
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrastable, cationic three-dimensional lead bromide frameworks that intrinsically emit broadband white-light†
Chengdong
Peng‡
,
Zewen
Zhuang‡
,
Huimin
Yang
,
Guiyang
Zhang
and
Honghan
Fei
 *
*
Shanghai Key Laboratory of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, 1239 Siping Rd., Shanghai 200092, P. R. China. E-mail: fei@tongji.edu.cn
First published on 19th December 2017
Abstract
Herein, we report the unusual broadband white-light emission as an intrinsic property from two cationic lead bromide frameworks. This is the first time that the metal halide materials adopting a purely inorganic positively-charged three-dimensional (3D) topology have been synthesized, thus affording highly distorted PbII centers. The single-component white-light emitters achieve an external quantum efficiency of up to 5.6% and a correlated color temperature of 5727 K, producing typical white-light close to that of fluorescent light sources. Unlike the air/moisture-sensitive 3D organolead halide perovskites, our cationic materials are chemically “inert” over a wide range of pH as well as aqueous boiling condition. Importantly, these long-sought ultrastable lead halide materials exhibit undiminished photoluminescence upon continuous UV-irradiation for 30 days under atmospheric condition (∼60% relative humidity, 1 bar). Our mechanistic studies indicate the broadband emission have contributions from the self-trapped excited states through electron-vibrational coupling in the highly deformable and anharmonic lattice, as demonstrated by variable-temperature photoluminescence/absorption spectra as well as X-ray crystallography studies. The chemical robustness and structural tunability of the 3D cationic bromoplumbates open new paths for the rational design of hybrid bulk emitters with high photostability.
Introduction
Solid-state lighting, an energy-saving alternative technology to conventional lighting sources, has attracted increasing attention in recent years.1 Among them, realizing white-light luminescence from light-emitting diodes (LEDs) is of particular interest for general illumination applications.2–4 A typical white-light LED device includes a blend of red, green and blue (RGB) LEDs or coating a blue (or near UV) LED with a yellow phosphor (or a mixture of RGB phosphors).5–7 These multi-color and/or multi-component strategies suffer from a variety of inevitable drawbacks, such as efficiency losses arising from self-absorption and long-term instability due to different degradation rates of the phosphors.8,9 Thus, a single-component broadband white-light emitter covering the entire visible spectrum is an attractive and challenging target in white-light LED research.10–12 However, very few known examples of single-source phosphors intrinsically emit white-light luminescence, thus hindering rational synthetic design.3,5,13Hybrid inorganic–organic lead halide perovskites containing vertex-sharing metal halide octahedra are an emerging class of photoactive materials, which have promising applications in photovoltaics and light-emitting devices.14,15 Among them, two-dimensional (2D) organolead halide perovskites usually exhibit narrow-band photoluminescence owing to their flat (100)-oriented layers and large exciton binding energy.16 Recently, a few instances of (110)-oriented 2D perovskites showed broadband photoemission as an intrinsic property, presumably ascribed to the formation of self-trapped excited states (e.g. Pb23+, Pb3+, X2−) (X = Cl, Br, I).17–24 Lowering the dimensionality of lead halide facilitates the self-trapping process and enhances the photoluminescence quantum efficiency (PLQE), albeit with a sacrifice in structural stability and photoluminescence tunability.25–29 The air/moisture-sensitive nature of organolead halide perovskites results in a gradual decrease of photoemission intensity upon UV-irradiation in air, thus hindering their industrial applications in LED technology.30–32 Therefore, the development of chemically ultrastable lead halide materials with efficient, tunable and broadband white-light emission is crucial to extend their photoactive applications.
Inorganic extended frameworks usually adopt a neutral or anionic inorganic host, including zeolites, aluminophosphates and metal halide perovskites.33–35 Purely inorganic structures bearing an overall positive charge are very rare, presenting merely <0.1% of over 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 crystal structures in the inorganic crystal structure database (ICSD). Layered double hydroxides are a widely studied class of cationic 2D inorganic materials, possessing trivalent-ion-substituted brucite layers intercalated with charge-balancing anions. Other examples include francisite minerals (Cu2BiSe2O8X, X = F, Cl, Br, I) and their derivatives,36,37 as well as layered heavy p-block hydroxides and fluorides.38–42 Until now, only two synthetic examples of purely inorganic cationic three-dimensional (3D) frameworks have been reported, namely a thorium borate and an ytterbium oxyhydroxide, respectively.43,44 Among the few examples of 3D lead halide inorganic frameworks, none of them bear a positively charge.45,46
000 crystal structures in the inorganic crystal structure database (ICSD). Layered double hydroxides are a widely studied class of cationic 2D inorganic materials, possessing trivalent-ion-substituted brucite layers intercalated with charge-balancing anions. Other examples include francisite minerals (Cu2BiSe2O8X, X = F, Cl, Br, I) and their derivatives,36,37 as well as layered heavy p-block hydroxides and fluorides.38–42 Until now, only two synthetic examples of purely inorganic cationic three-dimensional (3D) frameworks have been reported, namely a thorium borate and an ytterbium oxyhydroxide, respectively.43,44 Among the few examples of 3D lead halide inorganic frameworks, none of them bear a positively charge.45,46
Very recently, we reported a class of 2D (layered) cationic lead halide materials with high chemical resistance and high photoluminescence quantum efficiency.47 Herein, we report the synthesis, crystal structures and broadband photoluminescence of the first two 3D cationic metal halide frameworks, [Pb2Br2][O2C(CH2)4CO2] and [Pb3Br4][O2C(CH2)2CO2] (which we denote as TJU-6 for [Pb2Br2]2+ and TJU-7 for [Pb3Br4]2+, TJU = Tongji University). The unique positively charged 3D lead bromide networks define the arrays of unidimensional channels, in which reside the bridging organic anions. Intriguingly, both materials are intrinsically bulk white-light emitters spanning the entire visible-light spectrum. In contrast to lead halide perovskite and other hybrid bulk emitters, our cationic materials are essentially unaffected upon near-UV-irradiation (365 nm) for 30 days under ambient condition (∼60% relative humidity, 1 bar). The temperature-dependent photophysical studies (e.g. UV-vis absorption spectra and photoluminescence spectra) and X-ray crystallography studies attribute the broad emission to the self-trapped states from electron-vibrational coupling in the strongly deformable and anharmonic lattice.
Results and discussion
Hydrothermal reaction of PbBr2, adipic acid disodium salt (NaO2C(CH2)4CO2Na), and perchloric acid afforded colorless block-shaped crystals of TJU-6 (Fig. S1a†). Specifically, slow-cooling of the autoclaves at the rate of 10 °C h−1 after the static heating is necessary to obtain high phase purity of TJU-6. In addition, perchloric acid was discovered to tune the pH as well as act as a stabilizer, like the role of fluoride ion in zeolite synthesis.48 X-Ray crystallography reveals that TJU-6 is crystallized in the highly symmetric tetrahedral P41212 space group (Table S1†). TJU-6 consists of edge- and vertex-sharing PbBr3 units extending in three dimensions (Fig. 1a and S2†). Adipates covalently bridge and crosslink the inorganic extended connectivity, further enhancing the structural stability that will be discussed later. The crystallographically independent PbII atom occupies a highly distorted octahedral geometry in TJU-6, having three bridging bromines and three oxygens from two adipate anions (Fig. S3† inset). In metal halide materials (e.g. organolead halide perovskites), there is a strong correlation between the distortion of PbII centers and the self-trapped excitons from short-range electron–lattice interactions.14,18,19,23 Noting the Pb–O bond length is obviously shorter than Pb–Br, we sought to use the octahedral angle variance σoct2 to quantitatively evaluate the deformation of Oh symmetry in each PbBr3O3 octehedra:49 | (1) |
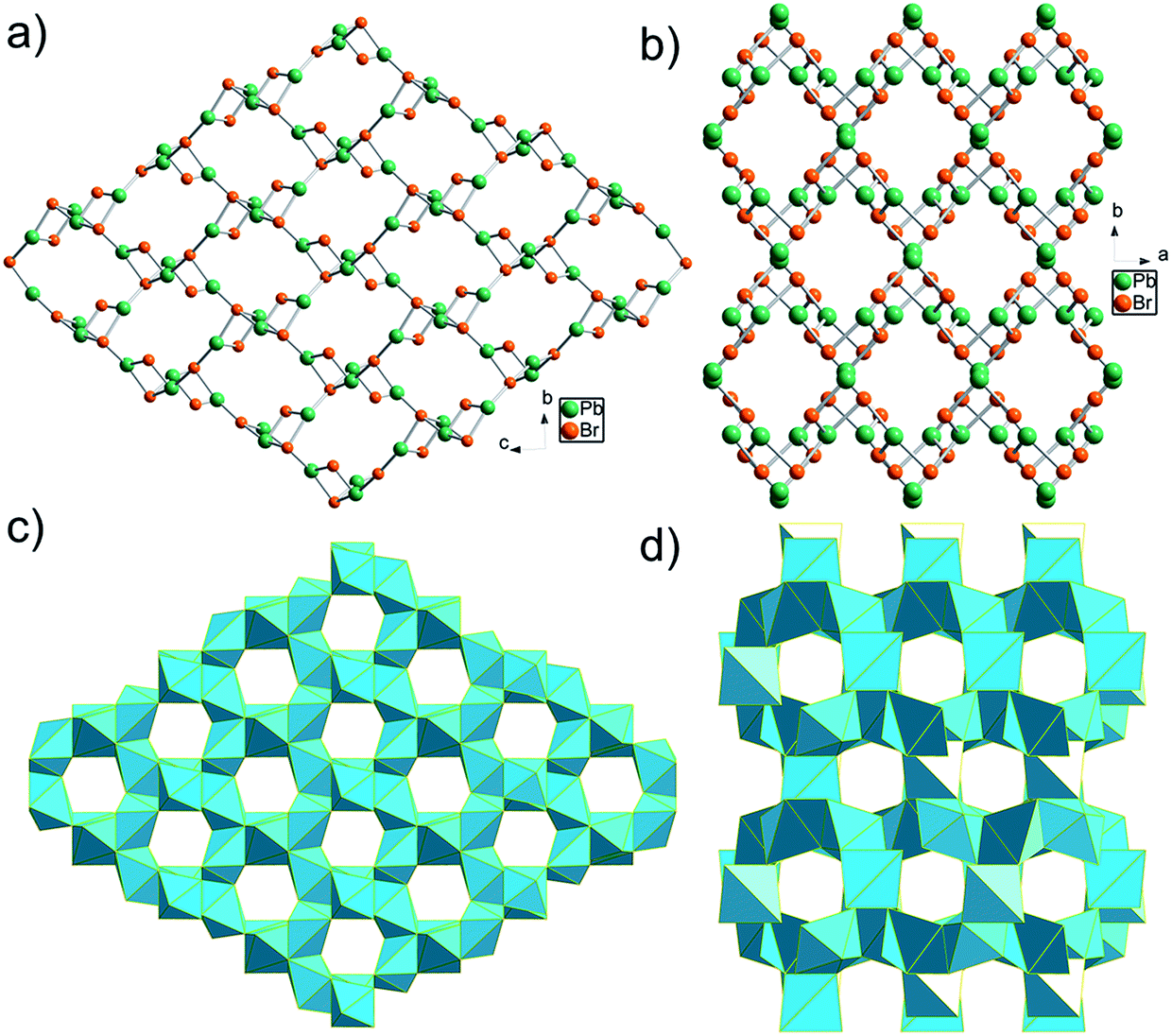 | ||
| Fig. 1 Crystallographic view of TJU-6 and TJU-7: (a, b) ball-and-stick view of TJU-6 (a) and TJU-7 (b), with organic anions omitted for clarity. (c, d) Polyhedral view of TJU-6 (c) and TJU-7 (d). Cyan polyhedra represents PbBr3O3 in TJU-6 and PbBr4O2/PbBr2O2 in TJU-7, respectively. Carbon and hydrogen atoms are omitted for clarity. | ||
The edge-sharing PbBr3O3 octahedra in TJU-6 are connected in both of the crystallographically (100) and (010) planes, defining the honeycomb arrays of 6-membered ring channels along the a- and b-axis, respectively (Fig. 1c and S3†). Adipates covalently bridge the lead centers within the hexagon-shaped channels, and the Pb–O bond lengths (2.505–2.664 Å) are well within the accepted Pb–O covalent range. The two crystallographically independent Br atoms are either vertex-bridging (μ2-Br) or quadruply bridging (μ4-Br) in a highly distorted tetrahedral geometry. Overall, the high-coordinated bridging Br atoms and the low-coordinated Pb centers collectively attribute to the cationic feature of a rare 3D metal halide topology.
Synthesis of the cationic 3D bromoplumbate framework was successfully extended to a more compact-packing inorganic topology via employing a shorter α,ω-alkanecarboxylate as the anionic structure-directing agent. Colorless block-shaped crystals of TJU-7 (Fig. S1b†) were prepared using succinate in place of adipate under otherwise identical synthetic conditions. TJU-7 crystallizes in the orthorhombic crystal system with Pbcn space group (Table S2†). The structure of TJU-7 consists of cationic [PbBr]+ chains along the c-axis and Pb2Br4 units vertex-bridge the adjacent chains (Fig. 1b and S4†). The crystallographically independent PbII center in the cationic chains adopts a distorted octahedral geometry, as for PbII in TJU-6. According to octahedral angle variance (σoct2) calculation (eqn (1)), the structural distortion of TJU-7 (σoct2 = 783) is close to that of TJU-6 (σoct2 = 735). The other crystallography independent PbII centers are in the Pb2Br4 pillars, which occupy a distorted tetrahedral geometry (Fig. S5† inset). The corner-sharing Pb2Br4O2 octahedra and vertex-sharing Pb2Br2O2 tetrahedra define 8-membered ring channels along the c-axis, in which reside the crosslinked succinates (Fig. 1d and S5†). It is worth noting that the shorter α,ω-alkanecarboxylate results in the inorganic framework of TJU-7 adopting a dense topology as well as a lower stoichiometric ratio of Pb:Br (3:4), compared to 1:1 in the open framework of TJU-6
Non-perovskite metal-halide hybrid materials usually have a low-dimensional inorganic extended structure (1D or 2D). In addition, all of the previously reported 3D lead halide examples occupy an anionic inorganic network hosting organoammonium cations.45,46,50,51 Overall, TJU-6 and TJU-7 are the first two lead halide materials with a cationic 3D M–X–M (M = metal, X = halogen) connectivity that have been unambiguously identified by single-crystal X-ray crystallography.
The high yield and phase purity of TJU-6 and TJU-7 was evidenced by Fourier-transform infrared spectroscopy (FTIR), elemental analysis and powder X-ray diffraction (PXRD), which matches well with the theoretical patterns simulated from single-crystal data (Fig. 2, S6 and S7†). Unlike organoammonium cations in perovskites, the anionic structure-directing agents (e.g. α,ω-alkanecarboxylate) covalently crosslink the metal centers within the pore channels of the cationic inorganic host. This structural feature plays a significant role to afford the chemical “inertness” of the resulting lead bromide materials. Stability tests were performed by treating the materials in water, ethanol, HCl solution (pH = 2), and NaOH solution (pH = 12) for 24 h. PXRD of the post-treated TJU-6 and TJU-7 remained intact, confirming the well-retained cationic topology (Fig. 2). In addition, no apparent loss in mass was observed during the chemical treatment, further proving the chemical inertness of TJU-6 and TJU-7. Moreover, thermogravimetric analysis (TGA) and ex situ thermodiffraction indicate that TJU-6 and TJU-7 are thermally stable up to 250 °C under air (Fig. 2, S8 and S9†). Overall, our cationic 3D bromoplumbate materials strikingly push forward the chemical-resistance and related photoactive applications of organolead halide hybrid materials.
 | ||
| Fig. 2 PXRD of TJU-6 (a) and TJU-7 (b) before and after heat or chemical treatment. | ||
Given the high robustness and lattice deformation of TJU-6 and TJU-7, we sought to investigate their photoluminescent properties (Table 1). Both materials show a typical semiconductive properties with an optical bandgap of 3.70 eV (335 nm) for TJU-6 and 3.50 eV (354 nm) for TJU-7, respectively, evidenced by UV-vis absorption spectra (Fig. 3a and b). The large band-gaps probably result from the open inorganic framework of [PbBr]+ and [Pb3Br4]3+, which leads to an increase in the bandgap and the energy level of conduction band.52 In addition, excitonic features (the shoulder peak at 3.99 eV for TJU-6 and 3.90 eV for TJU-7) are observed in the optical absorption spectra. The less-defined excitonic peak is largely ascribed to the moderate exciton binding energy (290 meV) from the absorption spectra of TJU-6 at 103 K (Fig. S10†).
| Material | λ abs/nm | λ ex/nm | λ em/nm | FWHM/nm | φ (%) | τ av/ns |
|---|---|---|---|---|---|---|
| a λ abs is the wavelength at absorbance maximum; λex is the excitation wavelength; λem is the wavelength at the emission maxima; φ; is the external photoluminescence quantum efficiency; τav is the PL lifetime. | ||||||
| TJU-6 | 335 | 360 | 530 | 124 | 5.6 | 1.7 |
| TJU-7 | 354 | 370 | 480 | 166 | 1.8 | 1.6 |
 | ||
| Fig. 3 (a, b) Emission spectra of the cm-sized bulk crystals (blue) and μm-sized microscopic crystals (red) of TJU-6 (excited at 360 nm) and TJU-7 (excited at 370 nm). (c) Photoimages of TJU-6, TJU-7, PbBr2 and adipate under 4 W 365 nm UV-light. (d) CIE chromaticity coordinates of TJU-6 and TJU-7. | ||
Interestingly, colorless block-shaped single crystals of TJU-6 show a pronounced white-light emission upon irradiation with a 4 W, 365 nm UV-lamp (Fig. 3c). The photoemission spectra (under 360 nm excitation) of TJU-6 demonstrates the unusual broadband photoluminescence spanning the entire visible spectrum (400–700 nm). The strongly Stokes-shifted broadband emission of TJU-6 has a maximum at 530 nm with a large FWHM up to 124 nm (0.56 eV), significantly overcoming the self-absorption problems in the near-UV region. In addition, μm-sized particles of TJU-6 were prepared via manual grinding, and showed nearly identical photoemission spectra (Fig. 3 and S11a†). These results confirm that the broadband emission arises from the bulk materials instead of from surface defect sites (which often contribute to the photoluminescence of lead halide perovskite nanocrystals53,54). The high-energy shoulder at 420 nm is more evident in microscale crystals, likely due to the strong self-adsorption in cm-sized single crystals.25 Since the two-band emission profiles are similar to those of lead halide perovskites,17,18 it is reasonable to attribute the high-energy shoulder to free excitons and the low-energy broadband emission to self-trapped excitons. The detailed photoemission mechanism will be discussed later. Time-resolved photoluminescence decay experiments indicate the lifetime of the emission measured at 537 nm is 1.7 ns for TJU-6 (Fig. 4a). The ns-ranged lifetimes are characteristic of fluorescence emission, while the longer lifetime of TJU-6 over the previously reported 2-D lead-based perovskites (τav = 0.23–1.39 ns)17–20 is presumably attributed to the populated self-trapped excitons. Moreover, the cm-sized single crystals and μm-sized particles of TJU-6 show similar lifetimes, again suggesting the broadband emission as an intrinsic effect. The strongly Stokes-shifted broadband photoemission covering the entire visible-light spectrum has also been observed in TJU-7. A more evident high-energy photoemission shoulder at 415 nm blue-shifts the maximum emission wavelength to 480 nm with a large FWHM up to 166 nm (0.88 eV).
 | ||
| Fig. 4 (a) The photoluminescence decay of TJU-6 (measured at 537 nm) at room temperature. (b) Photoemission spectra of TJU-6 before and after continuous UV-irradiation (4 W, 365 nm) under atmospheric condition (∼60% relative intensity, 1 bar, room temperature). (c) Temperature-dependent UV-vis diffuse reflectance micro-spectrum of TJU-6 from 133 to 293 K. (d) Temperature-dependent photoemission spectra of TJU-6 from 77 K to 473 K. (e) Integrated photoluminescence intensity (red) and FWHM (blue) as a function of temperature. (f) Schematic summarizing the main mechanism responsible for broadband emissions in the cationic lead bromide frameworks. | ||
Importantly, no apparent change to the color properties of the broadband emission is observed in TJU-6 when adjusting the excitation from 320 nm to 360 nm, confirming its nature as a broadband monochromatic emitter (Fig. S12†). The Commission International de l’Eclairage (CIE) chromaticity coordinates of the overall TJU-6 emission is determined to be (0.33, 0.48), corresponding to a color temperature of 5727 K (Fig. 3d). The emission is so-called “cold” white-light, and very close to that of conventional fluorescent light sources (∼5100 K). Meanwhile, the μm-sized crystals of TJU-6 exhibit a CIE coordinate of (0.30, 0.45) with a color temperature of 6464 K (Fig. 3d). The external quantum efficiency of the TJU-6 is measured to be 5.6%, which is significantly higher than most white-light emitters based on 2D organolead halide perovskites as well as a 3D anionic lead chloride framework (Table S3†). In addition, the photoemission band and intensity was monitored upon irradiation with a 4 W, 365 UV lamp under atmospheric condition (∼60% relative intensity, 1 bar, room temperature). Intriguingly, the broadband emission is remarkably stable after UV-irradiation in air for 30 days, achieving a substantial advance in hybrid lead halide white-light emitters (Fig. 4b).
The TJU-7 light emission was determined to have CIE chromaticity coordinates of (0.25, 0.32) for bulk cm-sized crystals and (0.25, 0.33) for μm-sized crystal particles, which are all closer to the coordinates of the sunlight source (0.33, 0.33) (Fig. 3d). The bluish white-light emission affords the correlated color temperature of 11967 K for cm-sized single crystals and 10640 K for μm-sized particles, respectively. The external PLQE of the TJU-7 was measured to be 1.8% with an expected lower efficiency from the dense [Pb3Br4]2+ framework.
In order to verify the broadband emission of TJU-6 arises from the self-trapped excited states attributable to the lattice deformation, we performed variable-temperature experiments of photoluminescence and UV-vis absorption spectra (Fig. 4d–f). In contrast to intrinsic white-light emitters of organolead halide perovskites,18,55,56 an obvious and gradual blue-shift of the maximum emission wavelength was noticed when the temperature was decreased from 473 K to 77 K (Fig. 4d). The overall photoluminescent peak position was shifted to higher energy by 27 nm (117 meV), while the integrated intensity of the photoemission was increased by 40 times (Fig. 4e). In order to investigate the origin of the photoluminescence blue-shift, we performed variable-temperature UV-vis absorption spectra of TJU-6 from 133 K to 298 K. In agreement with the photoluminescence experiments, a blue-shift of the absorption edge was clearly observed with decreasing temperature (Fig. 4c). Based on these variable-temperature photophysical studies, it is concluded that TJU-6 has a temperature-dependent (133–293 K) optical bandgap. The temperature dependence of the semiconductor bandgap, determined from the UV-vis absorption edge, is usually well described using Varshni’s equation:57
 | (2) |
In addition, broadening of the emission bandwidth was observed from 118 nm to 129 nm as the temperature was increased from 77 K to 493 K, further confirming the electron–phonon interactions (Fig. 4d and e). The temperature dependence of the FWHM in TJU-6 can be described using the following model:62
| Γ(T) = Γ0 + ΓLO(eELO/kBT − 1)−1 + Γinh(e−Eb/kBT) | (3) |
Here, Γ0 represents the emission FWHM at T = 0 K, ELO is the energy of the longitudinal optical phonon energy, and Eb represents the average binding energy of the defect states. ΓLO and Γinh give the relative contributions of exciton-phonon coupling and inhomogeneous broadening (induced by trap states), respectively. A fit to the data gives Γ0 = 219 ± 2 meV, ΓLO = 131 ± 7 meV, Γinh = 287 ± 3 meV, ELO = 13 ± 5 meV, and Eb = 12 ± 1 meV (Fig. 4e and S15†). The LO phonon energy obtained from the fitting corresponds to a frequency of ∼110 cm−1, which lies well within the range of Pb–Br stretching vibration frequencies from Raman spectroscopy (Fig. S16†). Based on our photophysical mechanistic studies, we confirm that the broadband emission and the self-trapped states of TJU-6 are mainly attributed to the electron–phonon coupling in a strongly deformable 3D lead halide lattice (Fig. 4f).63 Recent first-principle calculations of lead halide materials indicate the electron–phonon coupling in a deformable lead halide lattice forms self-trapped excitons (e.g. Pb23+, Pb3+, and Br2− species), which act as radiative color centers.64,65
Conclusions
We have discovered two hybrid bromoplumbate bulk emitters with an unusual broadband white-light emission spanning the entire visible-light region. These are the first two 3D lead halide frameworks possessing an overall positive charge, thus exhibiting high distortion of PbII centers and generated self-trapped states. Upon 360 nm excitation, the [PbBr]+ exhibit a moderately high PLQE of 5.6%, exceeding most of the perovskite-type white-light emitters (<1%). Importantly, the ultrastable nature of our materials affords undiminished photoemission throughout UV-irradiation for 30 days under air, largely overcoming the chemically labile problems of 3D lead perovskites. Our mechanistic studies confirm that the broadband emission arises from electron–phonon coupling in a strongly deformable and anharmonic lattice, which contributes to the formation of self-trapped states. The long-sought “inert” lead halide materials are amenable to synthetic design for enhancing PLQEs, thus serving as potential alternatives to commercial white-light LED phosphors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (21501136, 51772217), the Recruitment of Global Youth Experts by China, the Fundamental Research Funds for the Central Universities, and the Science & Technology Commission of Shanghai Municipality (14DZ2261100).References
- O. I. D. Association, The Promise of Solid State Lighting for General Illumination, US Department of Energy, Washington, DC, 2001 Search PubMed.
- A. Bergh, G. Craford, A. Duggal and R. Haitz, Phys. Today, 2001, 54, 42–47 CrossRef CAS.
- M. S. Wang, S. P. Guo, Y. Li, L. Z. Cai, J.-P. Zou, G. Xu, W. W. Zhou, F. K. Zheng and G. C. Guo, J. Am. Chem. Soc., 2009, 131, 13572–13573 CrossRef CAS PubMed.
- D. F. Sava, L. E. Rohwer, M. A. Rodriguez and T. M. Nenoff, J. Am. Chem. Soc., 2012, 134, 3983–3986 CrossRef CAS PubMed.
- X. H. Zhu, J. Peng, Y. Cao and J. Roncali, Chem. Soc. Rev., 2011, 40, 3509–3524 RSC.
- Y. Li, A. Rizzo, R. Cingolani and G. Gigli, Adv. Mater., 2006, 18, 2545–2548 CrossRef CAS.
- P. O. Anikeeva, J. E. Halpert, M. G. Bawendi and V. Bulović, Nano Lett., 2007, 7, 2196–2200 CrossRef CAS PubMed.
- J. M. Phillips, M. E. Coltrin, M. H. Crawford, A. J. Fischer, M. R. Krames, R. Mueller-Mach, G. O. Mueller, Y. Ohno, L. E. Rohwer and J. A. Simmons, Laser Photonics Rev., 2007, 1, 307–333 CrossRef CAS.
- S. Ye, F. Xiao, Y. Pan, Y. Ma and Q. Zhang, Mater. Sci. Eng., R, 2010, 71, 1–34 CrossRef.
- M. Shang, C. Li and J. Lin, Chem. Soc. Rev., 2014, 43, 1372–1386 RSC.
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 CrossRef CAS PubMed.
- M. C. Gather, A. Köhnen and K. Meerholz, Adv. Mater., 2011, 23, 233–248 CrossRef CAS PubMed.
- Y. Liu, M. Nishiura, Y. Wang and Z. Hou, J. Am. Chem. Soc., 2006, 128, 5592–5593 CrossRef CAS PubMed.
- S. D. Stranks and H. J. Snaith, Nat. Nanotechnol., 2015, 10, 391–402 CrossRef CAS PubMed.
- J. Berry, T. Buonassisi, D. A. Egger, G. Hodes, L. Kronik, Y. L. Loo, I. Lubomirsky, S. R. Marder, Y. Mastai and J. S. Miller, Adv. Mater., 2015, 27, 5102–5112 CrossRef CAS PubMed.
- F. Deschler, M. Price, S. Pathak, L. E. Klintberg, D.-D. Jarausch, R. Higler, S. Hüttner, T. Leijtens, S. D. Stranks and H. J. Snaith, J. Phys. Chem. Lett., 2014, 5, 1421–1426 CrossRef CAS PubMed.
- E. R. Dohner, E. T. Hoke and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 1718–1721 CrossRef CAS PubMed.
- E. R. Dohner, A. Jaffe, L. R. Bradshaw and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 13154–13157 CrossRef CAS PubMed.
- D. Cortecchia, S. Neutzner, A. R. Srimath Kandada, E. Mosconi, D. Meggiolaro, F. De Angelis, C. Soci and A. Petrozza, J. Am. Chem. Soc., 2017, 139, 39–42 CrossRef CAS PubMed.
- L. Mao, Y. Wu, C. C. Stoumpos, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 5210–5215 CrossRef CAS PubMed.
- K. Thirumal, W. K. Chong, W. Xie, R. Ganguly, S. K. Muduli, M. Sherburne, M. Asta, S. Mhaisalkar, T. C. Sum and H. S. Soo, Chem. Mater., 2017, 29, 3947–3953 CrossRef CAS.
- M. D. Smith, A. Jaffe, E. R. Dohner, A. M. Lindenberg and H. I. Karunadasa, Chem. Sci., 2017, 8, 4497–4504 RSC.
- Y. Li, C. Lin, G. Zheng, Z. Cheng, H. You, W. Wang and J. Lin, Chem. Mater., 2006, 18, 3463–3469 CrossRef CAS.
- Y. Li, G. Zheng and J. Lin, Eur. J. Inorg. Chem., 2008, 1689–1692 CrossRef CAS.
- Z. Yuan, C. Zhou, Y. Tian, Y. Shu, J. Messier, J. C. Wang, L. J. van de Burgt, K. Kountouriotis, Y. Xin, E. Holt, K. Schanze, R. Clark, T. Siegrist and B. Ma, Nat. Commun., 2017, 8, 14051 CrossRef CAS PubMed.
- B. Ma, C. Zhou, Y. Tian, M. Wang, A. Rose, T. Besara, N. K. Doyle, Z. Yuan, J. C. Wang, R. Clark, Y. Hu, T. Siegrist and S. Lin, Angew. Chem., Int. Ed., 2017, 56, 9018–9022 CrossRef PubMed.
- C. Zhou, Z. Yuan, Y. Tian, H. Lin, R. Clark, B. Chen, L. J. van de Burgt, J. C. Wang, K. Hanson and Q. J. Meisner, arXiv:1702.07200, 2017.
- M. I. Saidaminov, J. Almutlaq, S. Sarmah, I. Dursun, A. A. Zhumekenov, R. Begum, J. Pan, N. Cho, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2016, 1, 840–845 CrossRef CAS.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- E. Mosconi, J. M. Azpiroz and F. De Angelis, Chem. Mater., 2015, 27, 4885–4892 CrossRef CAS.
- R. K. Misra, S. Aharon, B. Li, D. Mogilyansky, I. Visoly-Fisher, L. Etgar and E. A. Katz, J. Phys. Chem. Lett., 2015, 6, 326–330 CrossRef CAS PubMed.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D’Haen, L. D’Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca and E. Mosconi, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- C. Baerlocher, L. B. McCusker and D. H. Olson, Atlas of zeolite framework types, Elsevier, 2007 Search PubMed.
- J. Yu and R. Xu, Chem. Soc. Rev., 2006, 35, 593–604 RSC.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- P. Millet, B. Bastide, V. Pashchenko, S. Gnatchenko, V. Gapon, Y. Ksari and A. Stepanov, J. Mater. Chem., 2001, 11, 1152–1157 RSC.
- K. M. Ok and P. S. Halasyamani, Inorg. Chem., 2002, 41, 3805–3807 CrossRef CAS PubMed.
- C. H. Swanson, H. A. Shaikh, D. L. Rogow, A. G. Oliver, C. F. Campana and S. R. J. Oliver, J. Am. Chem. Soc., 2008, 130, 11737–11741 CrossRef CAS PubMed.
- H. H. Fei and S. R. J. Oliver, Angew. Chem., Int. Ed., 2011, 50, 9066–9070 CrossRef CAS PubMed.
- H. H. Fei, C. H. Pham and S. R. J. Oliver, J. Am. Chem. Soc., 2012, 134, 10729–10732 CrossRef CAS PubMed.
- G. Zhang, G. Wei, Z. Liu, S. R. J. Oliver and H. Fei, Chem. Mater., 2016, 28, 6276–6281 CrossRef CAS.
- H. Yang and H. Fei, Chem. Commun., 2017, 53, 7064–7067 RSC.
- S. A. Wang, E. V. Alekseev, D. W. Juan, W. H. Casey, B. L. Phillips, W. Depmeier and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2010, 49, 1057–1060 CrossRef CAS PubMed.
- H. V. Goulding, S. E. Hulse, W. Clegg, R. W. Harrington, H. Y. Playford, R. I. Walton and A. M. Fogg, J. Am. Chem. Soc., 2010, 132, 13618–13620 CrossRef CAS PubMed.
- Z. J. Zhang, S. C. Xiang, G. C. Guo, G. Xu, M. S. Wang, J. P. Zou, S. P. Guo and J. S. Huang, Angew. Chem., Int. Ed., 2008, 47, 4149–4152 CrossRef CAS PubMed.
- G. E. Wang, G. Xu, M. S. Wang, L. Z. Cai, W. H. Li and G. C. Guo, Chem. Sci., 2015, 6, 7222–7226 RSC.
- Z. Zhuang, C. Peng, G. Zhang, H. Yang, J. Yin and H. Fei, Angew. Chem., Int. Ed., 2017, 56, 14411–14416 CrossRef CAS PubMed.
- D. L. Rogow, M. P. Russell, L. M. Wayman, C. H. Swanson, A. G. Oliver and S. R. J. Oliver, Cryst. Growth Des., 2010, 10, 823–829 CAS.
- K. Robinson, G. V. Gibbs and P. H. Ribbe, Science, 1971, 172, 567–570 CAS.
- A. B. Corradi, A. M. Ferrari, G. C. Pellacani, A. Saccani, F. Sandrolini and P. Sgarabotto, Inorg. Chem., 1999, 38, 716–721 CrossRef CAS.
- J. D. Martin and K. B. Greenwood, Angew. Chem., Int. Ed. Engl., 1997, 36, 2072–2075 CrossRef CAS.
- N. Zheng, X. Bu, H. Vu and P. Feng, Angew. Chem., Int. Ed., 2005, 44, 5299–5303 CrossRef CAS PubMed.
- M. B. Teunis, K. N. Lawrence, P. Dutta, A. P. Siegel and R. Sardar, Nanoscale, 2016, 8, 17433–17439 RSC.
- H. Wu, S. Xu, H. Shao, L. Li, Y. Cui and C. Wang, Nanoscale, 2017, 9, 16858–16863 RSC.
- T. Hu, M. D. Smith, E. R. Dohner, M.-J. Sher, X. Wu, M. T. Trinh, A. Fisher, J. Corbett, X. Y. Zhu, H. I. Karunadasa and A. M. Lindenberg, J. Phys. Chem. Lett., 2016, 7, 2258–2263 CrossRef CAS PubMed.
- A. Yangui, D. Garrot, J. S. Lauret, A. Lusson, G. Bouchez, E. Deleporte, S. Pillet, E. E. Bendeif, M. Castro, S. Triki, Y. Abid and K. Boukheddaden, J. Phys. Chem. C, 2015, 119, 23638–23647 CAS.
- Y. P. Varshni, Physica, 1967, 34, 149–154 CrossRef CAS.
- P. Sastry and B. Mtjlimani, Philos. Mag., 1969, 20, 859–862 CrossRef CAS.
- L. Barr and D. Dawson, Proc. Br. Ceram. Soc., 1971, 19, 151–160 Search PubMed.
- J. Kerssen, W. De Gruijter and J. Volger, Physica, 1973, 70, 375–396 CrossRef CAS.
- J. Wu, W. Walukiewicz, W. Shan, K. Yu, J. Ager Iii, S. Li, E. Haller, H. Lu and W. J. Schaff, J. Appl. Phys., 2003, 94, 4457–4460 CrossRef CAS.
- V. A. Fonoberov, K. A. Alim, A. A. Balandin, F. Xiu and J. Liu, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 165317 CrossRef.
- K. S. Song and R. T. Williams, Self-Trapped Excitons, Springer, Berlin, 2nd edn, 1996 Search PubMed.
- D. Cortecchia, J. Yin, A. Bruno, S. Z. A. Lo, G. G. Gurzadyan, S. Mhaisalkar, J. L. Bredas and C. Soci, J. Mater. Chem. C, 2017, 5, 2771–2780 RSC.
- J. Yin, H. Li, D. Cortecchia, C. Soci and J. L. Bredas, ACS Energy Lett., 2017, 2, 417–423 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional characterization. CCDC 1575434–1575436. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04118g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |