
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed regiodivergent hydroaminocarbonylation of alkenes to primary amides with ammonium chloride†
Bao
Gao
a,
Guoying
Zhang
a,
Xibing
Zhou
a and
Hanmin
Huang
 *ab
*ab
aDepartment of Chemistry, Hefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: hanmin@ustc.edu.cn
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, P. R. China
First published on 24th October 2017
Abstract
Palladium-catalyzed hydroaminocarbonylation of alkenes for the synthesis of primary amides has long been an elusive aim. Here, we report an efficient catalytic system which enables inexpensive NH4Cl to be utilized as a practical alternative to gaseous ammonia for the palladium-catalyzed alkene-hydroaminocarbonylation reaction. Through appropriate choice of the palladium precursors and ligands, either branched or linear primary amides can be obtained in good yields with good to excellent regioselectivities. Primary mechanistic studies were conducted and disclosed that electrophilic acylpalladium species were capable of capturing the NH2-moiety from ammonium salts to form amides in the presence of CO with NMP as a base.
Introduction
Ammonia is among the least expensive industrial chemicals, and has long been recognized as a cheap and ideal nitrogen-source for the synthesis of N-containing molecules via C–N bond formation reactions.1 However, in spite of the utility and abundance of ammonia, it has not frequently been used in transition-metal catalyzed reactions, although notable exceptions include reductive amination,2 hydroaminomethylation,3 hydroamination,4 allylic amination,5 aminocarbonylation,6 and coupling of ammonia with aryl halides.7 Besides the general technical complexities of using a gas, the potential reason for the scarcity of reports on exploiting ammonia in metal-catalyzed reactions may arise from its easy formation of unreactive Lewis acid-base adducts with transition-metals owing to its high basicity, small size and strong N–H bonds.8 An attractive and complementary approach would be to use ammonium salts as surrogates of ammonia gas for establishing transition-metal catalyzed reactions. In this respect, copper and palladium-catalyzed C–N bond formation reactions between aryl halides and ammonium salts have been established by Chang and Hartwig.9 However, super stoichiometric amounts of strong base were generally required in both of these systems to form the metal-amide species from the in situ released NH3, leading to wasteful by-product generation. Therefore, the design of new and efficient protocols which are capable of directly incorporating the nitrogen atom into the hydrocarbon from the ammonium salts in the absence of base is highly desirable.Transition-metal-catalyzed hydroaminocarbonylation of alkenes constitutes one of the most economical and efficient methods for the construction of aliphatic amides from simple alkenes, CO and amines.10 Early studies in this field with Co, Ni, Fe and Ru-catalysts required harsh reaction conditions and suffered from poor chemoselectivity.11 As a means of addressing the above synthetic problem, a palladium-catalyzed hydroaminocarbonylation reaction, initiated by palladium-hydride species, has been developed by Beller, Liu, our own group and others.12,13 While the utility of this process has been well demonstrated by the synthesis of various secondary and tertiary amides, it is surprising to note that the related reaction for the synthesis of aliphatic primary amides has never been realized, although they are more valuable building blocks in the synthesis of biologically active molecules and functional materials.14 One obvious reason for discouraging the study of this important unsolved synthetic problem is that the basic ammonia is thought to be necessarily utilized in the related hydroaminocarbonylation reaction, which is not only incompatible with the acids typically utilized for palladium-catalyzed hydroaminocarbonylation but also prone to forming unreactive Werner amine-complexes which deactivate the catalyst.8 For the past several years, our group has been focused on the development of palladium-catalyzed alkene hydroaminocarbonylation reactions.13 Crucial to the successful realization of these reactions is the formation of palladium-hydride species to initiate the catalysis. Consistent with this concept, we found that hydroxylamine hydrochloride (NH2OH·HCl) or alkylamine hydrochloride salts could act as co-catalysts or additives to generate palladium-hydride species via oxidative addition with Pd(0), leading to the establishment of efficient hydroaminocarbonylation reactions.13 In addition, Beller and coworkers also demonstrated that the alkylamine hydrochloride salts could be directly used as a nitrogen source for palladium-catalyzed hydroaminocarbonylation.12e Inspired by these results, we envisioned that the acidic ammonium salt would be a surrogate of ammonia for establishing an efficient catalytic hydroaminocarbonylation reaction to prepare primary amides. Herein, we report an efficient palladium-catalyzed regioselective hydroaminocarbonylation of alkenes to prepare aliphatic primary amides with NH4Cl as the amino source. The regioselectivity for this reaction is accomplished by the appropriate choice of palladium-catalysts and reaction conditions, and both of the linear and branched primary amides can be constructed in high yields with good regioselectivities (Scheme 1).
 | ||
| Scheme 1 Proposed hydroaminocarbonylation of alkenes to primary amides with NH4Cl. | ||
Results and discussion
Initially, to test whether the key palladium-hydride species could be formed by the reaction of Pd(0) with an acidic ammonium salt, the reaction of Pd(t-Bu3P)3 with NH4Cl was investigated and monitored by NMR. After heating the mixture of Pd(t-Bu3P)2 and NH4Cl in NMP at 120 °C for 30 min, a new signal at −16.7 ppm appeared in the 1H NMR spectra, which corresponded to the Pd–H resonance of HPd(t-Bu3P)2Cl.13c,15 After prolonging the reaction time, the Pd–H resonance signal increased accordingly (see ESI†). These results convincingly supported our hypothesis that palladium-hydride species can be produced by the reaction of Pd(t-Bu3P)2 and NH4Cl.The above results intrigued us to study the catalytic hydroaminocarbonylation of alkenes with NH4Cl as the nitrogen source according to the proposed reaction pathway. On the basis of our previous studies of Pd-catalyzed hydroaminocarbonylation reactions,13 the present reaction was initially investigated with styrene (1a) and NH4Cl in the presence of a catalytic amount of Pd(t-Bu3P)2 under 30 atm of CO in NMP at 120 °C. We were pleased to find that the reaction did indeed proceed leading to the desired primary amides in 95% combined yield with excellent regioselectivity for exclusively giving the branched amide 2a as the main product (Table 1, entry 1). Decreasing the pressure of CO to 10 atm led to a lower reactivity and selectivity (Table 1, entries 1–3). The simple Pd(PPh3)4 could also deliver the desired amide 2a as the main product in excellent yield albeit with a relatively lower regioselectivity. With Pd(t-Bu3P)2 as the catalyst, the reaction time could be shortened to 15 h. Further evaluation of various ammonium salts revealed that the transformation was sensitive to the counter anion of the ammonium salts and found that NH4Cl was a superb ammonium salt for this reaction (see ESI†). After identifying the optimized catalytic system for the synthesis of the branched amide 2a, we next sought to determine whether the regioselectivity could be switched by changing the catalyst precursors and reaction conditions. This proved to be highly successful: using PdI2 as a palladium source and Xantphos as the ligand, the regioselectivity was changed to the opposite direction, favoring the linear amide 3a when the reaction was conducted at 120 °C under 30 atm of CO (Table 1, entry 5). Encouraged by this result, we then checked different bidentate phosphine ligands with distinct bite angles, such as DPEphos, DPPF, and DPPB, and found that Xantphos was the most efficient for giving the linear amide 3a in terms of the reactivity and selectivity (Table 1, entries 6–12). With this optimal catalyst, we further screened other reaction parameters to maximize the efficiency of the reaction. This led to finding that the reaction time could be shortened to 4 hours (Table 1, entry 13). Reducing the CO-pressure resulted in a lower reactivity but the same regioselectivity was kept (Table 1, entries 13–15). Interestingly, while the reaction can still be carried out without Xantphos, a lower yield and regioselectivity were observed, revealing that the ligand is an important factor that contributes to a satisfactory result (Table 1, entry 16). In addition, we examined the effect of the solvent and observed that other solvents provided unsatisfactory results in terms of the reactivity and selectivity.
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Ligand | CO (atm) | Yield (%) |
2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a :3a |
|
a Reaction conditions: 1a (1.0 mmol), NH4Cl (2.0 mmol), [Pd] (5 mol%), NMP (5 mL), 120 °C, and 24 h. The combined yield based on the alkene and the ratio (2a:3a) of the crude reaction mixture was determined by GC and GC-MS analysis using n-cetane as the internal standard.
b [Pd] (2 mol%), ligand (2.5 mol%), NMP (3 mL), 120 °C, and 24 h.
c 4 h.
|
|||||
| 1 | Pd(t-Bu3P)2 | — | 30 | 95 | 96:4 |
| 2 | Pd(t-Bu3P)2 | — | 20 | 96 | 88:12 |
| 3 | Pd(t-Bu3P)2 | — | 10 | 82 | 85:15 |
| 4 | Pd(PPh3)4 | — | 30 | 90 | 81:19 |
| 5b | Pdl2 | Xantphos | 30 | 86 | 20:80 |
| 6b | Pdl2 | DPEphos | 30 | 34 | 22:78 |
| 7b | Pdl2 | DPPF | 30 | 25 | 36:64 |
| 8b | Pdl2 | DPPH | 30 | 27 | 51:49 |
| 9b | Pdl2 | DPPPen | 30 | 23 | 21:79 |
| 10b | Pdl2 | DPPB | 30 | <5 | — |
| 11b | Pdl2 | DPPE | 30 | <5 | — |
| 12b | Pdl2 | DPPM | 30 | <5 | — |
| 13b,c | Pdl2 | Xantphos | 30 | 90 | 17:83 |
| 14b,c | Pdl2 | Xantphos | 20 | 65 | 17:83 |
| 15b,c | Pdl2 | Xantphos | 10 | 60 | 17:83 |
| 16b,c | Pdl2 | — | 30 | 20 | 32:68 |
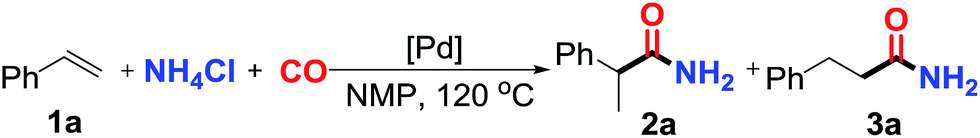
With the optimized reaction conditions in hand, we first examined the substrate scope for the synthesis of branched amides under the catalysis of Pd(t-Bu3P)2, and the results are summarized in Table 2. A variety of styrenes bearing mono-substitutents on the aryl ring were initially surveyed. Both electron-donating and -withdrawing groups are compatible with this transformation, and a series of functional groups, such as ether, halogen and ketone, are tolerated to give the desired products 2a–2j in moderate to excellent yields with good to excellent regioselectivities. The structure of 2e was confirmed by X-ray crystallographic analysis.16 For the styrenes with disubstituents on the aryl ring, including 3,4-dichlorostyrene and 2,6-dimethylstyrene, the reactions proceed smoothly to generate 2k and 2l in good yields with good selectivities. The reactions of 2-vinylnaphthalene and 1-vinylnaphthalene deliver 2m and 2n in 71% and 68% yields, respectively. In addition, the reaction of 9-vinylphenanthrene also provides product 2o in 68% yield with good regioselectivity. Hydroaminocarbonylation of an estrone derivative alkene bearing a steroid scaffold was also conducted to produce the linear amide 2p in 45% yield with a tolerating ketone group. This transformation indicated that the reaction system is amenable to functionalization and modification of complex alkenes bearing the skeleton of natural products, and shows high potential applications in biological evaluation. Unfortunately, aliphatic alkenes were less reactive and only 30% yield was obtained for allylbenzene when the reaction was conducted under 50 atm of CO for 24 h, which may be attributed to faster β-hydride elimination and slower CO insertion for aliphatic alkenes.

Inspired by the above results, we turned our attention to the investigation of the substrate scope for the synthesis of primary linear amides under the catalysis of PdI2/Xantphos in the presence of 30 atm CO pressure (Table 3). To our delight, a range of styrenes and vinylnaphthalenes are also suitable to react with NH4Cl and CO under the optimized reaction conditions, and a series of functional groups, such as ethers, halides and ketones, can survive to give the desired linear amides 3a–3l in 67–86% isolated yields with good regioselectivities. Notably, the regioselectivity is changed dramatically depending on the substitution. For instance, while the reaction took place with a high selectivity with substrates bearing a methyl group at the ortho-position (Table 3, entry 4), the selectivity was lower when it was located at the para- or meta- position (Table 3, entries 2 and 3). Similar excellent regioselectivities and good yields were observed for the reactions of 2-chlorostyrene, 2,6-dimethylstyrene and 1-vinylnaphthalene (Table 3, entries 9, 11 and 14). In addition to aryl alkenes, simple unactivated aliphatic alkenes are also suitable substrates for this reaction to give the corresponding linear amides in good yields and regioselectivities (Table 3, entries 15–19). Importantly, this reaction is not limited to simple aliphatic alkenes. For instance, functionalized aliphatic alkenes, bearing chlorine, bromine, ether, ester and phthalimide, can be successfully converted to the desired linear amides 3t–3y in satisfactory yields and good regioselectivities. An estrone derivative was employed to deliver the linear amide 2p in 65% yield under the catalysis of the Pd(COD)Br2/Xantphos system. The solid structure of 3h was further unambiguously confirmed by X-ray crystallographic analysis.16
|
|
||||
|---|---|---|---|---|
| Entry | R | Product | Yield (%) |
L:B |
|
a Reaction conditions: 1 (1.0 mmol), NH4Cl (2.0 mmol), Pdl2 (2 mol%), Xantphos (2.5 mol%), NMP (3 mL), CO (30 atm), 120 °C, and 4 h. The isolated yield based on the alkene and the ratio (L:B) of the crude reaction mixture was determined by GC and GC-MS.
b Pd(COD)Br2 (5 mol%), Xantphos (6 mol%), NMP (5 mL), 120 °C, and 24 h.
|
||||
| 1 | C6H5 | 3a | 72 | 83:17 |
| 2 | 4-CH3C6H4 | 3b | 73 | 80:20 |
| 3 | 3-CH3C6H4 | 3c | 69 | 82:18 |
| 4 | 2-CH3C6H4 | 3d | 86 | 94:6 |
| 5 | 4-t-BuC6H4 | 3e | 77 | 83:17 |
| 6 | 4-CH3OC6H4 | 3f | 69 | 79:21 |
| 7 | 4-FC6H4 | 3g | 67 | 82:18 |
| 8 | 4-ClC6H4 | 3h | 67 | 82:18 |
| 9 | 2-ClC6H4 | 3i | 77 | 95:5 |
| 10 | 4-CH3COC6H4 | 3j | 73 | 81:19 |
| 11 | 2,6-(CH3)2C6H3 | 3k | 85 | 96:4 |
| 12 | 3,4-Cl2C6H3 | 3l | 69 | 85:15 |
| 13 | 2-Naphthyl | 3m | 78 | 81:19 |
| 14 | 1-Naphthyl | 3n | 82 | 96:4 |
| 15 | t-Bu | 3o | 74 | >99:1 |
| 16 | C6H5CH2 | 3p | 54 | 79:21 |
| 17 | CyCH2 | 3q | 56 | 91:9 |
| 18 | CH3(CH2)9 | 3r | 59 | 89:11 |
| 19 | CH3(CH2)11 | 3s | 56 | 88:12 |
| 20 | Cl(CH2)4 | 3t | 63 | 81:19 |
| 21 | Br(CH2)5 | 3u | 51 | 81:19 |
| 22 | MeOOC(CH2)8 | 3v | 55 | 87:13 |
| 23 | C6H5O(CH2)2 | 3w | 63 | 89:11 |

|
||||

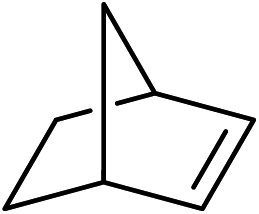
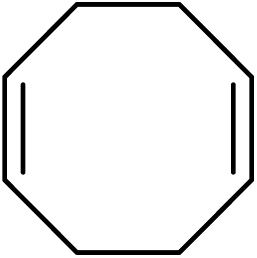
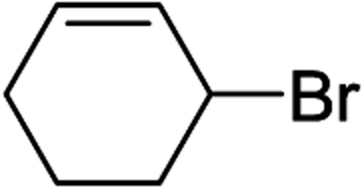
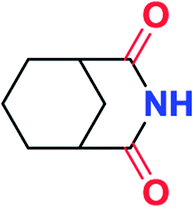
To further examine and expand the scope of the reaction, the use of disubstituted alkenes was considered as they allow access to considerably more structurally diverse amides but pose a greater challenge. Indeed, the reaction of α-methyl styrene failed to yield the desired amide with a low conversion under the catalysis of the Pd(t-Bu3P)2 or PdI2/Xantphos system. However, a slight change from PdI2 to Pd(COD)Br2 led to a successful hydroaminocarbonylation, allowing for a significant expansion of the scope (Table 4). Under these reaction conditions, the desired reaction was amenable to a broad range of α-methyl styrenes, 2-(prop-1-en-2-yl)naphthalene as well as 2-(prop-1-en-2-yl)thiophene, giving a range of primary amides in good to excellent yields with complete regioselectivities (Table 4, entries 1–9). Aside from simple α-methyl styrenes, many other α-substituted aromatic alkenes with different substituents at the α-position are also compatible with this novel reaction, generating the corresponding primary amides in 51–69% yields (5j–5l). The structure of the amide 5l was also confirmed by single-crystal X-ray crystallographic analysis.16 In addition, cyclic alkenes, such as norbornene and cyclohexene, can be smoothly transferred to the corresponding primary amides 5m and 5o in 77% and 79% yield, respectively. Notably, the 1,5-cyclooctadiene is also readily converted to the desired primary amide 5n in good yield with one double bond remaining. Interestingly, double carbonylation occurs when 3-bromocyclohex-1-ene is subjected to the present reaction conditions, providing the corresponding imide 5p in 65% yield.
|
|
||||
|---|---|---|---|---|
| Entry | R 1 | R 2 | Product | Yield (%) |
| a Reaction condition: 4 (1.0 mmol), NH4Cl (2.0 mmol), CO (30 atm), NMP (5 mL), 120 °C, and 24 h. The isolated yield. b 48 h. c dr of the crude reaction mixture determined by GC and GC-MS. | ||||
| 1 | C6H5 | CH3 | 5a | 82 |
| 2 | 4-CH3C6H4 | CH3 | 5b | 71 |
| 3 | 4-i-BuC6H4 | CH3 | 5c | 74 |
| 4 | 4-CH3OC6H4 | CH3 | 5d | 90 |
| 5 | 2-CH3OC6H4 | CH3 | 5e | 80 |
| 6 | 2-ClC6H4 | CH3 | 5f | 71 |
| 7 | 3,5-(CF3)2C6H3 | CH3 | 5g | 73 |
| 8 | 2-Naphthyl | CH3 | 5h | 71 |
| 9 | 2-Thiophene | CH3 | 5i | 52 |
| 10b | C6H5 | i-Pr | 5j | 69 |
| 11 | C6H5 | (CH2)7CH3 | 5k | 50 |
| 12b | C6H5 | C6H5 | 5l | 51 |
| 13 |
|
|
5m | 77(86:14)c |
| 14 |
|

|
5n | 62 |
| 15 |

|

|
5o | 79 |
| 16 |
|
|
5p | 65 |

The hydroaminocarbonylation of simple ethylene to propionamide (6) with NH4Cl on a 20 mmol scale was successfully realized in the presence of 0.01 mol% of Pd(t-Bu3P)2 (18% yield based on NH4Cl, ton = 1800). In addition, with Pd(CH3CN)2Cl2/Xantphos as the catalyst, the secondary amide N-propionylpropionamide (7) can be effectively assembled in 95% yield. Furthermore, the 15N-labeled primary amide (2m) was obtained when 15NH4Cl was utilized as the coupling partner, which provides a convenient method to incorporate 15N into amides. Finally, to demonstrate scalability, the reaction was performed on a preparative scale (15 mmol) with 1-(tert-butyl)-4-vinylbenzene (1e) as a substrate. The target reaction proceeded smoothly at a lower catalyst loading (0.1 mol%), affording the desired branched primary amide 2e in 81% yield with excellent regioselectivity (Scheme 2, eqn (4)).17
 | ||
| Scheme 2 Synthetic applications. | ||
To better understand the mechanism, a series of control experiments were performed. Initially, HPd(t-Bu3P)2Cl was prepared according to the reported method and utilized as a catalyst for the standard reaction.15 As expected, excellent reactivity and selectivity were observed for the desired reaction (Scheme 3, eqn (1)), suggesting the plausible intermediacy of palladium-hydride in the catalytic cycle. Moreover, the putative acylpalladium intermediate 9 was prepared via the reaction of 3-phenylpropanoyl chloride 8 with Pd(t-Bu3P)2 (Scheme 3, eqn (2)). The structure of complex 9 was confirmed by X-ray crystallography.16 This complex adopts a monomeric T-shaped geometry at palladium, with the acyl ligand trans to the open coordination site, as has been reported for related aroyl-palladium complexes.18 With the acylpalladium complex 9 in hand, we proceeded to execute a set of experiments to elucidate whether the acylpalladium 9 could be converted to an amide and how the NH2-moiety was installed into the desired amide from NH4Cl. The treatment of acylpalladium 9 with two equivalents of NH4Cl in the presence of CO in NMP at 120 °C afforded the linear amide 3a and the branched amide 2a in good yields (Scheme 3, eqn (3)). However, no desired amide was detected, but styrene was formed when the same reaction was conducted in the absence of CO under other identical reaction conditions (Scheme 3, eqn (4)). These results indicate that the acylpalladium 9 can be converted to the desired amides and CO is essential for facilitating the process.18a The formation of the branched amide 2a from the linear acylpalladium 9 suggests that CO-deinsertion/insertion and β-hydride elimination/hydropalladation occur and are reversible. However, the lower selectivity for getting the branched amide 2a from the linear acylpalladium 9 indicated that the reverse reaction is slow under the reaction conditions, which was further supported by the deuterium studies, in which only partial D-scrambling was observed (Scheme 3, eqn (7) and (8)). As expected, the acylpalladium 9 can catalyze the present hydroaminocarbonylation to give the desired product in high yield under the standard conditions, suggesting the plausible intermediacy of the acylpalladium 9 in the catalytic cycle (Scheme 3, eqn (5)). Further control experiments disclosed that the desired amide 3a could be facilely produced by the reaction of the acid chloride 8 with NH4Cl in NMP at 120 °C (Scheme 3, eqn (6)).19 This result together with previous reports18 that acid chloride could be generated by the reductive elimination of aroyl palladium complexes in the presence of CO suggested two possible pathways for the formation of amide from the acylpalladium 9: (1) the NH4Cl directly reacts with the acylpalladium 9 and (2) acid chloride is formed firstly from the acylpalladium 9, which then reacts with NH4Cl to generate the desired amide. In both pathways NMP may function as a base to capture the produced HCl (see ESI†).
 | ||
| Scheme 3 Preliminary mechanistic studies. | ||
To distinguish the two possibilities, kinetic reaction profiles for the two stoichiometric reactions of NH4Cl with the acylpalladium complex (9) (eqn (3)) or the acid chloride 8 (eqn (6)) were investigated. The rate of reaction in eqn (3) is faster than that in eqn (6) (Fig. 1). In principle, if a reaction is composed of multiple steps, the observed global kinetic rate should be slower than the rate of each step, or at least equal to the slowest step if it exists. If the desired amide is generated from the acid chloride, eqn (6) should be part of the reaction pathway, and the observed rate of reaction in eqn (6) should be faster or at least equal to that in eqn (3). However, opposite results were gained, which excluded acid chloride as an intermediate in the reaction of eqn (3), and supported that the desired amide was most likely generated by the direct reaction of NH4Cl with the acylpalladium complex in NMP.
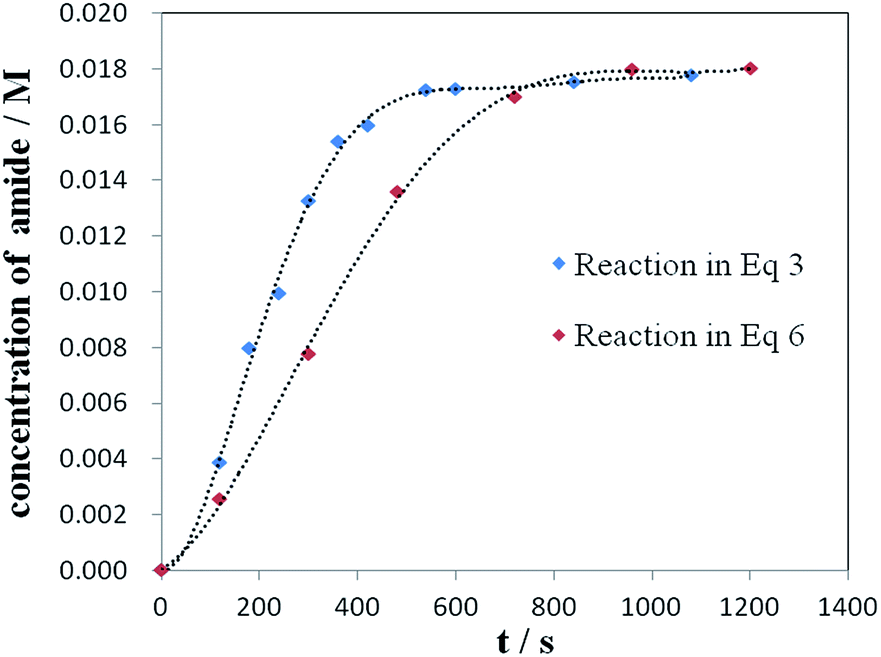 | ||
| Fig. 1 Kinetic plots of the reaction in eqn (3) [acylpalladium 9 (0.2 mmol), NH4Cl (0.4 mmol), CO (10 atm), and NMP (10 mL) at 120 °C] and the reaction in eqn (6) [8 (0.2 mmol), NH4Cl (0.4 mmol), and NMP (10 mL) at 120 °C]. | ||
While a precise reaction mechanism is not yet clear at the present stage, the most plausible mechanism in line with our experimental results and previous reports is illustrated in Fig. 2. First, an oxidative addition of NH4Cl to Pd(0) will produce the key palladium-hydride species. Reversible coordination and insertion of alkene into the palladium-hydride yield the Pd-alkyl intermediate A or C, which undergoes CO insertion to form B or D, respectively. Consequently, the resultant acylpalladium species B and D directly react with NH4Cl in the presence of CO to generate the desired amides. The key palladium-hydride species is simultaneously generated to enter the next catalytic cycle and the released HCl might be trapped by NMP solvent (see ESI†) to finish the catalytic cycle. The regioselectivity was found to be highly influenced by the ligand used and the branched amide is exclusively formed for the aromatic alkenes with monodentate ligands. The phenyl group can stabilize the palladium intermediate Cvia delocalization to form an η3-benzyl palladium-complex.20t-Bu3P appeared to be highly favorable to such stabilization due to its ability to create a coordinatively unsaturated palladium complex arising from its size and donor ability, thus facilitating the formation of C and/or the subsequent migratory insertion process to predominately give the branched amide 2. In contrast, bidentate Xantphos appeared to favor the linear palladium intermediate A likely due to the steric hindrance in the corresponding palladium catalyst created by the bidentate diphosphine ligand, consequently facilitating the formation of the linear amide.
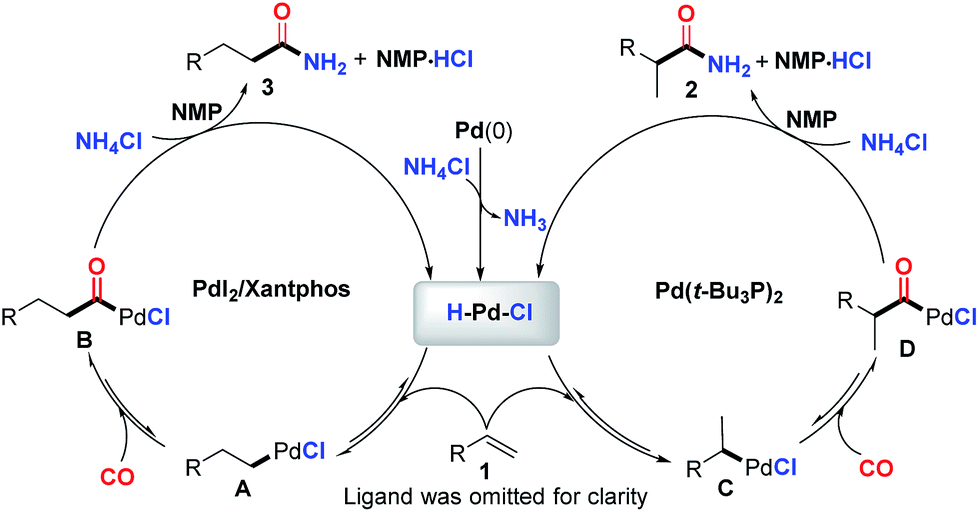 | ||
| Fig. 2 Plausible reaction mechanism. | ||
Conclusions
In summary, we have successfully developed a practical palladium-catalyzed protocol for the synthesis of primary aliphatic amides via hydroaminocarbonylation of alkenes with NH4Cl as a surrogate of ammonia in the presence of CO. A wide range of linear or branched primary amides have been obtained in high yields with good to excellent regioselectivities, which represents the first example of the direct conversion of NH4Cl to primary aliphatic amides in the absence of base. Apart from the synthetic value of this transformation, important mechanistic evidence regarding the origin of the palladium-hydride and the pathway for incorporation of an NH2-moiety from NH4Cl into the amide was provided. These results suggest that the reaction proceeds through formation of palladium-hydride species generated by oxidative addition of NH4Cl to Pd(0) and the acylpalladium species is capable of directly capturing the NH2-moiety from the ammonium salt under the assistance of CO in NMP, and has promise as a valuable strategy for utilizing ammonium salts as practical alternatives to gaseous ammonia in a variety of C–N bond-forming manifolds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (21672199 and 21702197), CAS Interdisciplinary Innovation Team, the Fundamental Research Funds for the Central Universities and the Anhui provincial Natural Science Foundation (1708085MB28).Notes and references
- (a) J. R. J. LeBlanc, S. Madhavan, R. E. Porter and P. Kellogg, Encyclopedia of Chemical Technology, 2nd edn, Wiley, 1963, vol. 2 Search PubMed; (b) Y. Aubin, C. Fischmeister, C. M. Thomas and J.-L. Renaud, Chem. Soc. Rev., 2010, 39, 4130 RSC; (c) J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 86 CrossRef CAS PubMed; (d) J. Kim, H. J. Kim and S. Chang, Eur. J. Org. Chem., 2013, 3201 CrossRef CAS; (e) H. Kim and S. Chang, Acc. Chem. Res., 2017, 50, 482 CrossRef CAS PubMed.
- (a) T. Gross, A. M. Seayad, M. Ahmad and M. Beller, Org. Lett., 2002, 4, 2055 CrossRef CAS PubMed; (b) S. Ogo, K. Uehara, T. Abura and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 3020 CrossRef CAS PubMed.
- B. Zimmermann, J. Herwig and M. Beller, Angew. Chem., Int. Ed., 1999, 38, 2372 CrossRef CAS PubMed.
- V. Lavallo, G. D. Frey, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 5224 CrossRef CAS PubMed.
- (a) M. J. Pouy, A. Leitner, D. J. Weix, S. Ueno and J. F. Hartwig, Org. Lett., 2007, 9, 3949 CrossRef CAS PubMed; (b) T. Nagano and S. Kobayashi, J. Am. Chem. Soc., 2009, 131, 4200 CrossRef CAS PubMed.
- (a) X.-F. Wu, H. Neumann and M. Beller, Chem.–Asian. J., 2010, 5, 2168 CrossRef CAS PubMed; (b) X.-F. Wu, J. Schranck, H. Neumann and M. Beller, ChemCatChem, 2012, 4, 69 CrossRef CAS; (c) X.-F. Wu, H. Neumann and M. Beller, Chem.–Eur. J., 2012, 18, 419 CrossRef CAS PubMed; (d) K. M. Driller, H. Klein, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 6041 CrossRef CAS PubMed.
- (a) Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 10028 CrossRef CAS PubMed; (b) D. S. Surry and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 10354 CrossRef CAS PubMed; (c) T. Schulz, C. Torborg, S. Enthaler, B. Schäffner, A. Dumrath, A. Spannenberg, H. Neumann, A. Börner and M. Beller, Chem.–Eur. J., 2009, 15, 4528 CrossRef CAS PubMed; (d) G. D. Vo and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11049 CrossRef CAS PubMed.
- (a) D. M. Roundhill, Chem. Rev., 1992, 92, 1 CrossRef CAS; (b) J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080 CrossRef CAS PubMed.
- (a) J. Kim and S. Chang, Chem. Commun., 2008, 3052 RSC; (b) R. A. Green and J. F. Hartwig, Org. Lett., 2014, 16, 4388 CrossRef CAS PubMed.
- For recent reviews on the synthesis of amides via carbonylation, see: (a) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986 RSC; (b) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS PubMed; (c) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041 CrossRef CAS PubMed.
- (a) P. Pino and P. Paleari, Gazz. Chim. Ital., 1951, 81, 64 Search PubMed; (b) P. Pino and R. Magri, Chim. Ind., 1952, 34, 511 CAS; (c) G. Natta, P. Pino and R. Ercoli, J. Am. Chem. Soc., 1952, 74, 4496 CrossRef CAS; (d) W. Reppe and H. Main, Chem. Abstr., 1953, 47, 5428 Search PubMed; (e) B. F. Crowe and O. C. Elmer, Chem. Abstr., 1956, 50, 16849 Search PubMed; (f) T. J. Kealy and R. E. Benson, J. Org. Chem., 1961, 26, 3126 CrossRef CAS; (g) A. Striegler and J. Weber, J. Prakt. Chem., 1965, 29, 281 CrossRef CAS; (h) N. S. Imyanitov and D. M. Rudkvsku, Chem. Abstr., 1967, 66, 10530 Search PubMed; (i) B. K. Nefedov, N. S. Sergeeva and Y. T. Eidus, Chem. Abstr., 1975, 83, 205731 Search PubMed; (j) Y. Tsuji, T. Ohsumi, T. Kondo and Y. Watanabe, J. Organomet. Chem., 1986, 309, 333 CrossRef CAS; (k) S. I. Lee, S. U. Son and Y. K. Chung, Chem. Commun., 2002, 1310 RSC.
- (a) X. Fang, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14089 CrossRef CAS PubMed; (b) H. Liu, N. Yan and P. J. Dyson, Chem. Commun., 2014, 50, 7848 RSC; (c) X. Fang, H. Li, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2014, 136, 16039 CrossRef CAS PubMed; (d) H. Li, K. Dong, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10239 CrossRef CAS PubMed; (e) J. Liu, H. Li, A. Spannenberg, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 13544 CrossRef CAS PubMed; (f) T. Xu, F. Sha and H. Alper, J. Am. Chem. Soc., 2016, 138, 6629 CrossRef CAS PubMed.
- (a) G. Zhang, B. Gao and H. Huang, Angew. Chem., Int. Ed., 2015, 54, 7657 CrossRef CAS PubMed; (b) G. Zhang, X. Ji, H. Yu, L. Yang, P. Jiao and H. Huang, Tetrahedron Lett., 2016, 57, 383 CrossRef CAS; (c) Y. Hu, Z. Shen and H. Huang, ACS Catal., 2016, 6, 6785 CrossRef CAS.
- (a) F. Matsuda, Chem. Technol., 1977, 7, 306 CAS; (b) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC.
- I. D. Hills and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 13178 CrossRef CAS PubMed.
- CCDC 1515391 (2e), 1515399 (3h), 1515405 (5l) and 1552470 (9) contain the supplementary crystallographic data for this paper.†.
- The utility of this catalytic reaction has been further demonstrated by streamlined synthesis of ibuprofen, nitriles and 3,4-dihydroquinolin-2(1H)-one from simple alkenes and NH4Cl. See ESI† for details.
- (a) J. S. Quesnel and B. A. Arndtsen, J. Am. Chem. Soc., 2013, 135, 16841 CrossRef CAS PubMed; (b) S. Korsager, R. H. Taaning and T. Skrydstrup, J. Am. Chem. Soc., 2013, 135, 2891 CrossRef CAS PubMed; (c) J. Tjutrins and B. A. Arndtsen, J. Am. Chem. Soc., 2015, 137, 12050 CrossRef CAS PubMed.
- No reaction occurredwhen the reaction was conducted in many other solvents such as toluene, CH2Cl2, THF, 1,4-dioxane and hexane, suggesting that basic NMP is essential for facilitating the reaction by capturing the released HCl (see ESI† for details).
- (a) W. Ren, W. Chang, Y. Wang, J. Li and Y. Shi, Org. Lett., 2015, 17, 3544 CrossRef CAS PubMed; (b) J. Li, W. Chang, W. Ren, J. Dai and Y. Shi, Org. Lett., 2016, 18, 5456 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1515391 (2e), 1515399 (3h), 1515405 (5l) and 1552470 (9). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04054g |
| This journal is © The Royal Society of Chemistry 2018 |