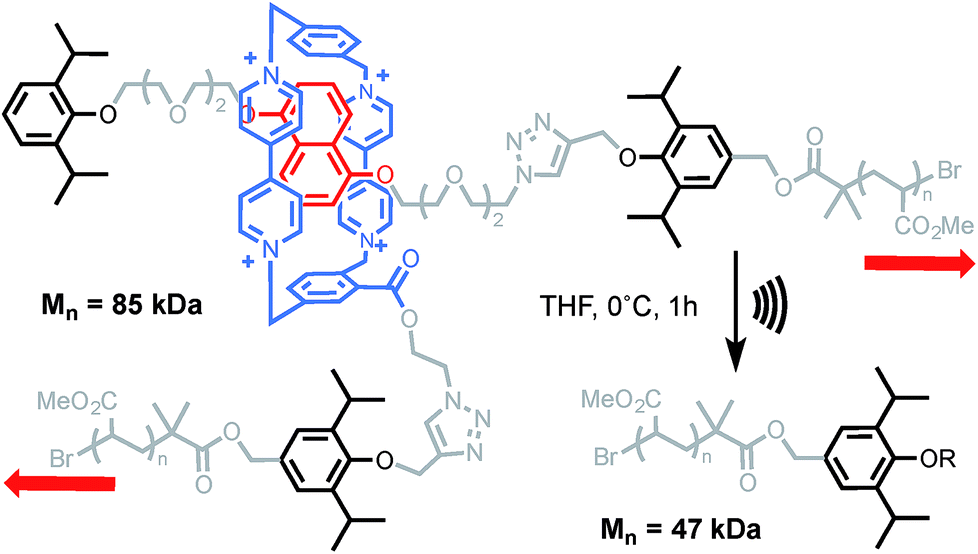
More recently, the group of Stephen Craig used the same technique to investigate the strength of a [2]catenane (2a) in comparison with its macrocyclic (2b) and linear (2c) counterparts (Scheme 9).50 Copolymers of gem-dichlorocyclopropanated-polybutadiene (gDCC-PB) with 5 mol% of either the catenane or a topological control were obtained by entropy-driven ring-opening metathesis polymerization (ED-ROMP).51 Each polymer was sonicated for a prolonged time in order to reach the limiting molecular weights (Mlim) below which mechanical activation does not occur. Hence, by comparing Mlim one can establish the relative mechanical strength of each topology. A narrow distribution of Mlim (28–30 kDa) is obtained for each polymer. Since the efficiency of mechanical activation actually depends on the length of the polymer rather than its molecular weight,52,53 the limiting contour lengths (Llim) were calculated for each polymer. Again, no significant difference was observed between the three systems and further comparison with a polymer containing only gDCC-PB units revealed no difference either. These striking results suggest that, at the high loading rate developed during sonication, the catenane, which can be seen as a model for polymer entanglement, is not significantly weaker than the covalent bonds composing the host polymer (although small differences in reactivity are difficult to detect by sonication).54 The ability to incorporate a mechanical bond without compromising the mechanical resistance of a polymer is a very exciting prospect indeed and, if confirmed at different strain rates, will certainly lead to new materials taking advantage of the dynamic properties of this mechanical bond.
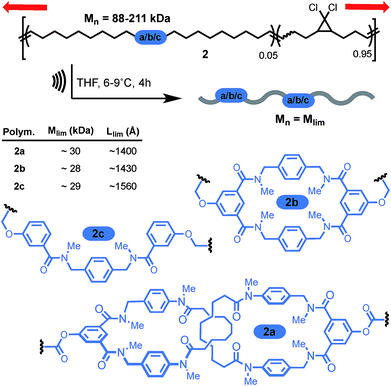 |
| | Scheme 9 Craig's sonomechanochemical approach to probe the mechanical strength of a catenane: limiting mass of a polymer containing Leigh's catenane 2a was compared to non-interlocked counterparts 2b–c. Red arrows indicate the direction of the force. | |
Conclusions
These examples offer an insight into a little-explored area of the chemistry of the mechanical bond and illustrate the complementarity of single molecule and bulk techniques for the investigation of these systems. With its ability to generate (rather low) forces in a controlled manner, AFM is well suited to interrogate molecular systems at and out of equilibrium. This characteristic is particularly desirable for the investigation of molecular machines and has proved effective in determining the force (and hence the energy) required for a macrocycle to escape the attraction of a binding site, and to cross electrostatic and steric barriers in molecular shuttles. More intriguing is the capacity of a sliding macrocycle to trigger secondary processes such as the unzipping of oligoGs and DNA hairpins. Mechanical force is a remarkably effective stimulus and we anticipate that the use of elongational techniques to generate directional forces should find more application in the activation and operation of molecular machines. These future developments will have to address the challenge of the additional structural complexity required to interface a mechanical bond with an external force and of applying mechanical force on intricate molecular constructs in a controlled fashion beyond the single molecule. Indeed, because higher forces and loading rates can be attained (albeit in a less controlled manner), the sonochemical approach is better suited for the activation of covalent bonds. This brings us back to our initial question: what is the strength of a mechanical bond? Perhaps surprisingly, it appears from Craig's example that the mechanical resistance of a [2]catenane, the archetypal mechanical bond, is not significantly weaker (or stronger) than its constitutional covalent bonds. This result, if confirmed at different strain rates, could have major implications for the development of new materials. More of these intriguing structures are waiting to be stretched to prove their strength and what even more elaborate structures, such as knots and higher order links, will reveal is anyone's guess, but it will certainly be of great interest. More than half a century after the first synthesis of a catenane, these fantastic molecules continue to amaze.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The author wishes to thank the Royal Society for a University Research Fellowship.
Notes and references
-
C. J. Bruns and J. F. Stoddart, in The Nature of the Mechanical Bond, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2016, pp. 55–268 Search PubMed.
- E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433–4434 CrossRef CAS.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
-
K. Ito, K. Mayumi and K. Kato, Polyrotaxane and Slide-Ring Materials, The Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- A. M. Saitta and M. L. Klein, J. Phys. Chem. B, 2000, 104, 2197–2200 CrossRef CAS.
- M. L. Klein, A. M. Saitta, P. D. Soper and E. Wasserman, Nature, 1999, 399, 46–48 CrossRef.
- T. Stauch and A. Dreuw, Angew. Chem., Int. Ed., 2016, 55, 811–814 CrossRef CAS PubMed.
- A. Janshoff, M. Neitzert, Y. Oberdörfer and H. Fuchs, Angew. Chem., Int. Ed., 2000, 39, 3212–3237 CrossRef CAS PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
-
Polymer Mechanochemistry, ed. R. Boulatov, Springer, Berlin, 2015, vol. 369 Search PubMed.
- W. Kauzmann and H. Eyring, J. Am. Chem. Soc., 1940, 62, 3113–3125 CrossRef CAS.
- B. Brough, B. H. Northrop, J. J. Schmidt, H.-R. Tseng, K. N. Houk, J. F. Stoddart and C.-M. Ho, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8583–8588 CrossRef CAS PubMed.
- B. A. Ashcroft, Q. Spadola, S. Qamar, P. Zhang, G. Kada, R. Bension and S. Lindsay, Small, 2008, 4, 1468–1475 CrossRef CAS PubMed.
- A. Dunlop, J. Wattoom, E. A. Hasan, T. Cosgrove and A. N. Round, Nanotechnology, 2008, 19, 345706 CrossRef PubMed.
- M. Janke, Y. Rudzevich, O. Molokanova, T. Metzroth, I. Mey, G. Diezemann, P. E. Marszalek, J. Gauss, V. Böhmer and A. Janshoff, Nat. Nanotechnol., 2009, 4, 225–229 CrossRef CAS PubMed.
- P. Lussis, T. Svaldo-Lanero, A. Bertocco, C.-A. Fustin, D. A. Leigh and A.-S. Duwez, Nat. Nanotechnol., 2011, 6, 553–557 CrossRef CAS PubMed.
- A. Van Quaethem, P. Lussis, D. A. Leigh, A.-S. Duwez and C.-A. Fustin, Chem. Sci., 2014, 5, 1449–1452 RSC.
- K. A. Bowman, O. A. Aarstad, B. T. Stokke, G. Skjåk-Bræk and A. N. Round, Langmuir, 2016, 32, 12814–12822 CrossRef CAS PubMed.
- P. R. Ashton, I. Baxter, M. C. T. Fyfe, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 2297–2307 CrossRef CAS.
- A. Affeld, G. Hubner, C. Seel and C. A. Schalley, Eur. J. Org. Chem., 2001, 2877–2890 CrossRef CAS.
- H. W. Gibson, S. H. Lee, P. T. Engen, P. Lecavalier, J. Sze, Y. X. Shen and M. Bheda, J. Org. Chem., 1993, 58, 3748–3756 CrossRef CAS.
- P. R. McGonigal, H. Li, C. Cheng, S. T. Schneebeli, M. Frasconi, L. S. Witus and J. F. Stoddart, Tetrahedron Lett., 2015, 56, 3591–3594 CrossRef CAS.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- G. De Bo, S. Kuschel, D. A. Leigh, B. Lewandowski, M. Papmeyer and J. W. Ward, J. Am. Chem. Soc., 2014, 136, 5811–5814 CrossRef CAS PubMed.
- G. De Bo, M. A. Y. Gall, M. O. Kitching, S. Kuschel, D. A. Leigh, D. J. Tetlow and J. W. Ward, J. Am. Chem. Soc., 2017, 139, 10875–10879 CrossRef CAS PubMed.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- C. Pezzato, M. T. Nguyen, C. Cheng, D. J. Kim, M. T. Otley and J. F. Stoddart, Tetrahedron, 2017, 73, 4849–4857 CrossRef CAS.
- C. F. A. Gómez-Durán, W. Liu, M. de L. Betancourt-Mendiola and B. D. Smith, J. Org. Chem., 2017, 82, 8334–8341 CrossRef PubMed.
- A. Martinez Cuezva, L. V. Rodrigues, C. Navarro, F. Carro-Guillen, L. Buriol, C. P. Frizzo, M. A. P. Martins, M. Alajarin and J. Berna, J. Org. Chem., 2015, 80, 10049–10059 CrossRef CAS PubMed.
- S. Saito, E. Takahashi, K. Wakatsuki, K. Inoue, T. Orikasa, K. Sakai, R. Yamasaki, Y. Mutoh and T. Kasama, J. Org. Chem., 2013, 78, 3553–3560 CrossRef CAS PubMed.
- T. Clifford, A. Abushamleh and D. Busch, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4830–4836 CrossRef CAS PubMed.
- C. Heim, A. Affeld, M. Nieger and F. Vögtle, Helv. Chim. Acta, 1999, 82, 746–759 CrossRef CAS.
- F. M. Raymo, K. N. Houk and J. F. Stoddart, J. Am. Chem. Soc., 1998, 120, 9318–9322 CrossRef CAS.
- J. W. Choi, A. H. Flood, D. W. Steuerman, S. Nygaard, A. B. Braunschweig, N. N. P. Moonen, B. W. Laursen, Y. Luo, E. DeIonno, A. J. Peters, J. O. Jeppesen, K. Xu, J. F. Stoddart and J. R. Heath, Chem.–Eur. J., 2006, 12, 261–279 CrossRef CAS PubMed.
- A. Coskun, P. J. Wesson, R. Klajn, A. Trabolsi, L. Fang, M. A. Olson, S. K. Dey, B. A. Grzybowski and J. F. Stoddart, J. Am. Chem. Soc., 2010, 132, 4310–4320 CrossRef CAS PubMed.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- A. Carrasco-Ruiz and J. Tiburcio, Org. Lett., 2015, 17, 1858–1861 CrossRef CAS PubMed.
- M. Hmadeh, A. C. Fahrenbach, S. Basu, A. Trabolsi, D. Benítez, H. Li, A. M. Albrecht-Gary, M. Elhabiri and J. F. Stoddart, Chem.–Eur. J., 2011, 17, 6076–6087 CrossRef CAS PubMed.
- G. Wenz, B.-H. Han and A. Müller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed.
- S. Qamar, P. M. Williams and S. M. Lindsay, Biophys. J., 2008, 94, 1233–1240 CrossRef CAS PubMed.
- J. Liphardt, B. Onoa, S. B. Smith, I. Tinoco and C. Bustamante, Science, 2001, 292, 733–737 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- R. D. Astumian, Am. J. Phys., 2006, 74, 683–688 CrossRef CAS.
- T. Hugel and M. Seitz, Macromol. Rapid Commun., 2001, 22, 989–1016 CrossRef CAS.
- C. Bustamante, Y. R. Chemla, N. R. Forde and D. Izhaky, Annu. Rev. Biochem., 2004, 73, 705–748 CrossRef CAS PubMed.
- B. Onoa, S. Dumont, J. Liphardt, S. B. Smith, I. Tinoco and C. Bustamante, Science, 2003, 299, 1892–1895 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- R. S. Stoll, D. C. Friedman and J. F. Stoddart, Org. Lett., 2011, 13, 2706–2709 CrossRef CAS PubMed.
- E. Evans, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 105–128 CrossRef CAS PubMed.
- B. Lee, Z. Niu and S. L. Craig, Angew. Chem., Int. Ed., 2016, 55, 13086–13089 CrossRef CAS PubMed.
- P. Hodge and S. D. Kamau, Angew. Chem., Int. Ed., 2003, 42, 2412–2414 CrossRef CAS PubMed.
- P. A. May, N. F. Munaretto, M. B. Hamoy, M. J. Robb and J. S. Moore, ACS Macro Lett., 2016, 5, 177–180 CrossRef CAS.
- M. Schaefer, B. Icli, C. Weder, M. Lattuada, A. F. M. Kilbinger and Y. C. Simon, Macromolecules, 2016, 49, 1630–1636 CrossRef CAS.
- S. Akbulatov and R. Boulatov, ChemPhysChem, 2017, 18, 1422–1450 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence