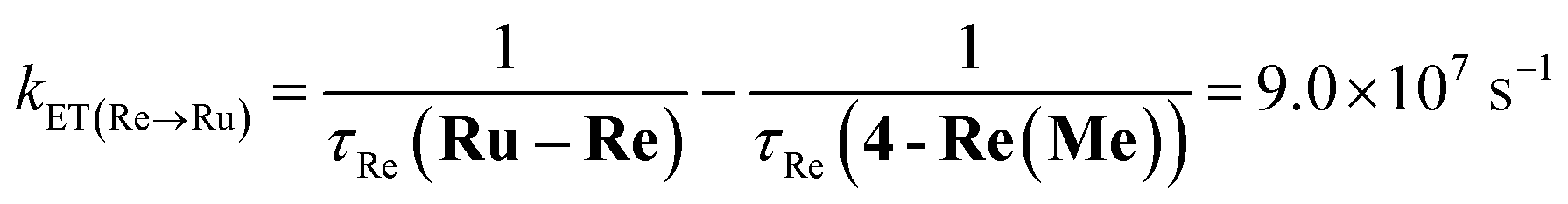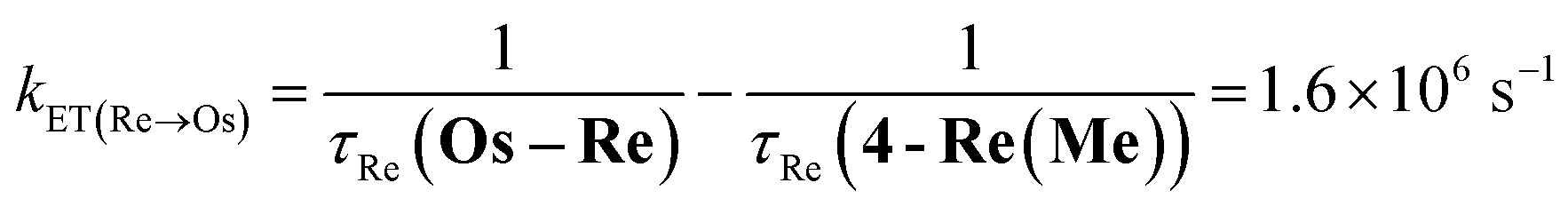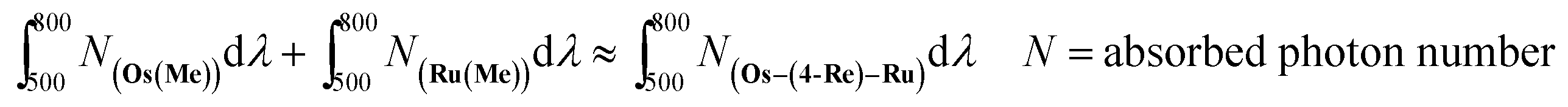DOI:
10.1039/C7SC04162D
(Edge Article)
Chem. Sci., 2018,
9, 1031-1041
Synthesis of Os(II)–Re(I)–Ru(II) hetero-trinuclear complexes and their photophysical properties and photocatalytic abilities†
Received
24th September 2017
, Accepted 29th November 2017
First published on 4th December 2017
Abstract
Photofunctional trinuclear complexes containing three different central metals, i.e. Os(II), Re(I) and Ru(II), were synthesised for the first time using stepwise Mizoroki–Heck reactions. The vinylene groups in the bridging ligands of the Os(II)–Re(I)–Ru(II) trinuclear complexes were selectively reduced by photochemical hydrogenation in moderate yield, affording novel supramolecular photocatalysts which can absorb a wide range of visible light up to 730 nm and induce CO2 reduction with high selectivity and durability. The turnover numbers of CO formation were over 4300. Details of the photophysical properties of these new trinuclear complexes, especially their intramolecular excitation-energy transfer phenomena, are also reported.
Introduction
Photofunctional multinuclear complexes are attracting attention in various research fields, such as light-harvesting systems and photocatalysis. Examples of reported light-harvesting systems include a linear Re(I) penta-nuclear complex whose centre unit is connected with an emissive Ru(II) complex1 as well as polymers connected with numerous Ru(II) complexes as light absorbers and a much smaller number of Os(II) complexes as accumulators of absorbed excitation energy.2 Hetero-dinuclear complexes, e.g. Ru(II)–Re(I),3–9 Os(II)–Re(I),10 Ru(II)–Ni(II),11–13 Zn(II)–Re(I),14,15 and Pd(II)–Re(I) complexes,16 can function as photocatalysts for CO2 reduction, and Ru(II)–Pd(II)17 and Ru(II)–Ru(II) complexes18,19 have been shown to photocatalyse the oligomerisation of olefins and oxygen evolution from water, respectively. When each metal-complex unit has a different function that can function synergistically, photofunctional multinuclear complexes can provide prominent functions which cannot be achieved using mononuclear complexes. For example, many photocatalytic systems using metal complexes for CO2 reduction are constructed with two different mononuclear metal complexes, i.e. a redox photosensitiser and a CO2 reduction catalyst, to convert photochemically induced one-electron transfer to multi-electron reduction of CO2. It has been reported that connecting a photosensitiser and catalyst by an alkyl chain can drastically improve the efficiency and durability of photocatalysis; these compounds are called supramolecular photocatalysts.20
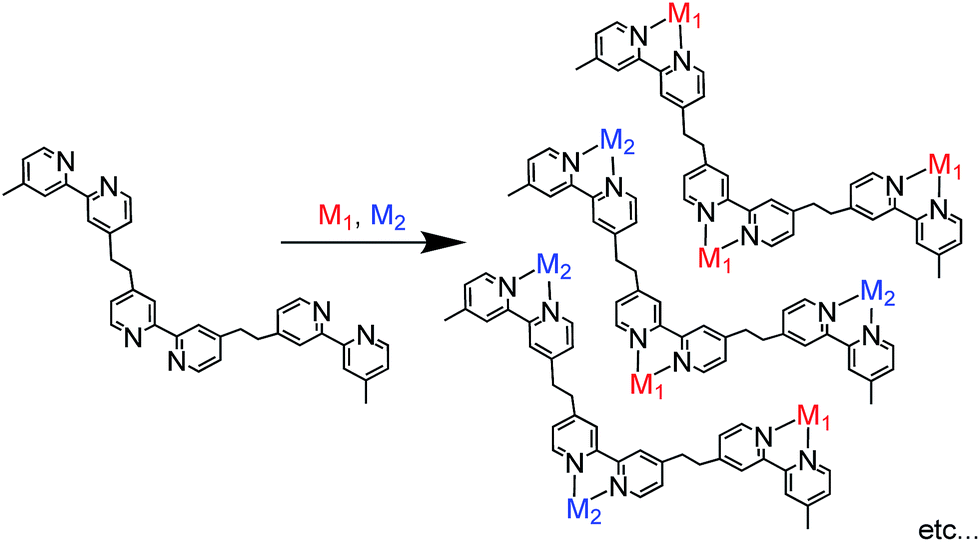
As described above, most photofunctional multinuclear complexes consist of only one or two metal centres, mainly because they are often synthesised via stepwise coordination of metal complexes to a bridging ligand.2 In this method, a mononuclear complex with a bridging ligand with coordination ability on one side is synthesised first; the complex is then reacted with another metal centre to afford the dinuclear complex. This method can be used to synthesise various photofunctional multinuclear metal diimine complexes containing metal ions such as Ru(II), Os(II), Re(I) and Pd(II). In principle, however, this synthesis method can be applied only for the connection of one metal complex or two different metal complexes with the same diimine units in the bridging ligand due to limitations of product selectivity. For example, if a bridging ligand with three different diimine moieties is used to synthesise a trinuclear complex with different metals, various products with different ratios of the metals are expected to form (Scheme 1).
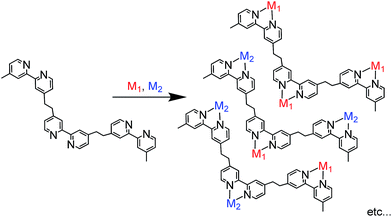 |
| | Scheme 1 Difficulty of the conventional method for synthesising trinuclear complexes with trisdiimine bridging ligands. | |
Recently, some coupling reactions between or among metal complexes have been applied for the synthesis of multinuclear complexes;21 metal complexes with proper functional groups as building blocks have been directly connected with each other using Sonogashira coupling,22–24 olefin metathesis,25,26 homo-coupling reactions,27 Suzuki–Miyaura coupling28–30 or Mizoroki–Heck reactions.31,32 Using these C–C coupling reactions, a metal complex can be linked with a different metal complex or complexes in a single step with relatively high selectivity and yield, which cannot be selectively accomplished using the conventional method described above.33 These coupling reactions can potentially be applied for the synthesis of hetero-multinuclear metal complexes containing more than two different complexes. If different photosensitisers can be introduced in one molecule, the resulting multinuclear complex can absorb a wider range of visible light compared to reported supramolecular photocatalysts with only one photosensitiser unit or two identical units.
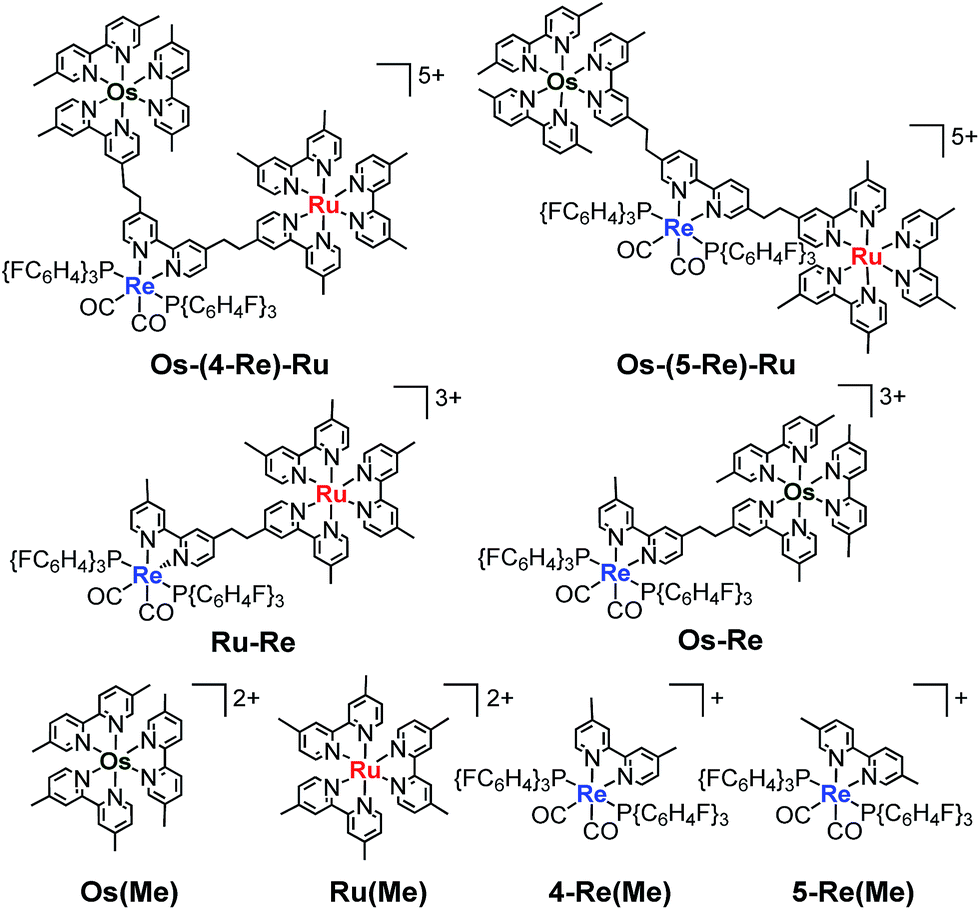
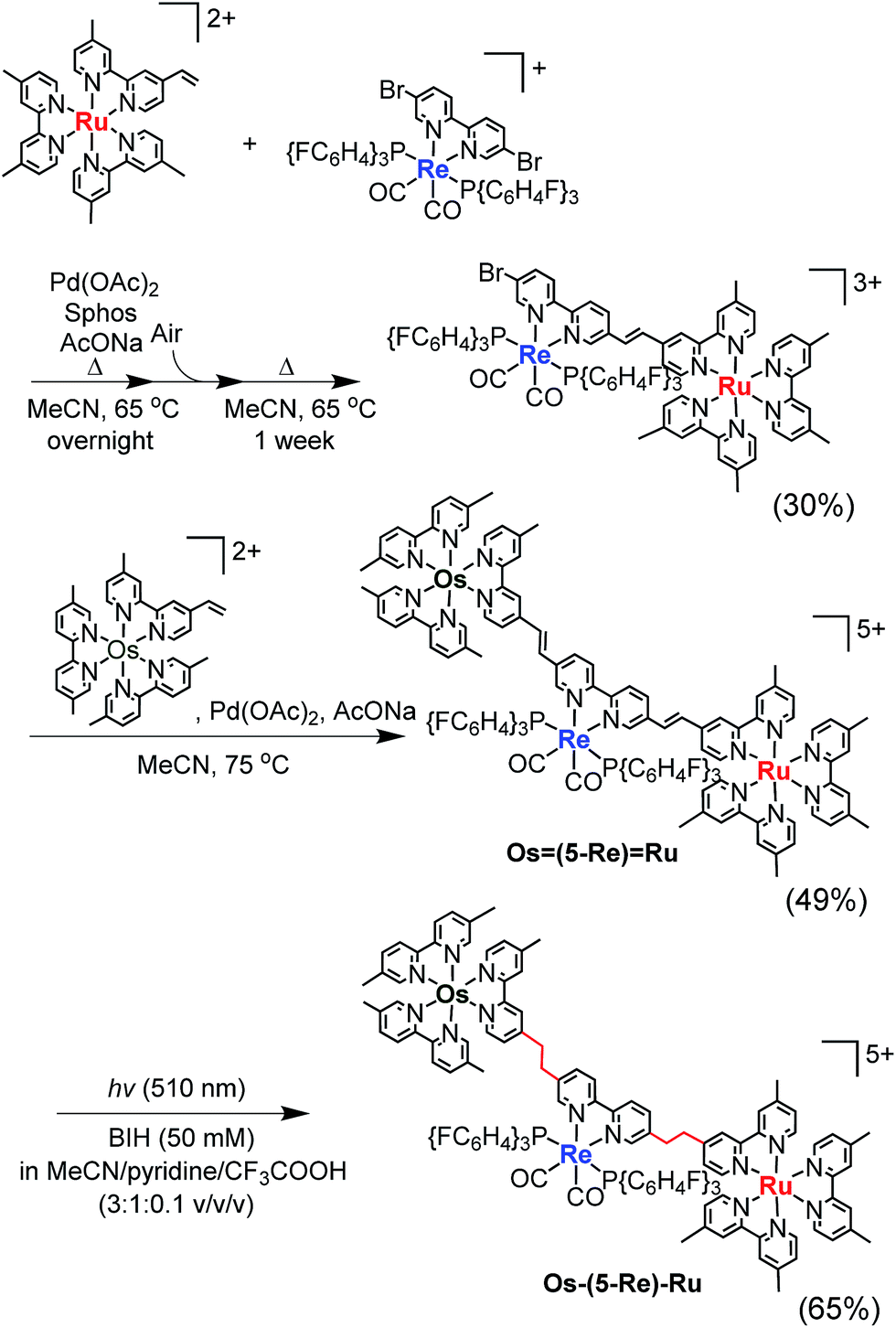
Herein, we report Os(II)–Re(I)–Ru(II) trinuclear complexes as the first examples of supramolecular photocatalysts containing three different metal-complex units; the complexes were synthesised using two separate Mizoroki–Heck reactions. The vinylene chains in the bridging ligands of the obtained complexes were successfully hydrogenated, affording the corresponding trinuclear complexes containing ethylene chains; these can absorb a wide range of visible light and function as durable photocatalysts for CO2 reduction, with the highest turnover number (TON) of CO formation (TONCO = 4347) among reported supramolecular photocatalysts containing Re(I)-catalytic units. Chart 1 shows the structures of the synthesised trinuclear complexes and the corresponding model mononuclear and dinuclear complexes.
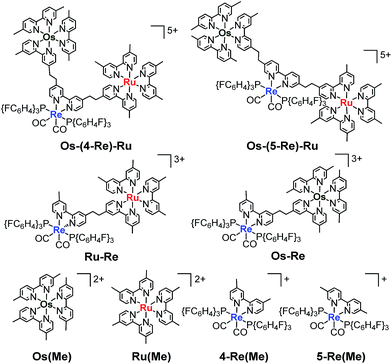 |
| | Chart 1 Structures of the Ru(II)–Re(I)–Os(II) trinuclear complexes and the corresponding model mononuclear and binuclear complexes. | |
Results and discussion
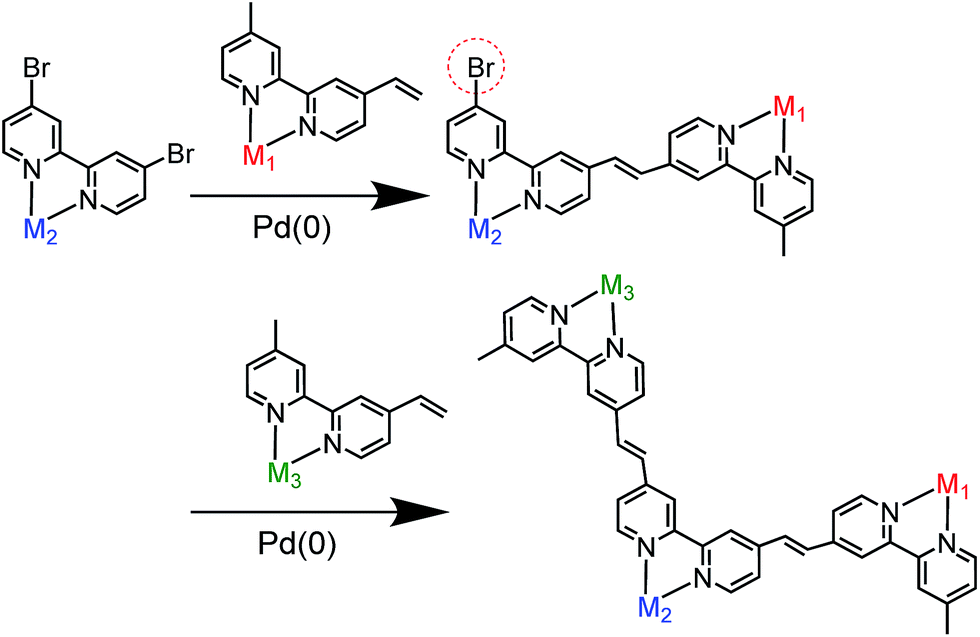
We selected stepwise Mizoroki–Heck reactions to construct the multinuclear complexes (Scheme 2); after the first coupling reaction, another metal complex can be introduced into the dinuclear complex by a second coupling reaction if an active bromo group remains in the ligand of the dinuclear complex.
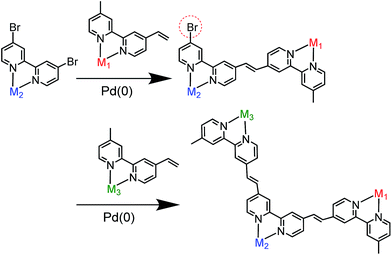 |
| | Scheme 2 Synthetic strategy for constructing hetero-multinuclear complexes using stepwise Mizoroki–Heck reactions. | |
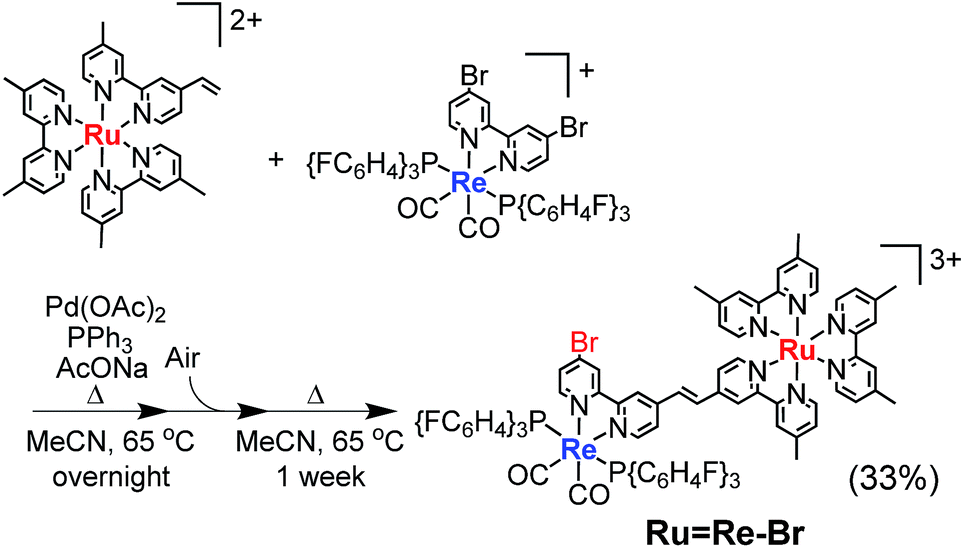
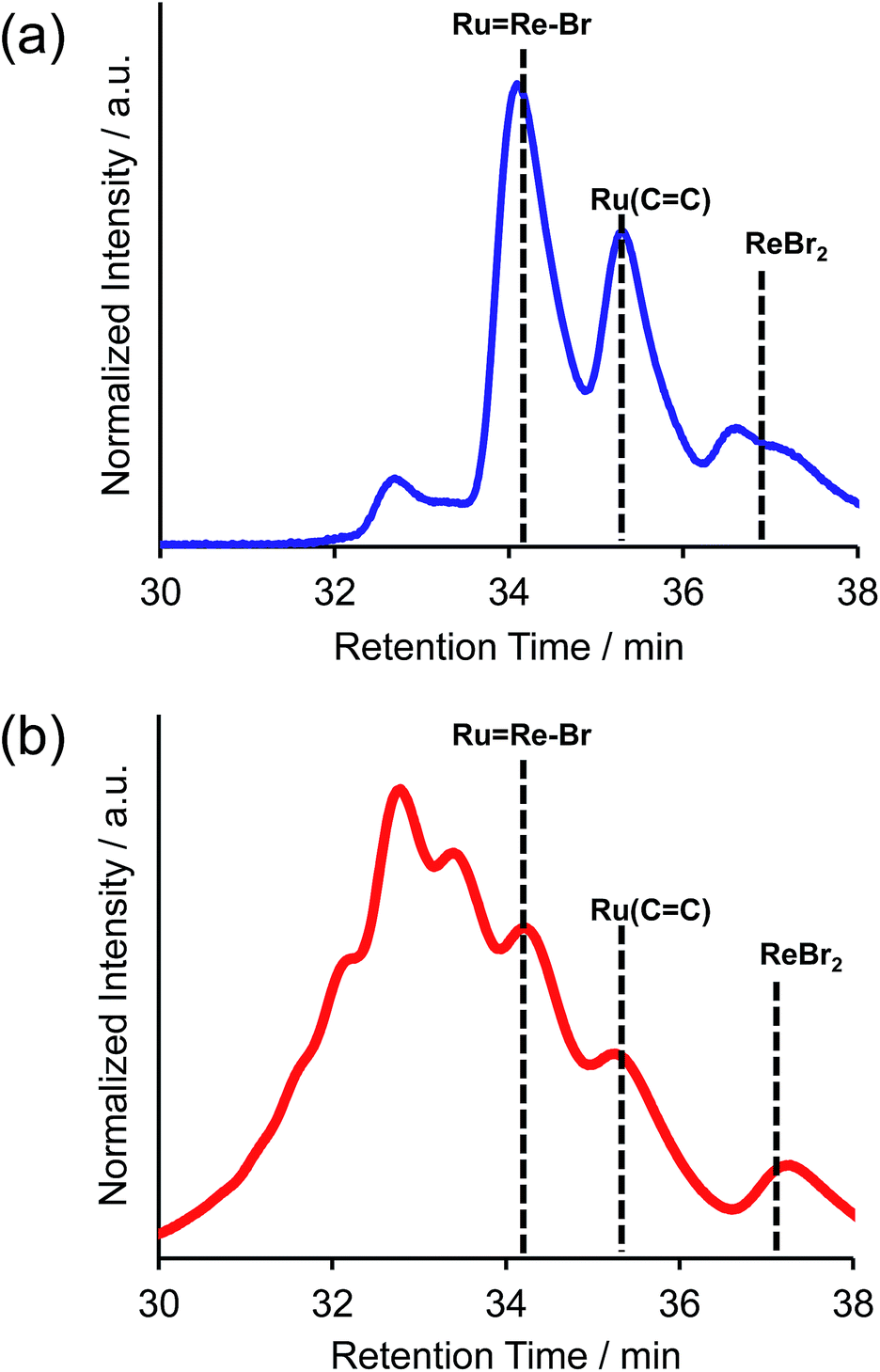
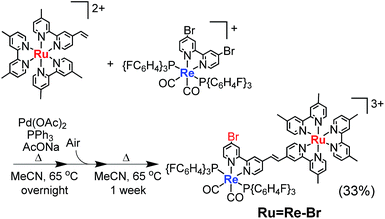
For example, to synthesise a dinuclear Ru(II)–Re(I) complex with a bromo group (Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Re–Br) (eqn (1)), an acetonitrile solution containing [Re(4,4′-dibromo-bpy)(CO)2{P(p-FC6H4)3}2](PF6) (ReBr2, bpy = 2,2′-bipyridine, 52 mg, 39 μmol), [Ru(4-dmb)2(vbpy)](PF6)2 (Ru(CC), 4-dmb = 4,4′-dimethyl-bpy, vbpy = 4-methyl-4′-vinyl-bpy, 19 mg, 20 μmol), Pd(OAc)2 (4.4 mg, 20 μmol), PPh3 (10 mg, 39 μmol) and AcONa (8.0 mg, 97 μmol) was heated at 75 °C under Ar atmosphere for 1 day. Air was introduced by opening the three-way cock attached to the reaction vessel to oxidise the PPh3 ligands on the Pd particles produced during the reaction.31,34 The solution was then heated for one week. In the size-exclusion chromatogram measured after heating, the targeted dinuclear complex RuRe–Br was detected as a major product (Fig. 1a, retention time (RT) = 34 min); this was isolated by preparative size-exclusion chromatography (SEC). The isolated yield was 33% based on Ru(CC).
Re–Br) (eqn (1)), an acetonitrile solution containing [Re(4,4′-dibromo-bpy)(CO)2{P(p-FC6H4)3}2](PF6) (ReBr2, bpy = 2,2′-bipyridine, 52 mg, 39 μmol), [Ru(4-dmb)2(vbpy)](PF6)2 (Ru(CC), 4-dmb = 4,4′-dimethyl-bpy, vbpy = 4-methyl-4′-vinyl-bpy, 19 mg, 20 μmol), Pd(OAc)2 (4.4 mg, 20 μmol), PPh3 (10 mg, 39 μmol) and AcONa (8.0 mg, 97 μmol) was heated at 75 °C under Ar atmosphere for 1 day. Air was introduced by opening the three-way cock attached to the reaction vessel to oxidise the PPh3 ligands on the Pd particles produced during the reaction.31,34 The solution was then heated for one week. In the size-exclusion chromatogram measured after heating, the targeted dinuclear complex RuRe–Br was detected as a major product (Fig. 1a, retention time (RT) = 34 min); this was isolated by preparative size-exclusion chromatography (SEC). The isolated yield was 33% based on Ru(CC).
| |  | (1) |
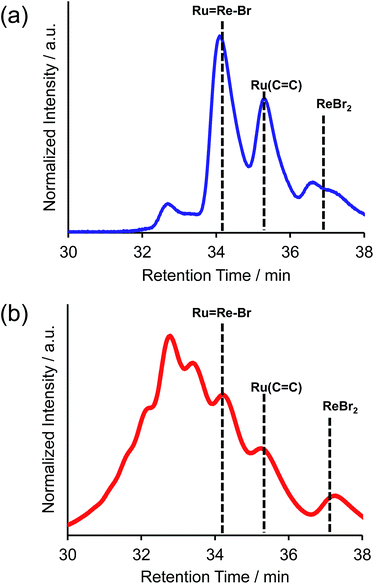 |
| | Fig. 1 Size-exclusion chromatogram measured after the reaction to obtain RuRe–Br (a) using PPh3 and subsequent air injection and (b) in the absence of PPh3. | |
It is noteworthy that under other reported reaction conditions for the Mizoroki–Heck reaction between metal complexes, i.e. usage of Pd(OAc)2 alone in the absence of phosphine,34 the reaction was not successful; various multinuclear complexes were produced as by-products via homo-coupling reactions between or among the complexes containing bromo groups (Fig. 1b). This result clearly indicates that, to suppress side reactions, the addition of phosphine and air is necessary for this type of Mizoroki–Heck reaction, where an excess amount of the metal complex with bromo groups is required. It has been reported that addition of both the phosphine compound and air during the first stage of the reaction controls the sizes of produced Pd particles in the reaction solution to be 600 to 8500 nm; these particles function as catalysts which are highly selective for the cross-coupling reaction over homo-coupling reactions between the metal complexes with bromo groups.34
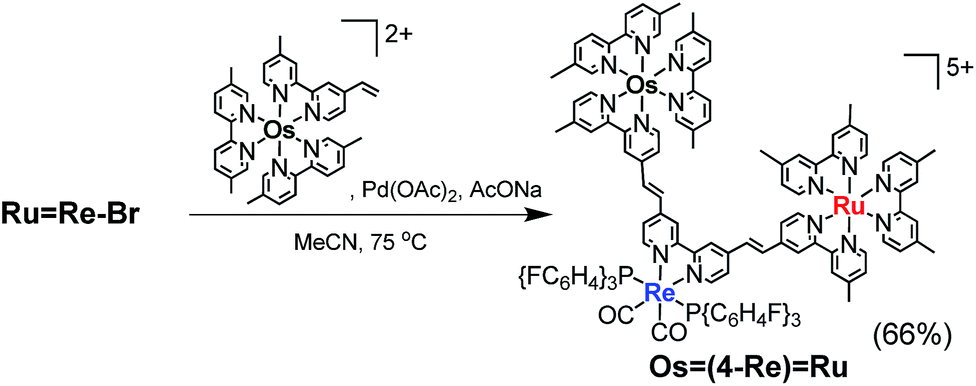
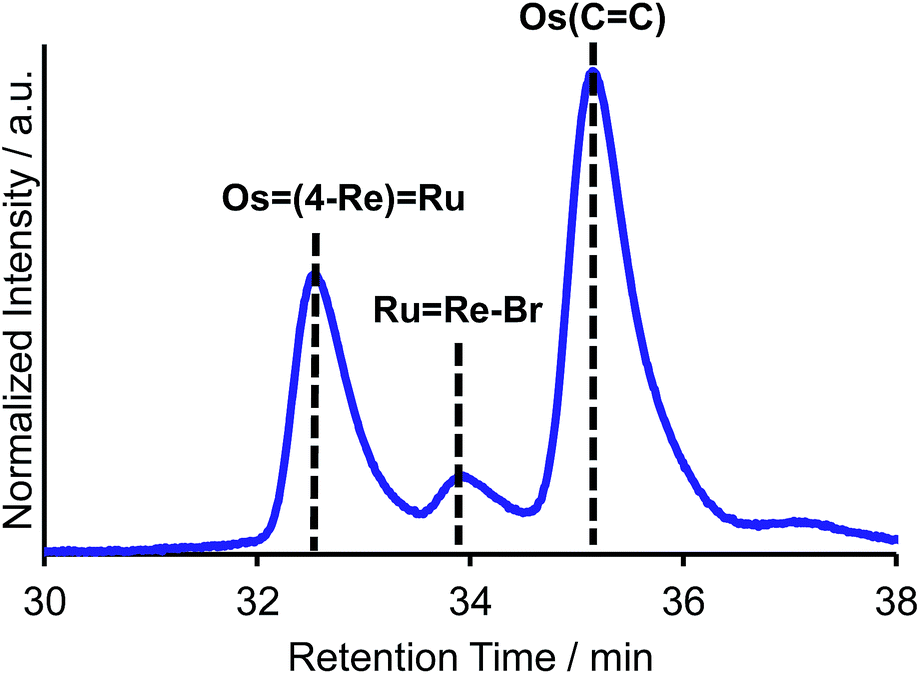
The trinuclear complex Os(4-Re)Ru was successfully synthesised by the Mizoroki–Heck reaction of RuRe–Br and [Os(5-dmb)2(vbpy)](PF6)2 (Os(CC), 5-dmb = 5,5′-dimethyl-bpy) under different reaction conditions (eqn (2)). An acetonitrile solution (2 mL) containing RuRe–Br (14 mg, 6.3 μmol), Os(CC) (26 mg, 25 μmol), Pd(OAc)2 (1.4 mg, 6.3 μmol) and AcONa (2.6 mg, 32 μmol) was heated at 65 °C under Ar atmosphere for 2 days. In this case, phosphine compounds and air were not added to the solution because an excess amount of Os(CC) compared to RuRe–Br was added to the reaction solution, which prevented the homo-coupling reaction of RuRe–Br with the Br group. Pd(OAc)2 (1.4 mg, 6.3 μmol) was additionally charged into the solution, which was heated for two more days. The size-exclusion chromatogram measured after the reaction showed three major peaks, which are respectively attributed to the target product Os(4-Re)Ru and the two starting complexes (Fig. 2). Os(4-Re)Ru, where the Re-complex unit was connected with Ru- and Os-complex units with vinylene chains, was isolated by preparative SEC with an isolated yield of 66% based on RuRe–Br.
| | 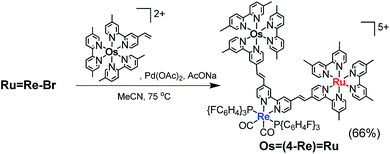 | (2) |
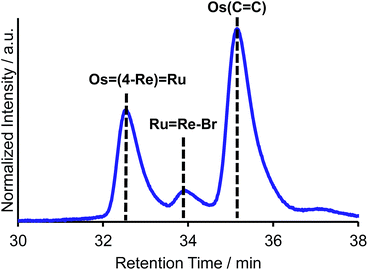 |
| | Fig. 2 Size-exclusion chromatogram measured after the reaction for the synthesis of Os(4-Re)Ru. | |
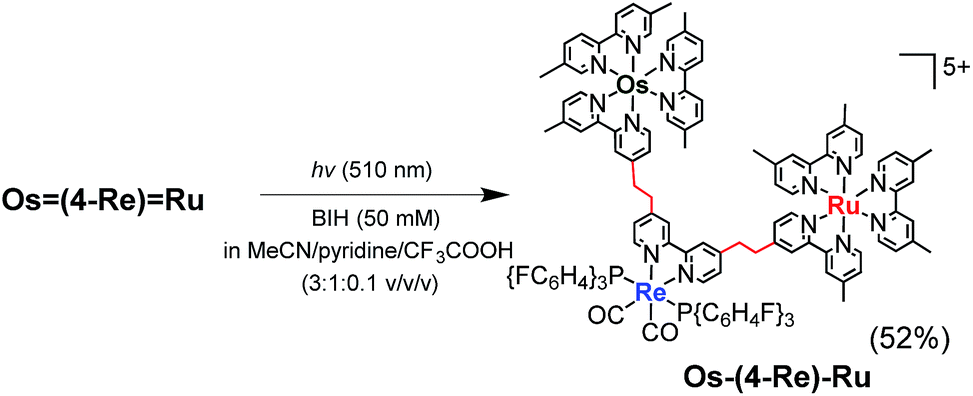
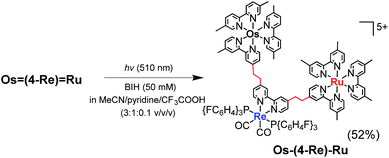
It has been reported that conjugation between the diimine moieties in the bridging ligand lowers the photocatalytic ability of supramolecular photocatalysts containing Re(I)-complex units as a catalyst for CO2 reduction due to a decrease in their reducing power.3,35,36 Therefore, we applied the photochemical hydrogenation reaction which was recently reported by our group33 to reduce the vinylene chains between the diimine moieties in the bridging ligands of Os(4-Re)Ru. An acetonitrile–pyridine–CF3COOH mixed solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.1 v/v/v, 4 mL) containing Os(4-Re)Ru (4.2 μmol) and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH, 0.1 M) as a reductant was irradiated at λex = 510 nm (light intensity = 4 × 10−8 einstein per s) for 15 h at ambient temperature (eqn (3)). During the irradiation, blue shifts of the absorption bands were observed in the visible region (Fig. S1†), which clearly indicates that the vinylene chains in the bridging ligand were converted to ethylene chains. This absorption change was fully accomplished within approximately 10 h of irradiation, and further irradiation did not cause any spectral changes. The compound was purified by ion-exchange chromatography; the isolation yield of Os–(4-Re)–Ru was 52%.
:1:0.1 v/v/v, 4 mL) containing Os(4-Re)Ru (4.2 μmol) and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH, 0.1 M) as a reductant was irradiated at λex = 510 nm (light intensity = 4 × 10−8 einstein per s) for 15 h at ambient temperature (eqn (3)). During the irradiation, blue shifts of the absorption bands were observed in the visible region (Fig. S1†), which clearly indicates that the vinylene chains in the bridging ligand were converted to ethylene chains. This absorption change was fully accomplished within approximately 10 h of irradiation, and further irradiation did not cause any spectral changes. The compound was purified by ion-exchange chromatography; the isolation yield of Os–(4-Re)–Ru was 52%.
| |  | (3) |
Similar synthesis and isolation methods were applied to synthesise another trinuclear complex, Os–(5-Re)–Ru, using [Re(5,5′-dibromo-bpy)(CO)2{P(p-FC6H4)3}2](PF6) as a starting material instead of ReBr2. The isolated yields of the corresponding Ru(II)–Re(I) dinuclear complex, Os(5-Re)Ru and Os–(5-Re)–Ru were 30%, 49% and 65%, respectively (Scheme 3).
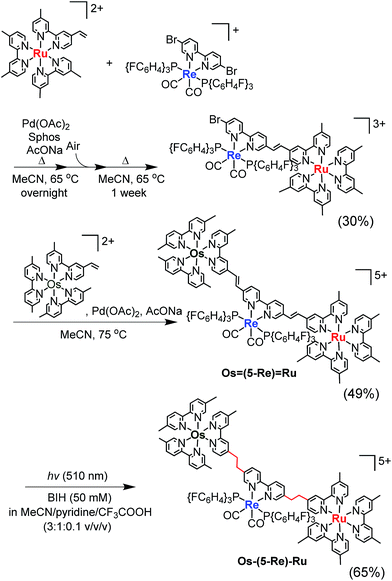 |
| | Scheme 3 Synthesis of Os–(5-Re)–Ru. | |
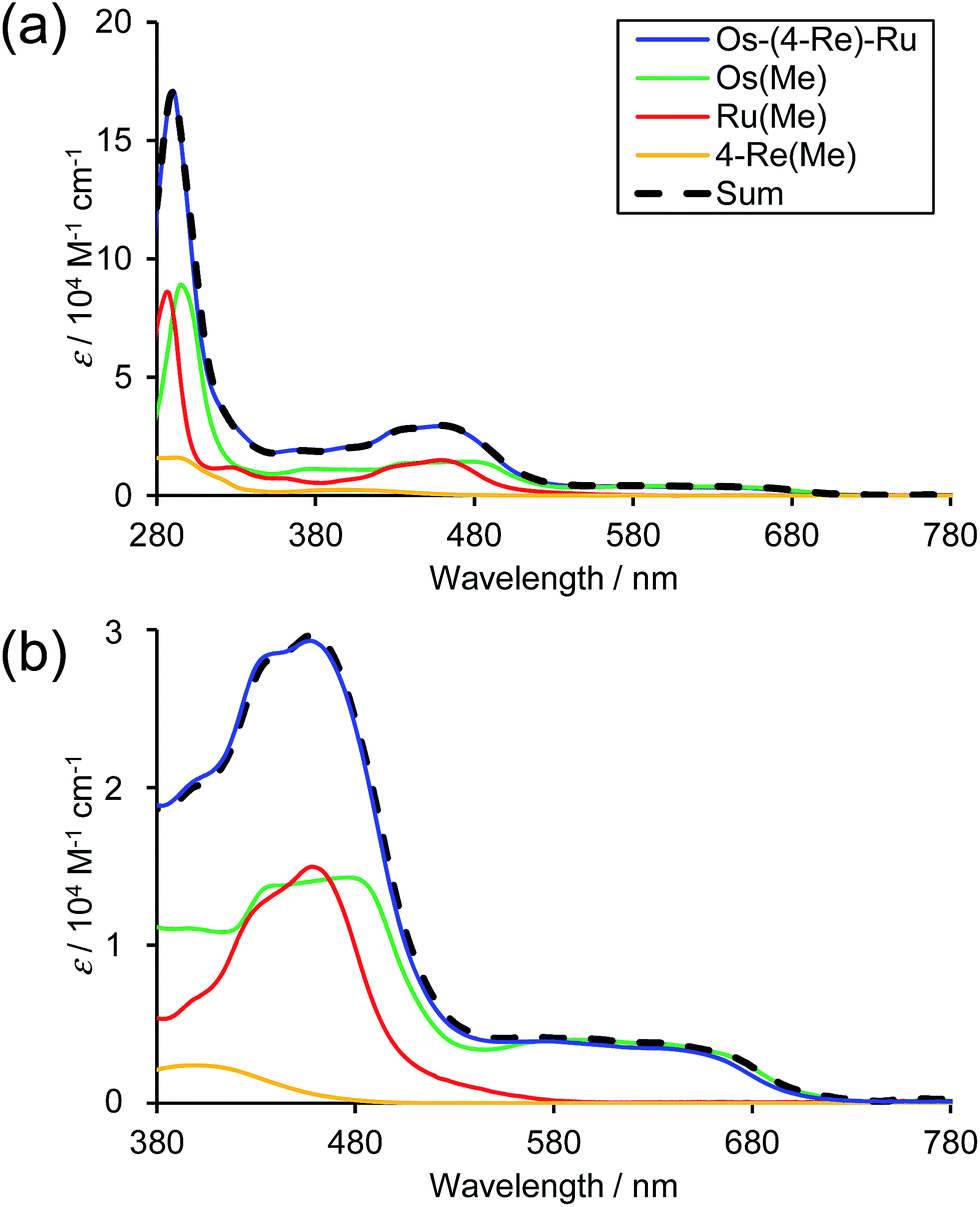
Fig. 3 shows the UV-Vis absorption spectra of Os–(4-Re)–Ru and the corresponding mononuclear complexes (Os(Me), 4-Re(Me) and Ru(Me) in Chart 1). The spectrum of Os–(4-Re)–Ru was almost identical to the 1:1:1 summation spectrum of Os(Me), 4-Re(Me) and Ru(Me). The spectrum of Os–(5-Re)–Ru was also very similar to the summation spectrum of Os(Me), 5-Re(Me) and Ru(Me) (Fig. S2†). These similarities indicate that no strong electronic interactions occur between the various metal-complex units through the ethylene chain. Notably, the Os(II)–Re(I)–Ru(II) complexes maintained molar extinction coefficients of over 15000 M−1 cm−1 up to 495 nm and showed absorptions up to 730 nm (Fig. 3 and S2†). In other words, these complexes can strongly absorb a wider range of visible light than even the dinuclear complexes, i.e.Ru–Re and Os–Re (their structures are shown in Chart 1).
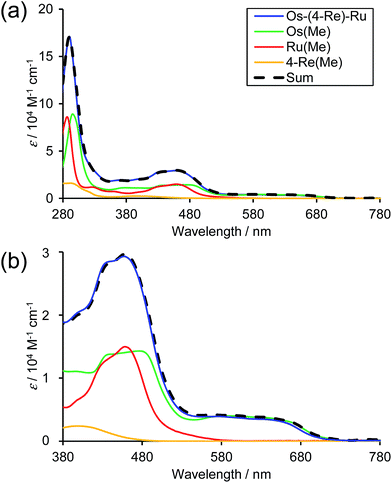 |
| | Fig. 3 UV-Vis absorption spectra of the obtained trinuclear complex (a) Os–(4-Re)–Ru (blue) and the corresponding mononuclear complexes, i.e.Os(Me) (green), Re(Me) (yellow) and Ru(Me) (red). The 1:1:1 summation spectrum of Os(Me), 4-Re(Me) and Ru(Me) is illustrated as a dotted line. The solvent was MeCN. (b) Enlarged spectra at λabs = 380 to 780 nm. | |
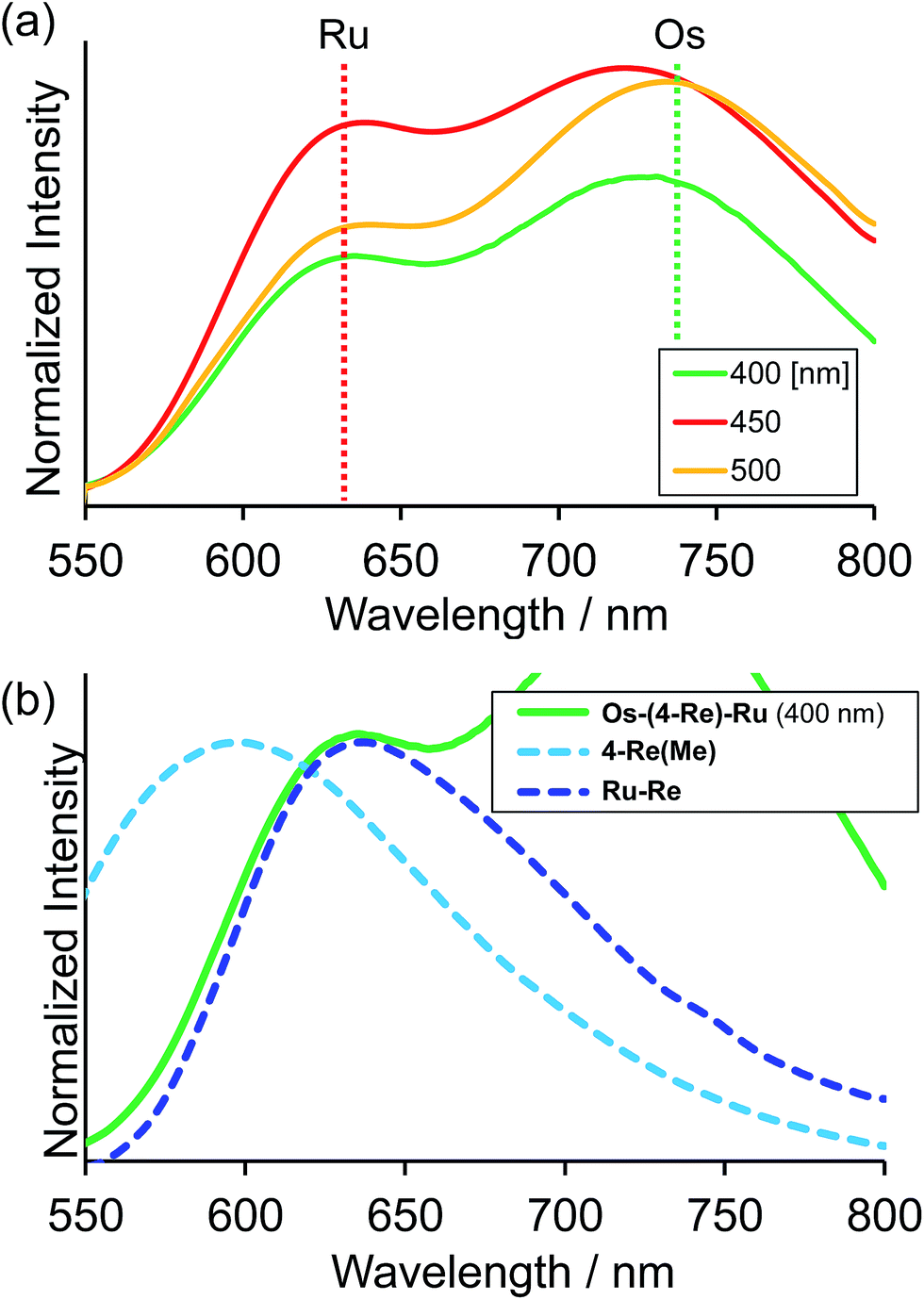
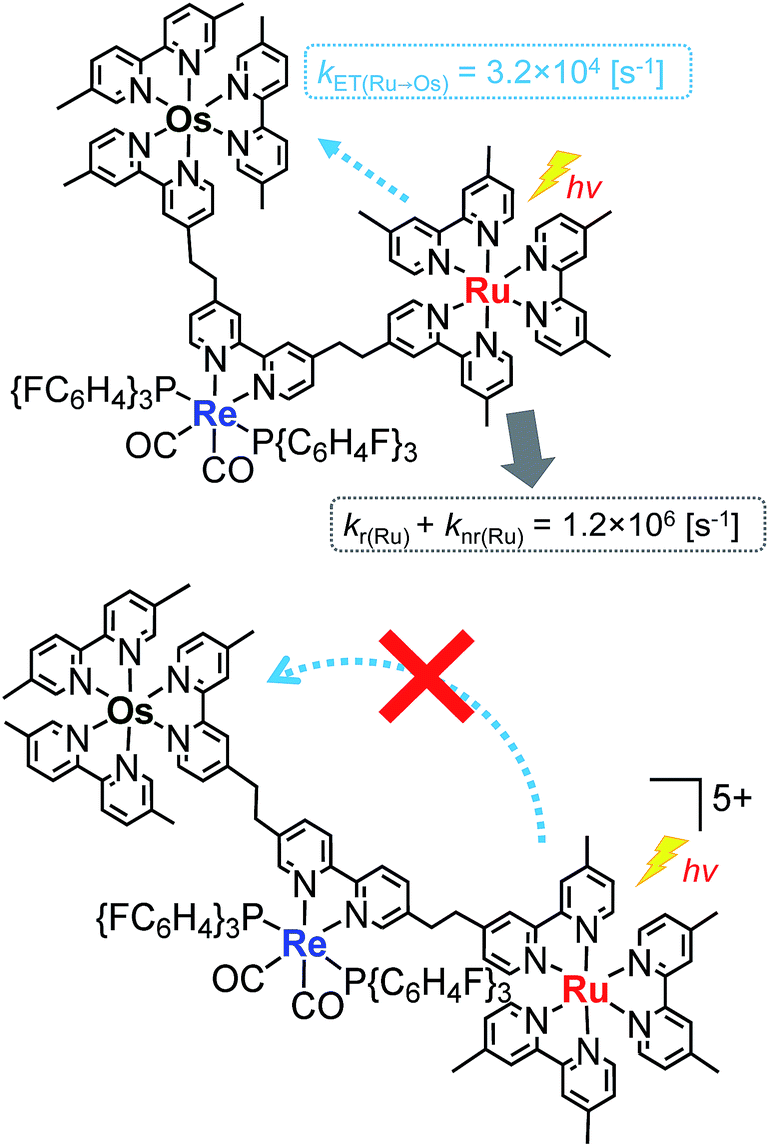
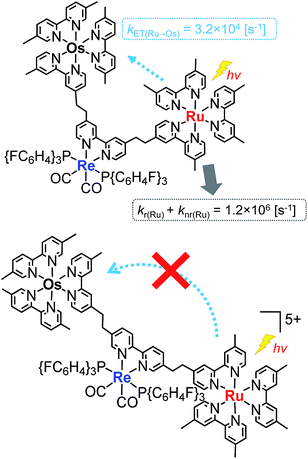
Fig. 4a shows the emission spectra of Os–(4-Re)–Ru under irradiation at three different excitation wavelengths, i.e. λex = 400 nm, 450 nm and 500 nm. The ratios of the excited units were expected to be very different at different excitation wavelengths because the emission spectra of the corresponding mononuclear complexes were very different from each other (Fig. S3†). However, the emission spectra of Os–(4-Re)–Ru at three different excitation wavelengths were similar, especially those at λex = 400 nm and 450 nm. In Fig. 4b, we can compare the emission spectrum of Os–(4-Re)–Ru excited at λex = 400 nm, which is close to the absorption maximum of the Re unit, with those of 4-Re(Me) and Ru–Re. It can be clearly seen that emission from the Re unit of Os–(4-Re)–Ru was not observed. It has been reported that in Ru(II)–Re(I) binuclear complexes with an alkyl linker, efficient intramolecular energy transfer (IEnT) occurs from the excited Re unit to the Ru unit, and most of the emission is produced from the excited Ru unit.37,38 We observed this phenomenon not only for Ru–Re but also for Os–(4-Re)–Ru (Fig. 5).
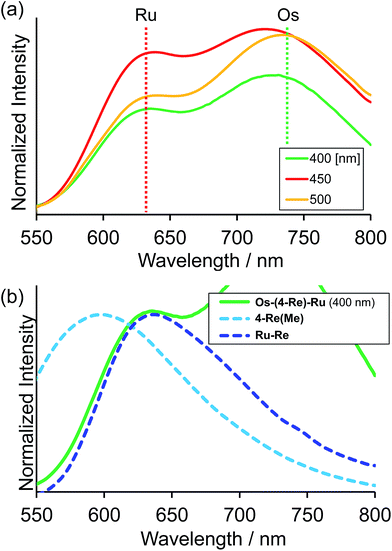 |
| | Fig. 4 (a) Emission spectra of Os–(4-Re)–Ru under irradiation at λex = 400 nm, 450 nm and 500 nm. The solvent was MeCN. (b) Enlarged emission spectrum of Os–(4-Re)–Ru at λex = 400 nm, which is mainly absorbed by the Re unit, with the emission spectra of 4-Re(Me) and Ru–Re. | |
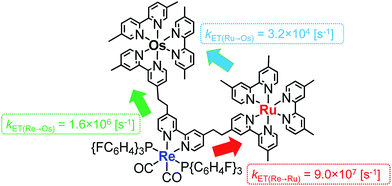 |
| | Fig. 5 Intramolecular energy transfer processes in Os–(4-Re)–Ru. | |
We expected to observe three different IEnT processes in Os–(4-Re)–Ru, i.e. from the Re unit to both the Os and Ru units and from the Ru unit to the Os unit (Fig. 4). We estimated the rate constants of these IEnT processes by comparing the emission lifetimes of the Ru and Re units with those of the corresponding mono- and dinuclear complexes (Table 1). The emission decays of Os–(4-Re)–Ru at λex = 401 nm and detection wavelength λdet = 650 nm could be fitted by a triple-exponential function with τ = 832 ns, 51 ns and 7.0 ns (eqn (4)). In contrast, at λex = 510 nm, which is not absorbed by the Re unit, and λdet = 725 nm, where the Re unit emits very weakly, the spectra can be reasonably analysed by a double-exponential function with τ = 834 ns and 50 ns. These results and a comparison with the model mono- and binuclear complexes (Table 1) enable us to conclude that the emissions with τ = 832, 51 and 7.0 ns originate from the Ru, Os and Re units, respectively.
| | | I(t) = A1e−t/τ1 + A2e−t/τ2 + A3e−t/τ3 | (4) |
Table 1 Emission lifetimes of Os–(4-Re)–Ru, Os–(5-Re)–Ru, Ru–Re, Os–Re and 4-Re(Me) measured in MeCN
| Complex |
τ
Ru/ns (A/%) |
τ
Os/ns (A/%) |
τ
Re/ns (A/%) |
|
λ
ex = 510 nm, detection wavelength (λdet) = 725 nm.
λ
ex = 401 nm, λdet = 650 nm.
λ
ex = 401 nm, λdet = 550 nm.
λ
ex = 510 nm, λdet = 800 nm.
λ
ex = 401 nm, λdet = 700 nm.
λ
ex = 439 nm, λdet = 587 nm.
|
|
Os–(4-Re)–Ru
|
834 (4) |
50 (96) |
|
|
Os–(4-Re)–Ru
|
832 (9) |
51 (14) |
7.0 (59) |
|
Os–(5-Re)–Ru
|
893 (1) |
47 (99) |
|
|
Os–(5-Re)–Ru
|
862 (9) |
39 (55) |
17 (36) |
|
Ru–Re
|
857 (100) |
|
|
|
Ru–Re
|
864 (73) |
|
11 (27) |
|
Os–Re
|
|
41 (100) |
|
|
Os–Re
|
|
41 (97) |
390 (3) |
|
4-Re(Me)
|
|
|
1003 (100) |
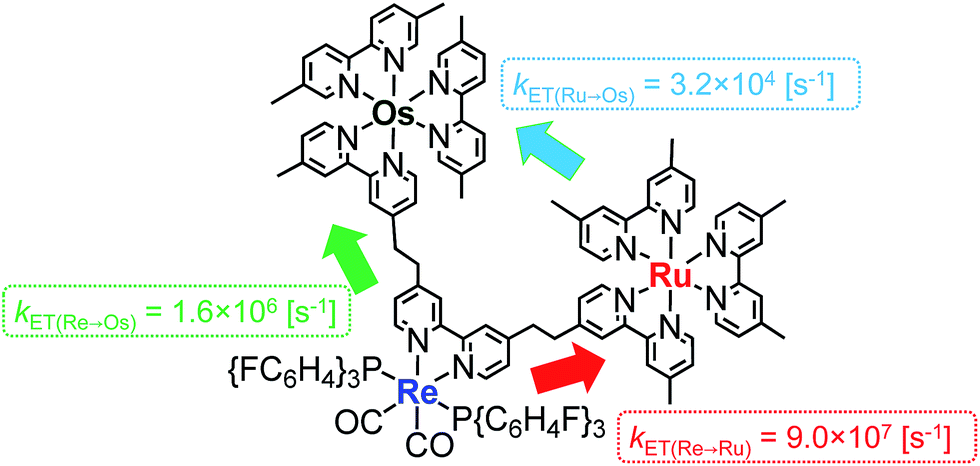
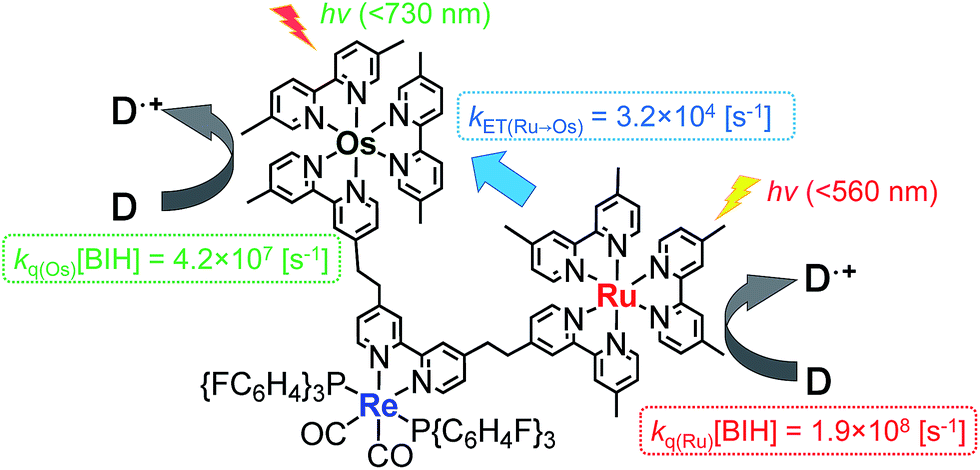
The rate constant of the IEnT process from the excited Ru unit to the Os unit was estimated to be kET(Ru→Os) = 3.2 × 104 s−1, as shown in eqn (5). This indicates that only approximately 3% of the excited Ru unit in Os–(4-Re)–Ru was quenched by the slow IEnT process to the Os. Similarly, the rates of IEnT from the Re unit to the Ru and the Os units could be estimated as kET(Re→Ru) = 9.0 × 107 s−1 and kET(Re→Os) = 1.6 × 106 s−1, respectively, using eqn (6) and (7) and by comparing the emission lifetimes of the Re units in Ru–Re (τRe(Ru–Re)) and Os–Re (τRe(Os–Re)) with that of 4-Re(Me) (τRe(4-Re(Me))).
| | 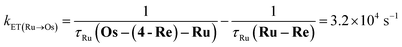 | (5) |
| | 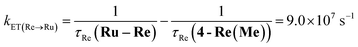 | (6) |
| | 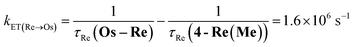 | (7) |
On the basis of these results, we can conclude that the excited state of the Re unit in Os–(4-Re)–Ru was almost entirely quenched by the Ru unit (≈98%), while the IEnT process to the Os unit is a minor process.
It is interesting that the IEnT processes from the excited Re unit to the Ru unit were much faster than those from both the excited Ru and Re units to the Os unit although the overlap between the absorption spectrum of the Os unit and the emission spectra of the Ru and Re units is much larger than that between the absorption spectrum of the Ru unit and the emission spectrum of the Re unit (Fig. S4†). These results suggest that the mechanisms of IEnT are different. Furue and his co-workers reported that IEnT from the excited Ru unit to the Os unit in Ru–Os dinuclear complexes proceeds via the Förster mechanism,37 while contrastingly, IEnT from the Re unit to the Ru unit in Ru–Re dinuclear complexes proceeds via the Dexter mechanism.38 In Os–(4-Re)–Ru, therefore, fast Dexter-type IEnT should proceed from the excited Re unit to the Ru unit, while Förster-type IEnT should proceed from the excited Ru unit to the Os unit. The speed of the IEnT from the excited Re unit to the Os unit was greater than that from the excited Re unit to the Ru unit and less than that from the excited Ru unit to the Os unit. We cannot clarify the reaction mechanism of the IEnT from the excited Re unit to the Os unit at this stage.
In the case of Os–(5-Re)–Ru, similar analysis can be applied to obtain τ = 862 ns, 39 ns and 17 ns, which are attributable to emissions from the Ru, Os and Re units, respectively. However, it is interesting to note that the emission lifetime of the excited Ru unit was almost the same as that of Ru–Re. This indicates that IEnT from the Ru unit to the Os unit proceeded very slowly or did not proceed (Fig. 6). This is likely because the distance between the Ru unit and the Os unit is greater than that in Os–(4-Re)–Ru.
 |
| | Fig. 6 Intramolecular energy transfer processes in Os–(4-Re)–Ru and Os–(5-Re)–Ru. | |
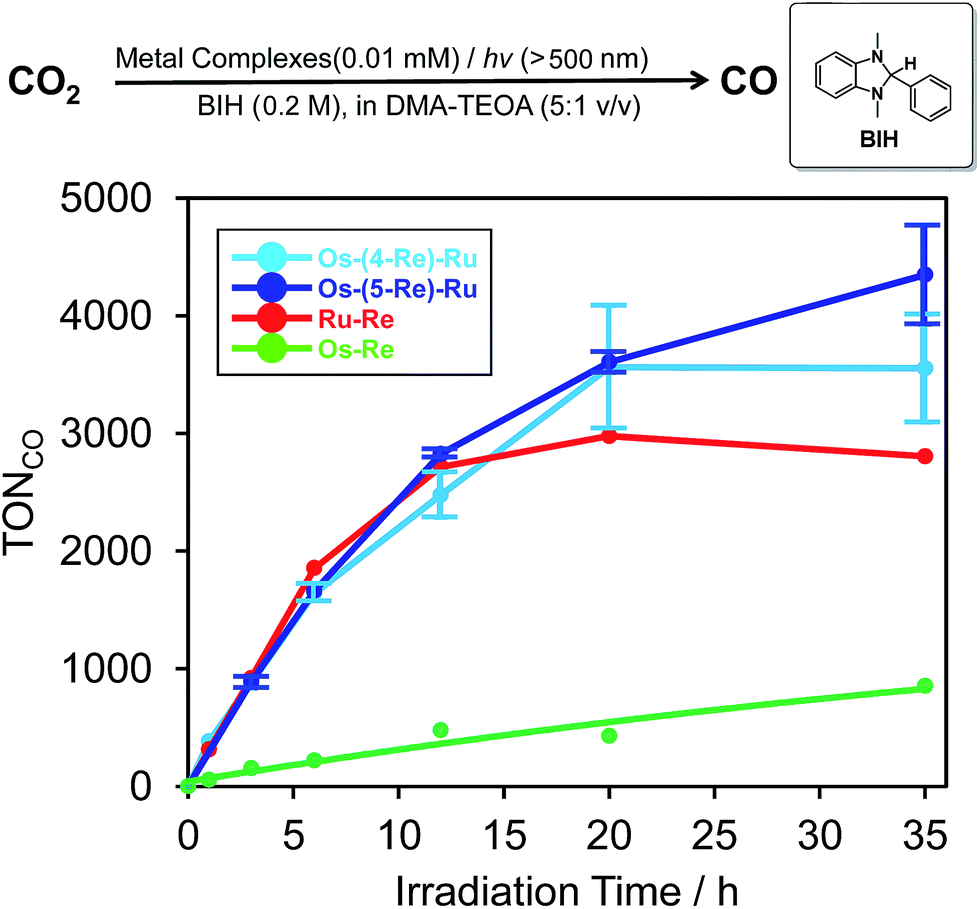
The dinuclear complexes Ru–Re6 and Os–Re,10 where two metal-complex units are connected to one another through an ethylene chain, are known as efficient supramolecular photocatalysts for CO2 reduction. Because the trinuclear complexes contain both Ru tris-diimine and Os tris-diimine complexes as potentially useful photosensitiser units and can absorb visible light more strongly and widely, as described above, we examined the photochemical reduction of CO2 using Os–(4-Re)–Ru as a photocatalyst. An N,N-dimethylacetamide (DMA)–triethanolamine (TEOA) (5:1 v/v) mixed solution containing 0.01 mM Os–(4-Re)–Ru and 0.2 M BIH as a reductant was irradiated at λex > 500 nm using a halogen lamp coupled with a K2CrO4 (30%, w/w, d = 1 cm) solution filter. Highly selective formation of CO was observed with very small amounts of H2 and HCOOH (TONH2, TONHCOOH < 1). The TON of CO formation (TONCO) reached 3552 ± 461 after 35 h irradiation, while those using the dinuclear photocatalysts were lower, i.e.Os–Re (TONCO = 854) and Ru–Re (TONCO = 2806), under the same reaction conditions. The Os–(5-Re)–Ru complex showed greater durability, and its TONCO reached 4347 ± 421 (Fig. 7), which is the highest TONCO among reported photocatalytic reactions to the best of our knowledge. It is noteworthy that the TONCO values using Os–(5-Re)–Ru were higher than the sum of those using Os–Re and Ru–Re. We also investigated the photocatalytic ability of Os–(5-Re)–Ru under red-light irradiation using a rhodamine B (0.2% w/v, d = 1 cm) solution filter (>620 nm). In this condition, only the Os unit should be excited. During the irradiation, catalytic CO formation was observed and a TONCO of 910 was reached after 35 h irradiation (Fig. S6†). This TONCO is higher than that with Os–Re under the same reaction condition (TONCO = 421).
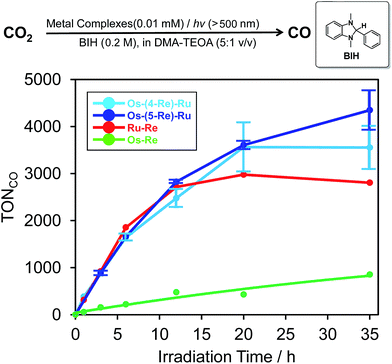 |
| | Fig. 7 Time courses of the TON of CO formation during photocatalytic reactions (λex > 500 nm) using a mixture of DMA and TEOA (5:1 v/v) containing 0.2 M BIH and metal complexes (sky blue: Os–(4-Re)–Ru, blue: Os–(5-Re)–Ru, red: Ru–Re, yellow-green: Os–Re). The concentration of each complex was 0.01 mM. | |
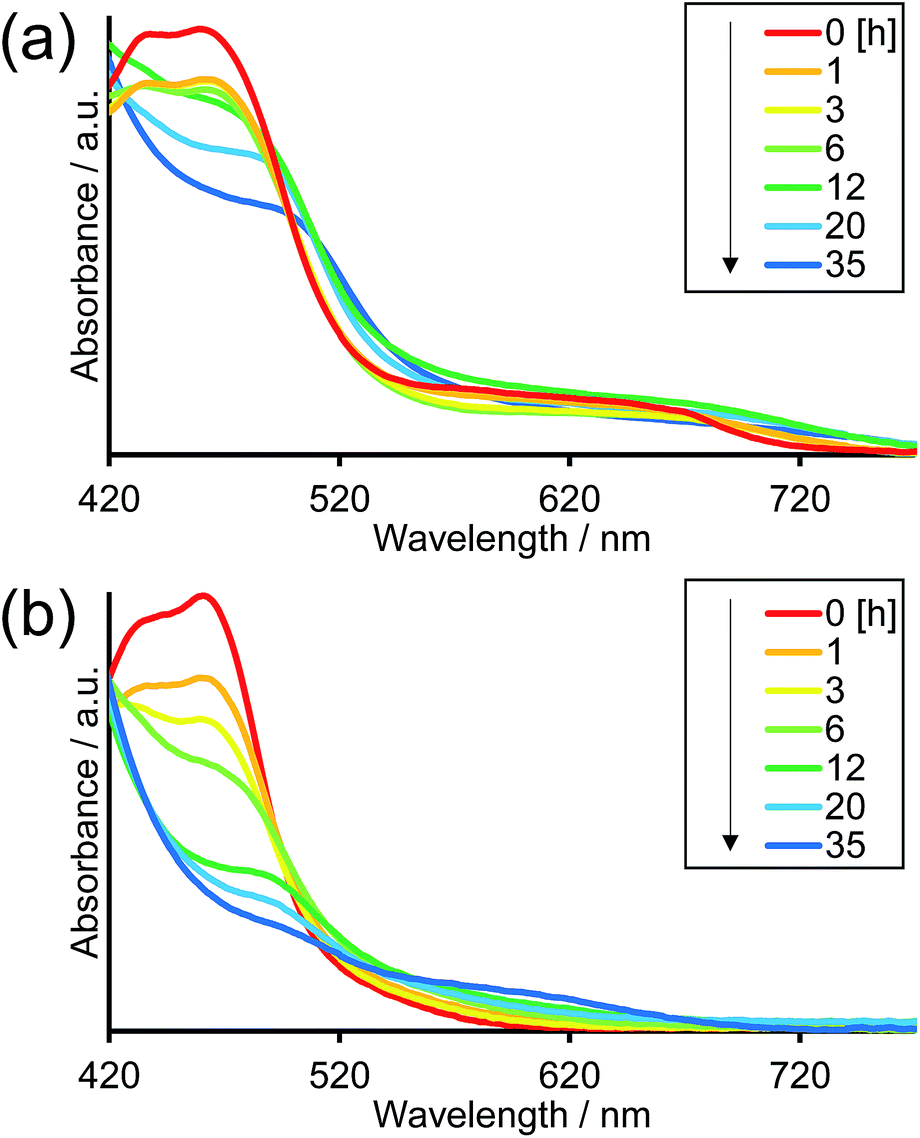
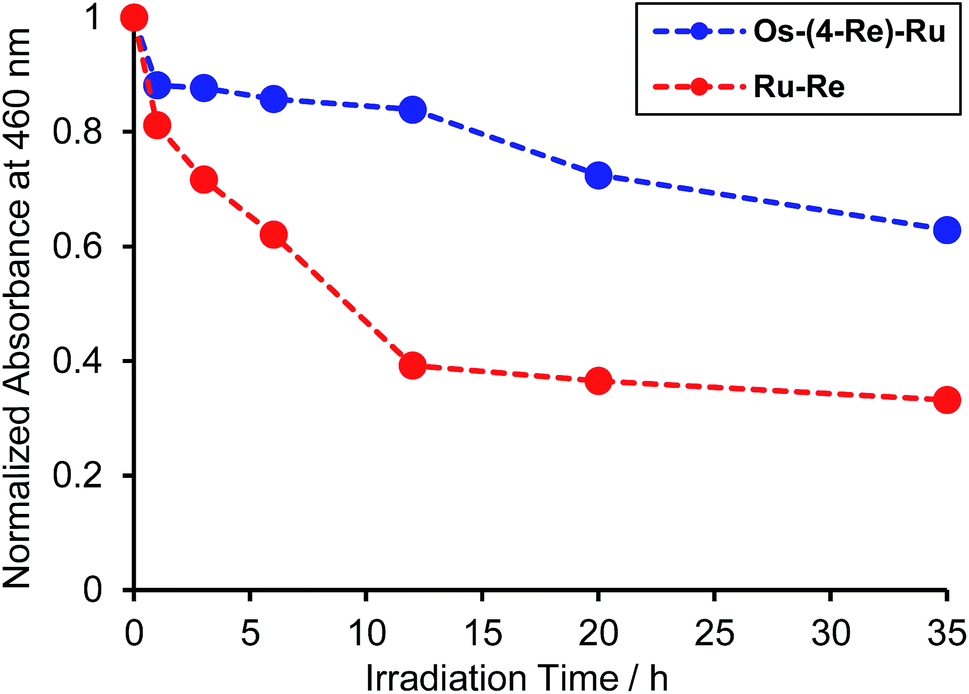
Fig. 8 illustrates the absorption spectra of the reaction solutions containing Os–(4-Re)–Ru and Ru–Re after irradiation at λex > 500 nm for various times. In the case of Os–(4-Re)–Ru, after a slight decrease of the MLCT absorption bands over 1 h, the shape of the spectrum was maintained for 6 h (Fig. 8a). After that, the absorption bands at approximately 420 to 500 nm gradually decreased. In contrast, in the case of Ru–Re, a rapid decrease of the MLCT absorption band of the Ru unit and an increase of the absorbance in the longer wavelength region (>550 nm) were observed (Fig. 8b). These absorption spectral changes were attributed to the ligand substitution reaction on the Ru unit,39 which deactivates the photosensitising ability of the Ru unit. The time courses of absorbance change at 460 nm are shown in Fig. 9. The Os and Ru units have similar molar extinction coefficients at 460 nm (Fig. 3). Because Os–Re and the decomposition products of Os–Re during the photocatalytic reaction using this dinuclear complex have similar absorbances at 460 nm, i.e. only a 16% decrease even after 35 h of photocatalytic reaction, as shown in Fig. S5,† the absorbance change at 460 nm should mainly reflect the decomposition of the Ru unit. In the case of Ru–Re, the absorbance change was almost complete within 12 h of irradiation, while in the case of Os–(4-Re)–Ru, the change was much slower and continued even after 20 h of irradiation. This decomposition reaction of the Ru unit matched the time courses of photocatalytic CO formation for each photocatalyst (Fig. 7). This indicates that the stability of the Ru unit in Os–(4-Re)–Ru during the photocatalytic reaction was improved due to the presence of the Os unit, which is expected to improve the durability of photocatalysis using Os–(4-Re)–Ru.
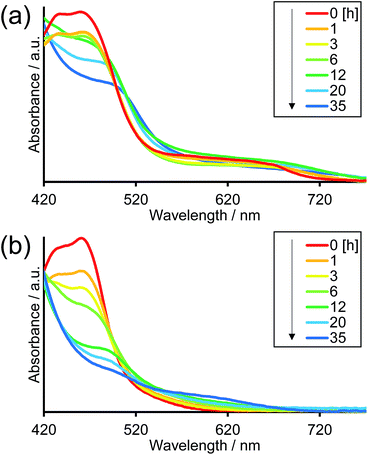 |
| | Fig. 8 Absorption spectral changes of reaction solutions of (a) Os–(4-Re)–Ru and (b) Ru–Re after irradiation. | |
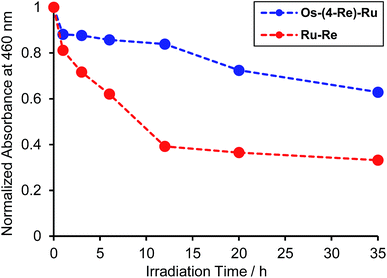 |
| | Fig. 9 Time courses of the absorbance changes at 460 nm in Fig. 8a (Os–(4-Re)–Ru, blue line) and in Fig. 8b (Ru–Re, red line). | |
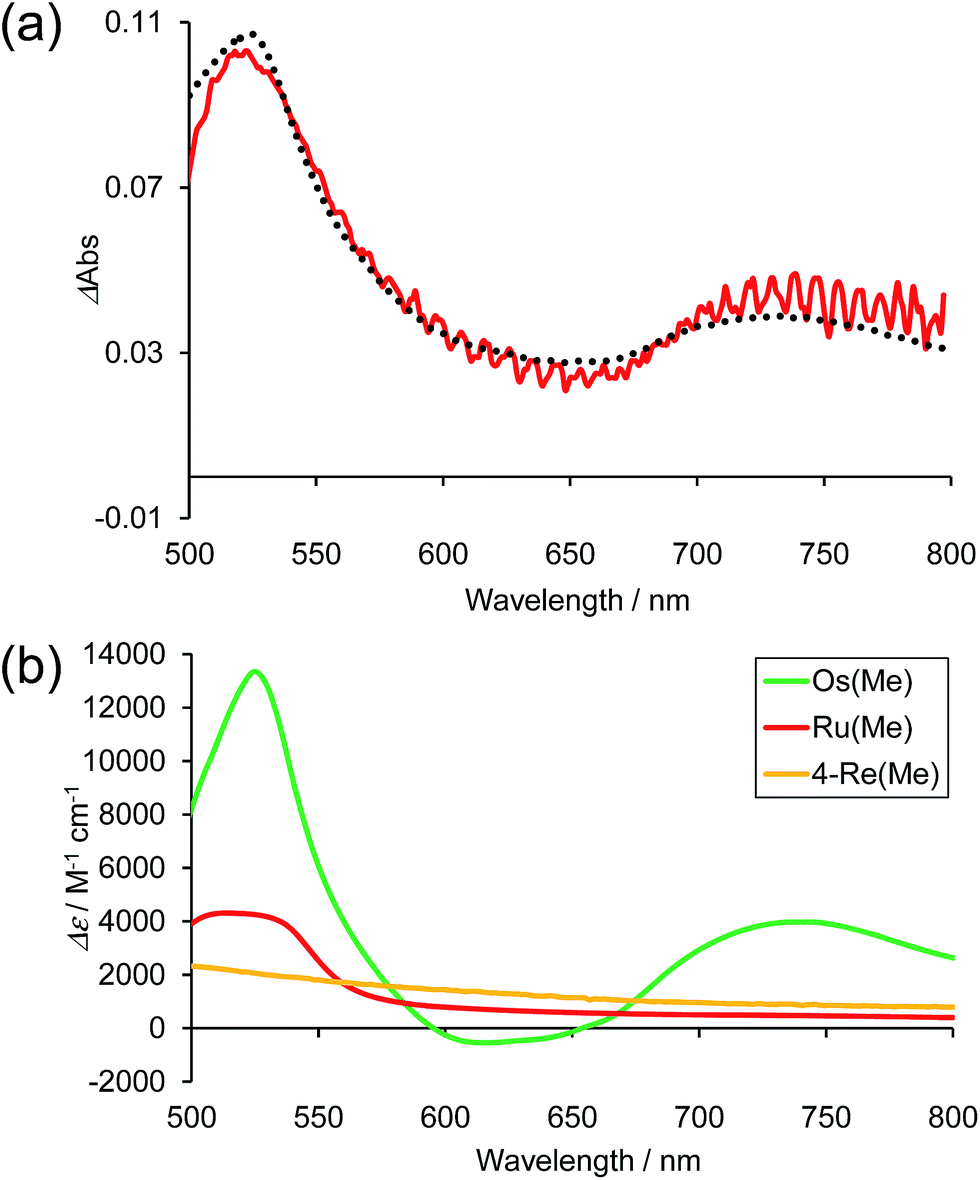
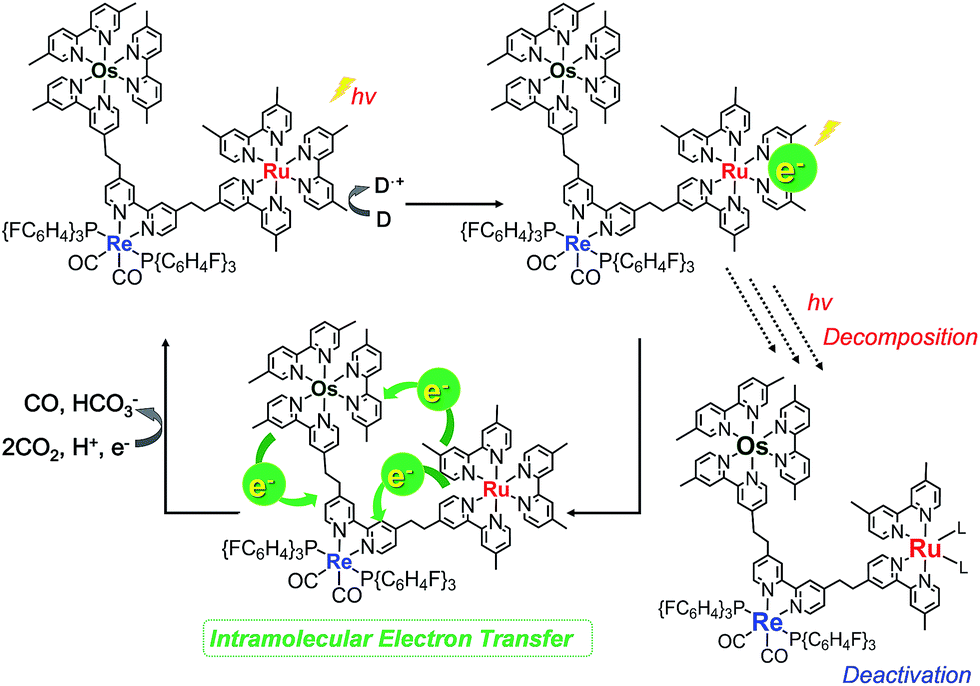
Fig. 10a shows the differential absorption spectrum of a DMA–TEOA mixed solution containing 0.01 mM Os–(4-Re)–Ru and 0.1 M BIH recorded after 1 min irradiation (λex = 480 nm) under an Ar atmosphere; a broad absorption with two obvious peaks at 525 and 730 nm was observed. This absorption spectrum can be fitted with the spectra of the one-electron reduced species (OERS) of both Os(Me) (15%) and 4-Re(Me) (85%) with correlation coefficients of 0.97.10,40 Therefore, the electron added via the reductive quenching processes of each excited metal unit should localise mainly on both the Os unit and the Re unit but not on the Ru unit during steady irradiation, even though both the Os and Ru photosensitiser units absorb the light and both excited states are quenched by BIH. In the case of Ru–Re, in contrast, approximately 10% of the added electrons are localised on the Ru unit during irradiation.40 This is the main reason for the increasing durability of the Ru unit in Os–(4-Re)–Ru compared to Ru–Re because the photochemical decomposition of Ru tris-diimine complexes mainly proceeds via OERS under photocatalytic reaction conditions (Scheme 4).20
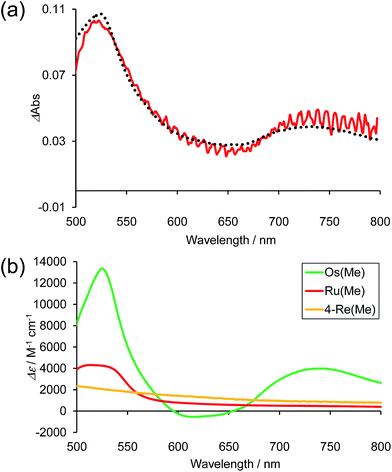 |
| | Fig. 10 (a) Difference absorption spectrum of DMA–TEOA (5:1 v/v) solution containing Os–(4-Re)–Ru (0.01 mM) and BIH (0.1 M) after irradiation at λex = 480 nm for 1 min under Ar atmosphere (red line), and the fitting spectrum with 15% Os(Me) and 85% 4-Re(Me) (black line). (b) The difference absorption spectra between the OERS and the non-reduced complexes of Os(Me) (green),10Ru(Me) (red)40 and 4-Re(Me) (orange).40 | |
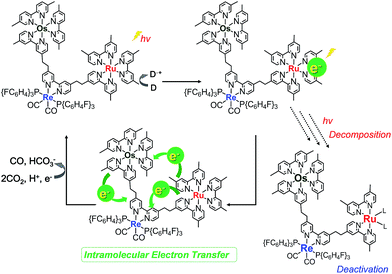 |
| | Scheme 4 Protection of the Ru unit during the photocatalytic reaction. | |
The CO formation rate using Os–(4-Re)–Ru or Os–(5-Re)–Ru over 6 h of irradiation (TOFCO = 280 h−1 (Os–(4-Re)–Ru), 281 h−1 (Os–(5-Re)–Ru) as determined by the slope of the fitting curve over 6 h, Fig. S7†) was similar to that of Ru–Re (TOFCO = 309 h−1) and slightly lower than the sum of those of Os–Re and Ru–Re (TOFCO = 361 h−1), despite the fact that the Os–Re–Ru trinuclear complexes can absorb more photons than Ru–Re and Os–Re. In this reaction system, the concentrations of the complexes were relatively low (absorbance of Os–(4-Re)–Ru < 0.14); thus, the number of photons absorbed by Os–Re–Ru trinuclear complexes can be approximated as the sum of those absorbed by Os(Me) and Ru(Me) (eqn (8)). Some photons absorbed by Os–Re–Ru trinuclear complexes may be wasted by some reaction paths, which does not occur with Ru–Re or Os–Re.
| | 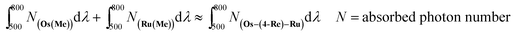 | (8) |
One possible reason for the lower efficiency in the case of Os–(4-Re)–Ru may be IEnT from the Ru unit to the Os unit. Because the quenching fraction (ηq)20 of Os–Re (ηq(Os–Re) = 51%)10 is lower than that of Ru–Re (ηq(Ru–Re) = 99%),40 IEnT to the Os unit may decrease the efficiency of the reductive quenching process of the excited Os–(4-Re)–Ru by BIH. However, because kET(Ru→Os) (3.2 × 104 s−1) is much smaller than the rate constant for the quenching by 0.2 M BIH, i.e. kq(Ru)[BIH] = 1.9 × 108 s−1 (Fig. 11), the excited state of the Ru unit should be quenched by BIH almost quantitatively under photocatalytic reaction conditions (>99%). Therefore, the IEnT process should not be the main reason for the decrease of the photocatalytic reaction rate for CO formation. This is also supported by the fact that the efficiency of CO formation using Os–(5-Re)–Ru, where IEnT from the Ru unit to the Os unit is negligible as described above, was similar to that using Os–(4-Re)–Ru. There are other possible reasons for the decrease of photocatalytic speed in the case of trinuclear photocatalysts, e.g. less localisation of the added electron in the Re catalyst unit due to the presence of the Os unit, slightly lower reduction power of the Re catalyst unit due to the electron-withdrawing properties of the photosensitizer units and steric hindrance around the Re catalyst unit.
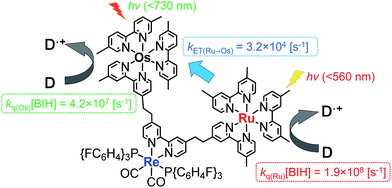 |
| | Fig. 11 Rate constants for the intramolecular energy transfer and reductive quenching by BIH. | |
We measured the quantum yield of CO formation (ΦCO) using Os–(5-Re)–Ru with irradiation at λex = 480 nm; ΦCO was 11%, which was lower than that using Ru–Re (ΦCO = 32%).41 This is mainly because the Ru unit can absorb only 40% of the absorbed photons in the case of Os–(5-Re)–Ru (ε480(Ru(Me)):ε480(Os(Me)):ε480(Re(Me)) = 98:143:2). It should be noted that the speed of the CO formation was comparable in both cases (Fig. S8†).
Conclusions
Two Os(II)–Re(I)–Ru(II) trinuclear complexes were synthesised using two separate Mizoroki–Heck reactions with different reaction conditions and subsequent photochemical hydrogenation of the bridging ligands. Both Os–(4-Re)–Ru and Os–(5-Re)–Ru selectively photocatalysed CO2 reduction to CO, where the Ru and Os units functioned as redox photosensitisers and the Re unit functioned as the catalyst. Increasing the number of photosensitiser units resulted in increased durability of the photocatalyst by 27% and 55%, respectively (TONCO(Os–(4-Re)–Ru) = 3552, TONCO(Os–(5-Re)–Ru) = 4347, TONCO(Ru–Re) = 2806 and TONCO(Os–Re) = 854); meanwhile, the speeds of the photocatalytic reaction were slightly lowered by 9%, compared to that of Ru–Re (TOFCO(Os–(4-Re)–Ru) = 280 h−1, TOFCO(Os–(5-Re)–Ru) = 281 h−1 and TOFCO(Ru–Re) = 309 h−1). Excitation of the Re units of the trinuclear complexes resulted in efficient IEnT, mainly to the Ru units. Although IEnT from the excited Ru unit to the Os unit was observed in the case of Os–(4-Re)–Ru, this did not contribute to the photocatalytic reaction because the process was much slower than reductive quenching of the excited Ru unit by the reductant BIH. This energy transfer was suppressed in Os–(5-Re)–Ru.
Experimental
General procedures
1H NMR and 31P NMR spectra were measured in acetone-d6 using a JEOL ECA400-II system at 400 MHz and 162 MHz, respectively. IR spectra were measured using a JASCO FT/IR-610 spectrometer at 1 cm−1 resolution in dichloromethane solution. Electrospray ionisation-mass spectroscopy (ESI-MS) was performed using a Shimadzu LC-MS-2010A system with acetonitrile as the mobile phase. Electrospray ionisation time-of-flight mass spectroscopy (ESI-TOFMS) was performed using a Waters LCT Premier mass spectrometer with acetonitrile as the mobile phase. For SEC analysis, we used a pair of Shodex PROTEIN KW-402J columns (300 mm × 8.0 mm i.d.) with a KW-LG guard column (50 mm × 6.0 mm i.d.), a JASCO 880-51 degasser, an 880-PU pump, an MD-2010 Plus UV-Vis photodiode-array detector and a Rheodyne 7125 injector. The column temperature was maintained at 40 °C using a JASCO 860-CO oven. The eluent was a 1:1 (v/v) mixture of methanol and acetonitrile with 0.5 M CH3COONH4, and the flow rate was 0.2 mL min−1. Separation of the multinuclear complexes was achieved by SEC using a pair of Shodex PROTEIN KW-2002.5 columns (300 mm × 20.0 mm i.d.) with a KW-LG guard column (50 mm × 8.0 mm i.d.) and a recycling preparative HPLC apparatus with a JASCO 870-UV detector. The eluent was a 1:1 (v/v) mixture of methanol and acetonitrile with 0.15 M CH3COONH4, and the flow rate was 6.0 mL min−1. UV-Vis absorption spectra were measured with a JASCO V-670 instrument. Each compound was dissolved in MeCN and degassed using the freeze–pump–thaw method prior to emission measurements. Emission spectra were recorded at 25 °C using either a JASCO FP-8600 spectrofluorometer or a Hamamatsu C9920-02 system. The absolute emission quantum yields were evaluated using the Hamamatsu C9920-02 system equipped with an integrating sphere and a multichannel photodetector (PMA-12). Emission lifetimes were obtained using a HORIBA TemPro fluorescence lifetime system with an emission monochromator. The excitation light source was a NanoLED-560 pulse lamp and the instrumental response time was less than 0.1 ns. All emission decays were fitted by single, double or triple exponential functions within appropriate χ2 (0.9–1.2). The irradiation light for the photochemical hydrogenation reactions was 510 nm light derived from a Xe lamp in a Max 303 system equipped with band-pass filters (FWHM = 10 nm) purchased from Asahi Spectra Co. UV-Vis absorption spectral changes during the photochemical hydrogenation reactions and absorbed photon numbers were recorded using a Shimadzu QYM-01 system.
Photocatalytic reactions
The photocatalytic reactions were performed in an 11 mL test tube (i.d. = 8 mm) containing a 2 mL DMA–TEOA (5:1 v/v) solution of Os–(4-Re)–Ru, Os–(5-Re)–Ru, Ru–Re or Os–Re (0.01 mM) and BIH (0.2 M) after purging with CO2 for more than 20 min. The solution was irradiated using a merry-go-round irradiation apparatus at λ > 500 nm and >620 nm with a halogen lamp combined with a Pyrex water jacket and a K2CrO4 (30%, w/w, d = 1 cm) and a rhodamine B (0.2% w/w, d = 1 cm) solution filter, respectively. During irradiation, the tube was cooled with a thermostatic bath (25 °C). The gaseous reaction products (CO and H2) were analysed using a GC-TCD instrument (GL Science GC323). HCOOH was analysed using a capillary electrophoresis system (Otsuka Electronics Co., CAPI-3300I).
Measurement of the absorption spectrum of the OERS of Os–(4-Re)–Ru
4 mL DMA–TEOA (5:1 v/v) solution containing Os–(4-Re)–Ru (0.01 mM) and BIH (0.1 M) in an 11 mL quartz cell (light-pass length: 1 cm) was purged with Ar for over 20 min. The solution was irradiated with 480 nm light derived from a Xe lamp in a Max 303 system equipped with a band-pass filter (FWHM = 10 nm) purchased from Asahi Spectra Co. UV-Vis absorption spectral changes during irradiation were recorded using a Shimadzu QYM-01 system.
Measurement of quantum yields of photocatalytic CO formation
A DMA–TEOA (5:1 v/v) solution (3 mL) containing metal complexes (0.01 mM) and BIH (0.1 M) in a quartz cell (light-pass length: 1 cm, volume: 11 mL, light intensity: 3 × 10−8 einstein per s) was purged with CO2 for 20 min. The solution was irradiated at λex = 480 nm derived from a 300 W Xe lamp in a Max 303 system equipped with a band-pass filter (FWHM = 10 nm, Asahi Spectra Co.). UV-Vis absorption spectral changes during irradiation were recorded using a Shimadzu QYM-01 system. During irradiation, the cell was cooled at 25 °C with a thermostatic bath. The gaseous reaction products (CO and H2) were analysed using a GC-TCD instrument (GL Science GC323).
Materials
All reagents were of reagent grade and were used without further purification. ReBr2,314-Re(Me),42Ru(Me),43Os(Me),10Ru–Re,40Os–Re,10Ru(CC)31 and BIH7 were prepared according to reported methods. Os(CC), 5-Re(Me) and [Re(5,5′-dibromo-bpy)(CO)2{P(p-C6H4F)3}2](PF6) were synthesised using vbpy, 5-dmb and 5,5′-dibromo-bpy instead of the corresponding diimine ligands according to the methods for Os(Me), 4-Re(Me) and ReBr2, respectively, with some modifications.
Synthesis
Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) Re–Br
.
ReBr2 (52 mg, 39 μmol), Ru(CC) (19 mg, 20 μmol), Pd(OAc)2 (4.4 mg, 20 μmol), PPh3 (10 mg, 39 μmol) and AcONa (8.0 mg, 97 μmol) were dissolved in acetonitrile (4 mL, degassed by N2). The solution was heated to approximately 75 °C under Ar for 1 d (in dim light). The atmosphere was changed to air by opening the three-way cock attached to the reaction vessel, and the solution was heated for one week. The solvent was removed in vacuo, and a red solid was obtained. The solid was purified by SEC. The solvent of the obtained red solution was evaporated under reduced pressure. The residue was dissolved in CH2Cl2 and washed twice with water containing NH4PF6. After evaporation of the solvent, a red solid was obtained. This compound was used in the next reaction without further purification. Yield: 14 mg (33%). ESI-MS (in acetonitrile) m/z: 591 ([M − 3PF6−]3+).
Re–Br
.
ReBr2 (52 mg, 39 μmol), Ru(CC) (19 mg, 20 μmol), Pd(OAc)2 (4.4 mg, 20 μmol), PPh3 (10 mg, 39 μmol) and AcONa (8.0 mg, 97 μmol) were dissolved in acetonitrile (4 mL, degassed by N2). The solution was heated to approximately 75 °C under Ar for 1 d (in dim light). The atmosphere was changed to air by opening the three-way cock attached to the reaction vessel, and the solution was heated for one week. The solvent was removed in vacuo, and a red solid was obtained. The solid was purified by SEC. The solvent of the obtained red solution was evaporated under reduced pressure. The residue was dissolved in CH2Cl2 and washed twice with water containing NH4PF6. After evaporation of the solvent, a red solid was obtained. This compound was used in the next reaction without further purification. Yield: 14 mg (33%). ESI-MS (in acetonitrile) m/z: 591 ([M − 3PF6−]3+).
Os–(4-Re)–Ru
.
RuRe–Br (14 mg, 6.3 μmol), Os(CC) (26 mg, 25 μmol), Pd(OAc)2 (1.4 mg, 6.3 μmol) and AcONa (2.6 mg, 32 μmol) were dissolved in acetonitrile (2 mL, degassed with N2). The solution was heated to approximately 65 °C under Ar for 2 d (in dim light). Pd(OAc)2 (1.4 mg, 6.3 μmol) was added to the solution as a second charge. After heating for another 2 d, the solvent was removed under reduced pressure, and a brown solid was collected. This solid was purified by SEC after filtration through a membrane filter (Millex LG 0.20 μm). The solvent of the obtained dark red solution was evaporated. The residue was dissolved in CH2Cl2 and washed twice with water containing NH4PF6. After evaporation of the solvent, a dark red solid (Os(4-Re)Ru) was collected and washed with water and Et2O. Os(4-Re)Ru (13 mg, 4.2 μmol) and BIH (90 mg, 400 μmol) were dissolved in an acetonitrile–pyridine–CF3COOH mixed solution (4 mL, 3:1:0.1 v/v/v). After bubbling with Ar for 30 min, the solution was irradiated with a Xe lamp equipped with a band-pass filter (510 ± 10 nm) for 15 h. The crude product was purified by ion-exchange chromatography (CM Sephadex C-25, eluent: acetonitrile–water (1:1 v/v) containing NH4PF6 (0 to 16 mM)). Some acetonitrile was evaporated under reduced pressure. The green precipitate was collected by filtration and washed with water and ether. The precipitate was dissolved in CH2Cl2 and washed with water. The solvent was removed under reduced pressure to obtain a green solid which was dried in vacuum at 60 °C. Yield: 6.9 mg (34%). 1H NMR (400 MHz, acetone-d6): δ/ppm, 8.77 (s, 1H), 8.72 (s, 1H), 8.63 (s, 5H), 8.57 (s, 3H), 8.55 (s, 2H), 8.38 (s, 2H), 7.99 (d, J = 6.0 Hz, 1H), 7.98 (d, J = 7.6 Hz, 1H), 7.92 (d, J = 6.0 Hz, 1H), 7.82–7.67 (m, 15H), 7.52 (dd, J = 2.0, 8.4 Hz, 1H), 7.43 (dd, J = 2.0, 7.6 Hz, 1H), 7.36–7.27 (m, 18H), 7.10–7.06 (m, 14H), 3.19–3.10 (m, 8H, –C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–C2–), 2.60–2.52 (m, 18H), 2.18 (s, 12H). 31P NMR (400 MHz, acetone-d6): δ/ppm, 20.8 (s, 2P, 2
2–C2–), 2.60–2.52 (m, 18H), 2.18 (s, 12H). 31P NMR (400 MHz, acetone-d6): δ/ppm, 20.8 (s, 2P, 2![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) (C6H4F)3), −143.6 (sep, JP–F = 707 Hz, 5P, 5F6−). FT-IR (in CH2Cl2) νCO/cm−1: 1939, 1869. ESI-MS (in acetonitrile) m/z: 490 ([M − 5PF6−]5+), 649 ([M − 4PF6−]4+), 914 ([M − 3PF6−]3+). HRMS (ESI-TOF) m/z: [M − 5PF6−]5+ calcd for C122H104F6N14O2OsP2ReRu 490.3214; found 490.3212. Retention time for HPLC (SEC) was 32 min with methanol–acetonitrile (1:1 v/v) containing 0.5 M CH3COONH4 as an eluent.
(C6H4F)3), −143.6 (sep, JP–F = 707 Hz, 5P, 5F6−). FT-IR (in CH2Cl2) νCO/cm−1: 1939, 1869. ESI-MS (in acetonitrile) m/z: 490 ([M − 5PF6−]5+), 649 ([M − 4PF6−]4+), 914 ([M − 3PF6−]3+). HRMS (ESI-TOF) m/z: [M − 5PF6−]5+ calcd for C122H104F6N14O2OsP2ReRu 490.3214; found 490.3212. Retention time for HPLC (SEC) was 32 min with methanol–acetonitrile (1:1 v/v) containing 0.5 M CH3COONH4 as an eluent.
Os–(5-Re)–Ru was synthesised from [Re(5,5′-dibromo-bpy)(CO)2{P(p-C6H4F)3}2](PF6), Ru(CC) and Os(CC) by a similar method to that for Os–(4-Re)–Ru. Yield: 10% (over 3 steps). 1H NMR (400 MHz, acetone-d6): δ/ppm, 8.73 (s, 1H), 8.70 (s, 1H), 8.66 (m, 4H), 8.61 (s, 1H), 8.59 (m, 3H), 8.58 (s, 1H), 8.56 (s, 1H), 8.38 (m, 2H), 8.02–8.00 (m, 4H), 7.93 (d, J = 5.2 Hz, 1H), 7.85–7.64 (m, 15H), 7.44–7.31 (m, 20H), 7.16–7.12 (m, 12H), 2.98–2.89 (m, 8H, –C2–C2–), 2.67 (s, 3H), 2.56 (s, 15H), 2.23–2.20 (m, 12H). 31P NMR (400 MHz, acetone-d6): δ/ppm, 21.4 (s, 2P, 2(C6H4F)3), −143.6 (sep, JP–F = 707 Hz, 5P, 5F6−). FT-IR (in CH2Cl2) νCO/cm−1: 1945, 1874. ESI-MS (in acetonitrile) m/z: 490 ([M − 5PF6−]5+), 649 ([M − 4PF6−]4+), 914 ([M − 3PF6−]3+). HRMS (ESI-TOF) m/z: [M − 5PF6−]5+ calcd for C122H104F6N14O2OsP2ReRu 490.3214; found 490.3212 (Fig. S9†). Retention time for HPLC (SEC) was 32 min with methanol–acetonitrile (1:1 v/v) containing 0.5 M CH3COONH4 as an eluent.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Strategic International Collaborative Research Program (SICORP) of JST, JSPS KAKENHI Grant Number 17K14526, and JSPS Grant-in-Aid for Scientific Research on Innovative Areas “Elucidation of the molecular mechanism of photosynthesis at high time and space resolution and development of artificial photosynthetic systems”.
Notes and references
- Y. Yamamoto, Y. Tamaki, T. Yui, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2010, 132, 11743–11752 CrossRef CAS PubMed.
- V. Balzani, S. Campagna, G. Denti, A. Juris, S. Serroni and M. Venturi, Acc. Chem. Res., 1998, 31, 26–34 CrossRef CAS.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- S. Sato, K. Koike, H. Inoue and O. Ishitani, Photochem. Photobiol. Sci., 2007, 6, 454–461 CAS.
- K. Koike, S. Naito, S. Sato, Y. Tamaki and O. Ishitani, J. Photochem. Photobiol., A, 2009, 207, 109–114 CrossRef CAS.
- Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Faraday Discuss., 2012, 155, 115–127 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- K. Ohkubo, Y. Yamazaki, T. Nakashima, Y. Tamaki, K. Koike and O. Ishitani, J. Catal., 2016, 343, 278–289 CrossRef CAS.
- E. Kato, H. Takeda, K. Koike, K. Ohkubo and O. Ishitani, Chem. Sci., 2015, 6, 3003–3012 RSC.
- Y. Tamaki, K. Koike, T. Morimoto, Y. Yamazaki and O. Ishitani, Inorg. Chem., 2013, 52, 11902–11909 CrossRef CAS PubMed.
- E. Fujita, S. J. Milder and B. S. Brunschwig, Inorg. Chem., 1992, 31, 2079–2085 CrossRef CAS.
- E. Kimura, S. Wada, M. Shionoya and Y. Okazaki, Inorg. Chem., 1994, 33, 770–778 CrossRef CAS.
- E. Kimura, X. Bu, M. Shionoya, S. Wada and S. Maruyama, Inorg. Chem., 1992, 31, 4542–4546 CrossRef CAS.
- K. Kiyosawa, N. Shiraishi, T. Shimada, D. Masui, H. Tachibana, S. Takagi, O. Ishitani, D. A. Tryk and H. Inoue, J. Phys. Chem. C, 2009, 113, 11667–11673 CAS.
- Y. Kou, S. Nakatani, G. Sunagawa, Y. Tachikawa, D. Masui, T. Shimada, S. Takagi, D. A. Tryk, Y. Nabetani, H. Tachibana and H. Inoue, J. Catal., 2014, 310, 57–66 CrossRef CAS.
- J. Schneider, K. Q. Vuong, J. A. Calladine, X. Z. Sun, A. C. Whitwood, M. W. George and R. N. Perutz, Inorg. Chem., 2011, 50, 11877–11889 CrossRef CAS PubMed.
- A. Inagaki, S. Yatsuda, S. Edure, A. Suzuki, T. Takahashi and M. Akita, Inorg. Chem., 2007, 46, 2432–2445 CrossRef CAS PubMed.
- R. L. House, N. Y. M. Iha, R. L. Coppo, L. Alibabaei, B. D. Sherman, P. Kang, M. K. Brennaman, P. G. Hoertz and T. J. Meyer, J. Photochem. Photobiol., C, 2015, 25, 32–45 CrossRef CAS.
- F. Li, Y. Jiang, B. Zhang, F. Huang, Y. Gao and L. Sun, Angew. Chem., Int. Ed., 2012, 51, 2417–2420 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- T. Ren, Chem. Rev., 2008, 108, 4185–4207 CrossRef CAS PubMed.
- P. J. Connors, D. Tzalis, A. L. Dunnick and Y. Tor, Inorg. Chem., 1998, 37, 1121–1123 CrossRef CAS.
- D. Tzalis and Y. Tor, Chem. Commun., 1996, 1043–1044 RSC.
- S. Goeb, A. De Nicola and R. Ziessel, J. Org. Chem., 2005, 70, 6802–6808 CrossRef CAS PubMed.
- R. M. Haak, M. Martinez Belmonte, E. C. Escudero-Adan, J. Benet-Buchholz and A. W. Kleij, Dalton Trans., 2010, 39, 593–602 RSC.
- R. M. Haak, A. M. Castilla, M. Martinez Belmonte, E. C. Escudero-Adan, J. Benet-Buchholz and A. W. Kleij, Dalton Trans., 2011, 40, 3352–3364 RSC.
- L. Cassidy, S. Horn, L. Cleary, Y. Halpin, W. R. Browne and J. G. Vos, Dalton Trans., 2009, 3923–3928 RSC.
- K. J. Arm and J. A. G. Williams, Dalton Trans., 2006, 2172–2174 RSC.
- V. L. Whittle and J. A. G. Williams, Dalton Trans., 2009, 3929–3940 RSC.
- S. Welter, N. Salluce, P. Belser, M. Groeneveld and L. De Cola, Coord. Chem. Rev., 2005, 249, 1360–1371 CrossRef CAS.
- Y. Yamazaki, T. Morimoto and O. Ishitani, Dalton Trans., 2015, 44, 11626–11635 RSC.
- O. B. Locos and D. P. Arnold, Org. Biomol. Chem., 2006, 4, 902–916 CAS.
- Y. Yamazaki, A. Umemoto and O. Ishitani, Inorg. Chem., 2016, 55, 11110–11124 CrossRef CAS PubMed.
- Y. Yamazaki and O. Ishitani, Dalton Trans., 2017, 46, 4816–4823 RSC.
- Z.-Y. Bian, S.-M. Chi, L. Li and W. Fu, Dalton Trans., 2010, 39, 7884–7887 RSC.
- Z.-Y. Bian, H. Wang, W.-F. Fu, L. Li and A.-Z. Ding, Polyhedron, 2012, 32, 78–85 CrossRef CAS.
- M. Furue, T. Yoshidzumi, S. Kinoshita, T. Kushida, S. Nozakura and M. Kamachi, Bull. Chem. Soc. Jpn., 1991, 64, 1632–1640 CrossRef CAS.
- M. Furue, M. Naiki, Y. Kanematsu, T. Kushida and M. Kamachi, Coord. Chem. Rev., 1991, 111, 221–226 CrossRef CAS.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef CAS.
- Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Faraday Discuss., 2012, 155, 115–127 RSC.
- The reaction condition, especially the concentration of the complex and the light intensity, was different from the reactions in this report; previously reported reaction conditions using Ru–Re as the photocatalyst: [Ru–Re] was 0.3 mM (light intensity: 4.3 × 10−9 einstein per s, ΦCO = 45%, ref. 7). On the other hand, [Ru–Re] was 0.01 mM in this study. The light intensity in the previous report was much lower compared to that in this study (3 × 10−8 einstein per s). Since the quantum yields in the systems using supramolecular photocatalysts containing a [Re(N^N)(CO)2{P(FC6H4)3}2]+-type catalyst unit is strongly
depended on such reaction condition (ref. 40), the obtained quantum yield in the cases using Ru–Re in this study should be different from the previous study.
- H. Tsubaki, A. Sekine, Y. Ohashi, K. Koike, H. Takeda and O. Ishitani, J. Am. Chem. Soc., 2005, 127, 15544–15555 CrossRef CAS PubMed.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Chem. Commun., 2014, 50, 1491–1493 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption spectra changes during photochemical hydrogenation of Os(5-Re)Ru, UV-Vis absorption spectra of Os–(5-Re)–Ru, emission spectra of 4-Re(Me) and Ru–Re, absorption spectra of Os–Re before and after the photocatalysis, time courses of TONCO using Os–(5-Re)–Ru and Os–Re under red-light irradiation, and ESI-mass spectrum of Os–(5-Re)–Ru. See DOI: 10.1039/c7sc04162d |
|
| This journal is © The Royal Society of Chemistry 2018 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*