
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nitric oxide activatable photosensitizer accompanying extremely elevated two-photon absorption for efficient fluorescence imaging and photodynamic therapy†
Wenbo
Hu‡
 ab,
Meng
Xie
a,
Hui
Zhao
a,
Yufu
Tang
a,
Song
Yao
c,
Tingchao
He
c,
Chuanxiang
Ye
c,
Qi
Wang
b,
Xiaomei
Lu
b,
Wei
Huang
bd and
Quli
Fan
*a
ab,
Meng
Xie
a,
Hui
Zhao
a,
Yufu
Tang
a,
Song
Yao
c,
Tingchao
He
c,
Chuanxiang
Ye
c,
Qi
Wang
b,
Xiaomei
Lu
b,
Wei
Huang
bd and
Quli
Fan
*a
aKey Laboratory for Organic Electronics and Information Displays, Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing University of Posts & Telecommunications, Nanjing 210023, China. E-mail: iamqlfan@njupt.edu.cn
bKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University (NanjingTech), Nanjing 211816, China. E-mail: wei-huang@njtech.edu.cn
cKey Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Physics Science & Technology, Shenzhen University, Shenzhen 518060, China
dShaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), Xi’an 710072, China
First published on 27th November 2017
Abstract
Elevated nitric oxide (NO) levels perform an important pathological role in various inflammatory diseases. Developing NO-activatable theranostic materials with a two-photon excitation (TPE) feature is highly promising for precision imaging and therapy, but constructing such materials is still a tremendous challenge. Here, we present the first example of a NO-activatable fluorescent photosensitizer (DBB-NO) accompanying extremely NO-elevated two-photon absorption (TPA) for efficient fluorescence imaging and photodynamic therapy (PDT). Upon responding to NO, DBB-NO shows not only a remarkably enhanced fluorescence quantum yield (ΦF, 0.17% vs. 9.3%) and singlet oxygen quantum yield (ΦΔ, 1.2% vs. 82%) but also an extremely elevated TPA cross-section (δ, 270 vs. 2800 GM). Simultaneous enhancement of ΦΔ, ΦF and δ allows unprecedented two-photon fluorescence brightness (δ × ΦF = 260.4 GM) and two-photon PDT (TP-PDT) efficiency (δ × ΦΔ = 2296 GM) which precedes the value for a commercial two-photon photosensitizer by two orders of magnitude. With these merits, the proof-of-concept applications of NO-activatable two-photon fluorescence imaging and TP-PDT in activated macrophages (in which NO is overproduced) were readily realized. This work may open up many opportunities for constructing two-photon theranostic materials with other pathological condition-activatable features for precise theranostics.
Introduction
Nitric oxide (NO) is an endogenous gaseous cellular messenger molecule which plays a pivotal role in the cardiovascular, nervous, and immune systems for various physiological functions.1 However, an elevated NO level will induce pathological conditions including glaucoma, age-related macular degeneration, and autoimmune and inflammatory diseases.2 This feature makes NO a promising target to develop NO-activatable theranostic materials not only for deep insights into the pathological activities of NO, but also for the innovative therapeutic approach to precisely treat these diseases.3 Despite considerable efforts centred on the design of a NO-activatable fluorescent probe to monitor the NO concentration produced either in vitro or in vivo,4 NO-activatable therapeutic materials have heretofore never been reported.Photodynamic therapy (PDT), whereby a cytotoxic singlet oxygen (1O2) is generated by a photosensitizer (PS), is a clinically approved yet non-invasive therapeutic approach.5 A particular interest of novel PDT is to develop a fluorescent PS (FPS) as a theranostic material for simultaneous fluorescence imaging and PDT,6 which intrinsically circumvents the complicated fabrication procedure of the traditional theranostic materials. Recently, FPSs that can only be activated in pathological conditions have become very appealing for precise imaging and selective killing of target cells.7 The design of the activatable FPS generally requires the conjugation of a stimulus-responsive quencher with the FPS to switch off its fluorescence and PDT ability via intramolecular photoinduced electron transfer (PeT).7 To ensure the PeT process, the energy level alignment between the quencher and the FPS is of particular importance. However, the energy level of an available NO-responsive moiety, such as typical o-phenylenediamine (OPD), can align only to those FPSs with ultraviolet (UV) or visible light absorption,8 which is apparently unfavorable for theranostics in deep tissues. One attractive approach to address this problem is to make use of a two-photon absorbing FPS (TP-FPS),9 which can not only ensure the required energy level alignment but also permit a better penetration depth and spatial selectivity that is unattainable with traditional one-photon excitation.10 Although conceptually impressively, the ultralow two-photon absorption cross-section (δ) of the available TP-FPS with values of 1–100 GM (GM: Goeppert–Mayer units) makes two-photon excited PDT (TP-PDT) in practical applications more challenging.11
Herein, we present a rational design to construct a NO-activatable TP-FPS with extremely NO-elevated δ to realize efficient TP-imaging and TP-PDT (Scheme 1). We coupled a zwitterionic bis(phenylethynyl)benzene derivative (DBB) with OPD through a typical amidation reaction to form the target TP-FPS (DBB-NO). Such a molecule design depends on the following considerations. First, the bis(phenylethynyl)benzene-based conjugated backbone, owing to its excellent absorption in the UV region,12 ensures a desirable energy level alignment with OPD for efficient PeT to maintain quenched fluorescence and PDT. Second, our previous work demonstrates that bis(phenylethynyl)benzene with zwitterionic groups exhibits an ultrahigh δ and 1O2 quantum yield (ΦΔ).13 Therefore, we envision that the release of zwitterionic DBB after the cleavage of OPD from DBB-NO by NO can simultaneously switch on the δ and ΦΔ, both of which are beneficial for realizing efficient TP-PDT. Third, coupling an amino group of OPD with the carboxyl group within DBB can improve the selectivity of DBB-NO to NO, effectively avoiding the “false positive” fluorescence signals or unexpected phototoxicity toward healthy cells. Finally, DBB-NO shows an extremely elevated δ (270 vs. 2800 GM) and ΦΔ (2.2% vs. 82%) as well as a slightly enhanced fluorescence quantum yield (ΦF, 0.17% vs. 9.3%) upon responding to NO, thus enabling the first example of the proof-of-concept applications of NO-activatable TP-imaging and TP-PDT in activated macrophages (in which NO is overproduced) and even in a more complex inflamed mouse model.
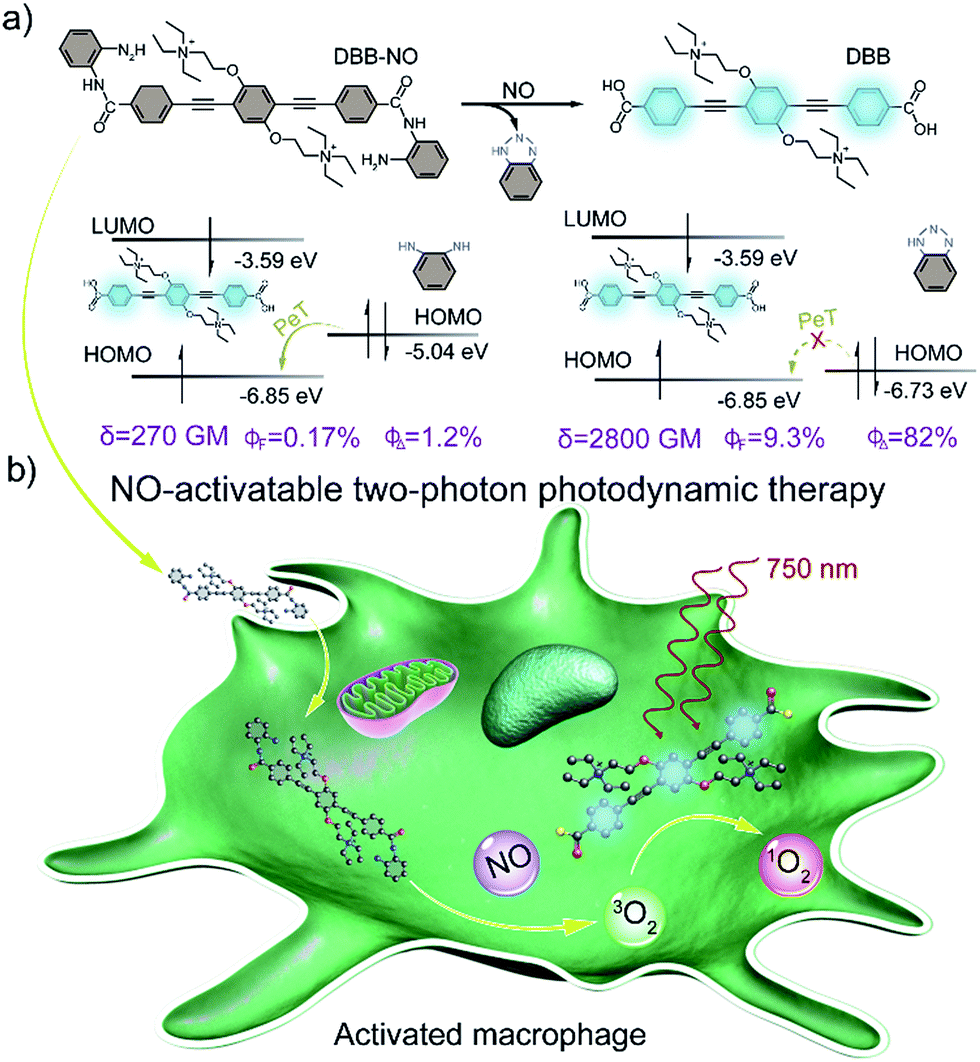 | ||
| Scheme 1 (a) Design principle and molecular structure of the NO-activatable TP-FPS (DBB-NO). LUMO, lowest unoccupied molecular orbital; HOMO, highest occupied molecular orbital; PeT, photoinduced electron transfer; δ, TPA cross-section; ΦF, absolute fluorescence quantum yield; ΦΔ, singlet oxygen quantum yield. (b) Schematic illustration of the NO-activatable TP-FPS for TP-imaging and TP-PDT in an activated macrophage (in which NO is upregulated). | ||
Results and discussion
Design and characterizations
The synthetic route to the NO-activatable TP-FPS of DBB-NO was presented in Scheme S1.† Built on the abovementioned considerations, we first synthesized the zwitterionic DBB structure with an end-capped carboxyl group (Scheme 1) as a potential TP-FPS. Then to avoid the unwanted response of the OPD-based probe toward other competitive species including ascorbic acid (AA), dehydroascorbic acid (DHA) and methylglyoxal (MGO) that have been observed in other reports,4a,14 the carboxyl group of DBB was directly coupled with an amino group of OPD through a typical amidation reaction. This is because OPD via this kind of connection will lose reaction ability with other competitive species.15 The detailed synthetic procedures, 1H NMR, 13C NMR, and high-resolution mass spectrum (HRMS) are given in ESI (Scheme S1 and Fig. S1–S9†).Spectral response of DBB-NO to NO
The photophysical properties of DBB-NO were studied in pH 7.4 phosphate buffer saline (PBS) solution (without other specifications, all the spectra were recorded in this solution). As expected, DBB-NO displays a broadband one-photon absorption spectrum with an excellent molar extinction coefficient (ε = 1.3 × 105 M−1 cm−1) at 361 nm (Fig. S10†) and a very weak fluorescence signal centred at 415 nm with an ΦF of 0.17%. We ascribe this low ΦF to the intramolecular PeT from the HOMO of OPD to the HOMO of DBB (Scheme 1), which significantly quenches the excited singlet exciton of DBB and thus results in a low ΦF. Upon addition of NO (DEA·NONOate was used as the source of NO), the absorption maximum remained almost unchanged while the ΦF exhibits an 18-fold enhancement from 0.17% to 9.3% at 415 nm (Fig. 1a). This is because, upon reacting with NO, OPD could be transformed into the benzotriazole unit whose HOMO (−6.73 eV) is considerably close to the HOMO of DBB, which is unfavorable for efficient intramolecular PeT. Meanwhile, the as-produced benzotriazole is a leaving group,16 which disables intramolecular PeT owing to the increased distance with DBB. These results facilitate the competitive radiative transition from the excited singlet state of DBB into the ground state for fluorescence emission. Fig. 1b presents the time-dependent fluorescence changes of DBB-NO toward NO. The kinetics profiles indicate a high reaction efficiency of DBB-NO with NO.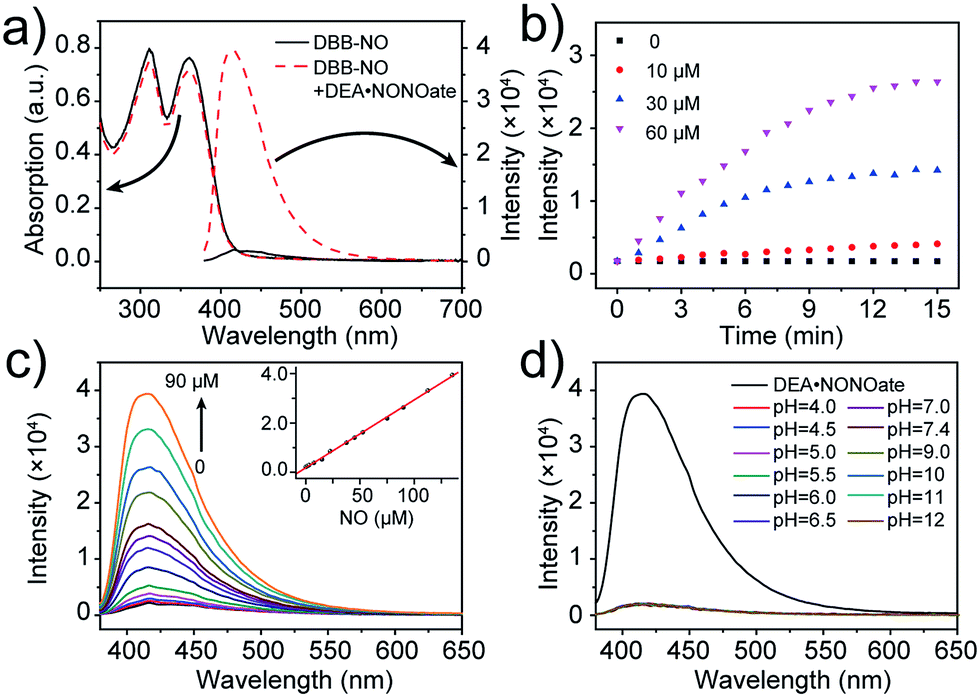 | ||
| Fig. 1 (a) Absorption and fluorescence spectra of DBB-NO (5 μM) in the absence and presence of DEA·NONOate. (b) Kinetic profiles of DBB-NO with varying DEA·NONOate concentrations. (c) Fluorescence spectra of DBB-NO upon the addition of DEA·NONOate (0–90 μM) over a period of 15 min. Inset: the fluorescence intensity at 415 nm against the NO concentration (0–135 μM) is plotted. (d) pH-Dependent fluorescence spectra of DBB-NO. The fluorescence spectrum upon the addition of DEA·NONOate (90 μM) over a period of 15 min was used as a reference. | ||
Next, we performed a fluorescence titration experiment of DBB-NO toward NO. The gradual fluorescence intensity enhancement of DBB-NO at 415 nm exhibits an excellent linear relationship (R2 = 0.998) with the concentration of DEA·NONOate (Fig. 1c). Based on this, the detection limit (S/N = 3) of DBB-NO was determined to be 14 nM, suggesting an excellent sensitivity for fluorescence imaging or activatable PDT.
High selectivity of DBB-NO toward NO
Given the pH variation and existence of other competitive reactive oxygen/nitrogen species (ROS/RNS) in complicated inflammatory conditions, the high selectivity of DBB-NO toward NO is of particular importance to avoid the “false positive” fluorescence signals or unexpected phototoxicity toward healthy cells. The negligible fluorescence of DBB-NO in a wide pH range demonstrates its excellent pH-stability (Fig. 1d). Furthermore, to verify the selectivity of DBB-NO, we screened a wide array of possible competitive reactive oxygen species (H2O2, HClO, ˙OH, and 1O2), nitrogen species (NO2−, ONOO−, and NO3−), biological reductants (cysteine (Cys) and glutathione (GSH)), metal ions (Na+, Ca2+, K+, and Fe3+) and other analytes (AA, DHA, and MGO). As anticipated, all these competitive species do not lead to any obvious fluorescence enhancement (Fig. 2a and S11†), suggesting an outstanding selectivity of DBB-NO toward NO. From these results, it is reasonable to expect that DBB-NO can work in approximate physiological conditions with negligible background fluorescence interference or phototoxicity.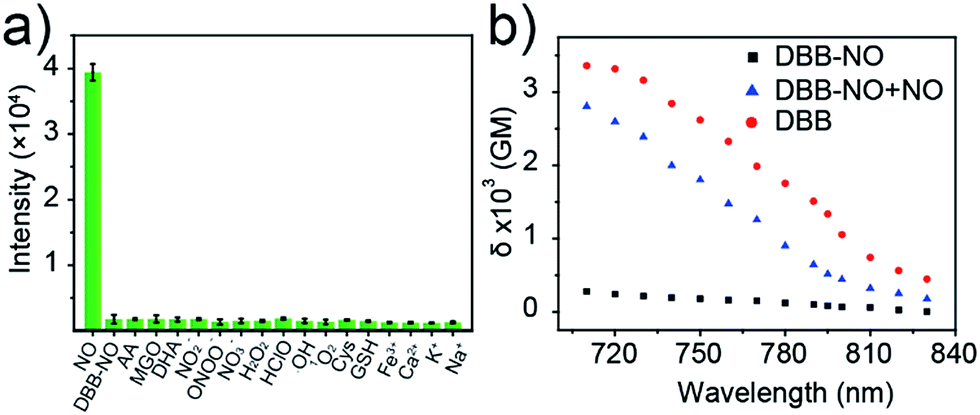 | ||
| Fig. 2 (a) Fluorescence intensity at 415 nm of DBB-NO (5 μM) in the presence of various competitive species after 15 min. (b) Two-photon absorption spectra of DBB-NO in the absence and presence of DEA·NONOate in pH = 7.4 PBS solution. DBB in pH = 7.4 PBS solution was used as a control. | ||
To verify the mechanism for the high selectivity of DBB-NO, we analyzed the reaction of DBB-NO to NO in PBS solution. The HRMS clearly showed that OPD converted into a cyclized benzotriazole unit with a mass peak of m/z 119.13 ([M + H]+) (Fig. S12†), which is in agreement with previous reports.16,17 Meanwhile, the occurrence of the characteristic mass peak of DBB ([M − 2CH3]+m/z 624.88) in HRMS indicates the successful release of the fluorescent and phototoxic DBB from DBB-NO.
Two-photon absorption cross-section spectra
δ is a key parameter to evaluate the TP-imaging quality and TP-PDT efficiency. To be specific, larger δ × ΦF and δ × ΦΔ indicate a better TP-imaging quality and TP-PDT efficiency. Thus, we quantify the δ of DBB-NO in the absence and presence of NO by a typical Z-scan technique (Fig. 2b).18 As anticipated, pure DBB-NO possesses a maximum δ of 270 GM at 710 nm. Although such a value is comparable to the TP-imaging contrast in other reports,19 the ultralow ΦF makes DBB-NO unusable for TP-imaging due to its low brightness (δ × ΦF). In sharp contrast, upon responding to NO, an extremely elevated δ (2800 GM at 710 nm) was observed, which nearly reached the maximum δ of DBB (3300 GM at 710 nm). This remarkably elevated δ of DBB in comparison with that of DBB-NO should be attributed to the formation of the zwitterionic structure of DBB. The zwitterionic structure in a strong polar solvent can provide an additional electric field to energetically increase its δ.13 Such a remarkably elevated δ, accompanying the intrinsic switch on ΦF and ΦΔ, affords DBB-NO enormous potential for effective NO-activatable TP-imaging and TP-PDT purposes.TP-imaging of exogenous and endogenous NO in living cells
Prior to bioapplications, the low cytotoxicity of DBB-NO and excellent biocompatibility of DBB-NO were initially verified as shown in Fig. S13 and 14.† For examining the response of DBB-NO to NO in the cellular environment, TP-imaging of exogenous NO in HeLa cells was performed (Fig. S15†). In the control group, no discernible fluorescence in the absence of DEA·NONOate demonstrates a negligible background fluorescence of DBB-NO. This phenomenon is attributed to the low brightness (δ × ΦF) of DBB-NO. After adding 10.0 equiv. DEA·NONOate, a strong fluorescence was observed in living cells within approximately 5 min. Such remarkable fluorescence enhancement indicates that DBB-NO can detect exogenous NO even in living cells.After that, we further examine the response of DBB-NO to endogenous NO in living cells using the activated RAW 264.7 macrophage. This is because activated macrophages have upregulated endogenous NO (micromolar) in comparison to normal ones. Typically, a normal macrophage could transform into an activated one by the stimulation of lipopolysaccharide (LPS) and interferon-γ (IFN-γ).20 Moreover, limiting the proliferation of the activated macrophages is a promising means of controlling inflammation.21 As shown in Fig. 3, the normal RAW 264.7 macrophages were incubated with DBB-NO and no obvious intracellular fluorescence was detected. In sharp contrast, after the pre-treatment with LPS and IFN-γ, the DBB-NO stained RAW 264.7 macrophages (activated macrophages) exhibit a bright intracellular fluorescence. These results clearly show that DBB-NO is also capable of detecting endogenous NO in living cells. Remarkably enhanced fluorescence within activated macrophages in comparison with that of the normal macrophages should be attributed to the enhanced TP-fluorescence brightness (δ × ΦF) that is induced by the simultaneous enhancement of ΦF and δ. More importantly, the different TP-fluorescence brightness in the abovementioned two kinds of macrophage makes DBB-NO a potential smart probe to distinguish activated macrophages from normal ones, which is of significant importance in the diagnosis and treatment of human diseases with an inflammatory etiology. To highlight the important role of NO in distinguishing activated macrophages from normal ones, the activated RAW 264.7 macrophages were treated with a powerful NO scavenger (N-acetylcysteine, NAC) to eliminate the concentration of endogenous NO. In this context, the intracellular fluorescence of the DBB-NO stained RAW 264.7 macrophage was heavily suppressed. From these results, it is evident that DBB-NO is endogenous NO-activatable and can serve as a smart probe to distinguish activated macrophages from normal ones.
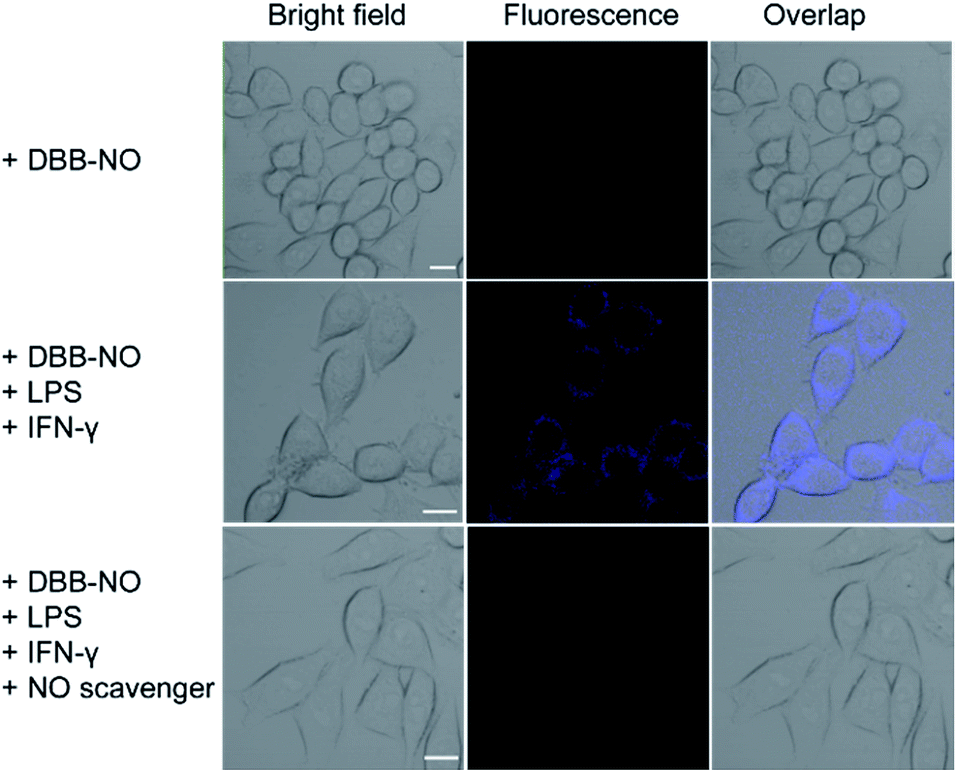 | ||
| Fig. 3 TP-images of RAW 264.7 macrophages stained with DBB-NO in the absence or presence of NO stimulants and inhibitor. Fluorescence was recorded at 410–450 nm upon TPE at 750 nm. Scale bar: 10 μm. | ||
Encouraged by the potential in vitro distinguishing ability of DBB-NO toward activated and normal macrophages, in vivo TP-imaging of NO using DBB-NO was then tested in an inflamed mouse model. Using a typical protocol (details in ESI†), we obtained an inflamed mouse model (Fig. 4a) in which inflammatory lumps (yellow circle) in the right rear paw were observed clearly in comparison with the control group (left rear paw, blue circle). Then, DBB-NO (150 μL, 10 μM) was intravenously injected into the mouse and the mouse was anesthetized after 1 h. The left and right rear paws were sectioned in different glass slides for subsequent TP-imaging. Pleasingly, the blue channel in Fig. 4b exhibits a distinct fluorescence enhancement in inflamed tissues in comparison with that of normal tissues. Meanwhile, immunostaining of histological sections was performed using macrophage marker CD11b to stain the activated macrophage, in which the activated macrophage can transform nonfluorescent CC11b into a red-emissive dye.22 The strong red fluorescence in the red channel suggests the generation of activated macrophages only in the inflamed paw. Moreover, the signals from DBB-NO and CD11b are largely overlapped (merge channel) in inflamed tissues. These results indicate not only the specific in vivo TP-imaging capability of DBB-NO toward endogenous NO but also the in vivo distinguishing ability between activated and normal macrophages.
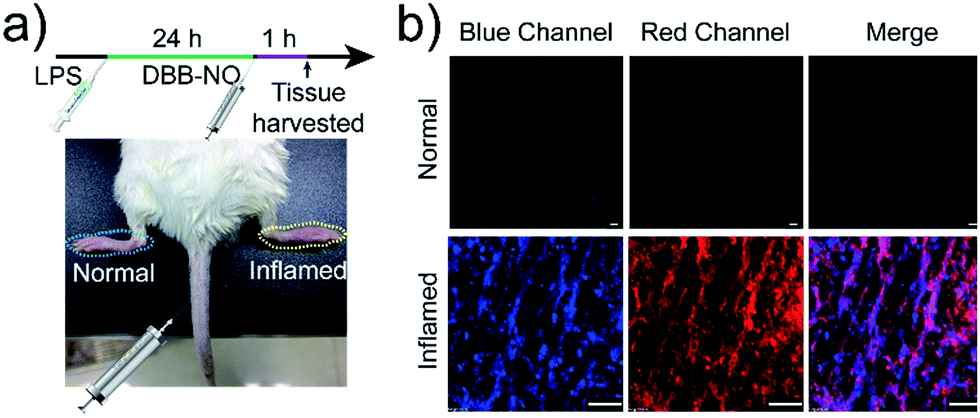 | ||
| Fig. 4 (a) Illustration of the construction of the inflamed mouse model. (b) TP-images of DBB-NO in the normal and inflamed tissues. Blue channel was collected at 410–450 nm upon TPE at 750 nm. Red channel was collected at 570–650 nm upon one-photon excitation at 559 nm. Scale bar: 60 μm. | ||
TP-PDT of activated RAW 264.7 macrophages
As a proof-of-concept for NO-activatable TP-PDT using DBB-NO, we conducted a set of experiments which involved the ROS generation by the well-established photodegradation of anthracene-9,10-diyl-bis-methylmalonate (ADMA) in the presence of DBB-NO and DBB (Fig. 5a).23DBB exhibits an exceptional large ΦΔ (89%) which is superior to that of commercial TMPyP4. The negligible ADMA consumption of DBB-NO demonstrates a completely quenched 1O2 generation (ΦΔ = 1.2%). Upon addition of excess DEA·NONOate into the mixture of DBB-NO and ADMA solution, the ADMA consumption is comparable to that of DBB. After the exclusion of DEA·NONOate interference in ADMA consumption, it is reasonably concluded that the ΦΔ of DBB-NO can recover to 82%. Such simultaneous enhancement of δ and ΦΔ allows an unprecedented large TP-PDT efficiency (δ × ΦΔ = 2296 GM), which precedes the value for the commercial TP-PS by two orders of magnitude.24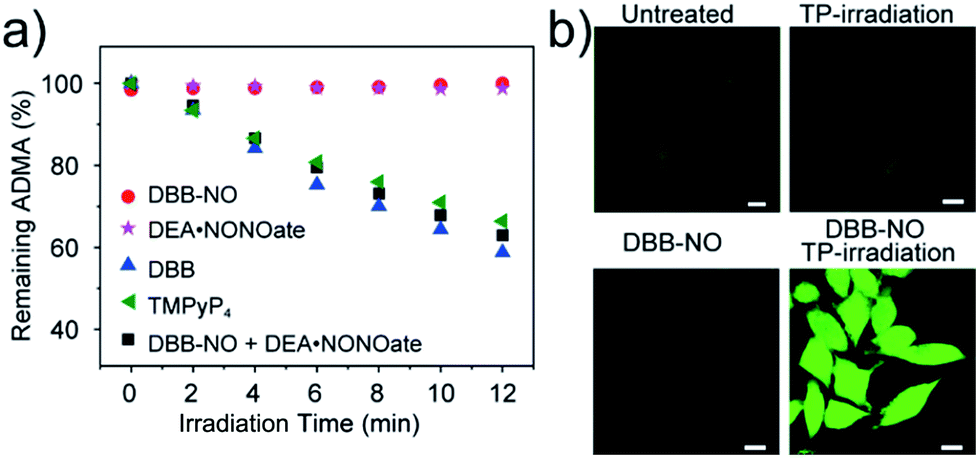 | ||
| Fig. 5 (a) Percentage of unreacted 1O2 chemical trap (ADMA) in the presence of different samples. 5,10,15,20-Tetrakis(1-methyl-4-pyridinio)porphyrin tetra(p-toluenesulfonate) (TMPyP4) is a frequently-used commercial PS with an 1O2 quantum yield of 74%. (b) Detection of intracellular 1O2 generation using a ROS probe (scale bar: 10 μm), the nonfluorescent ROS probe could generate green-fluorescence in the presence of ROS. | ||
For proof-of-concept applications, we further checked NO-activatable 1O2 generation in the activated RAW 264.7 macrophage by fluorescence imaging of a ROS tracker and DBB-NO co-stained macrophage. 2,7-Dichlorofluorescein diacetate (DCFH-DA), as a frequently-used ROS tracker, is nonfluorescent and can be oxidized by ROS into a green-emissive DCF. The weak DCF fluorescence within the activated macrophage of the untreated group should be attributed to the endogenous H2O2, because H2O2 is also overexpressed in the activated macrophage.21 When the activated macrophage was treated with DBB-NO or TP-irradiation alone, there is no obvious enhancement of DCF fluorescence in comparison with the untreated group. However, after the co-treatment of the activated macrophage with DBD-NO and TP-irradiation, a remarkable enhancement of DCF fluorescence was observed, indicating abundant 1O2 generation. Combining endogenous NO detection of DBB-NO in the activated macrophage, we thus concluded that DBB-NO has the capability for NO-activatable ROS (mainly 1O2) generation even in living cells.
The in vitro TP-PDT effect of DBB-NO was then evaluated by assessing the cellular phototoxicity. Here, we utilized the calcein AM (living cell) and propidium iodide (PI, dead cell) cellular viability kit to distinguish the dead cells from the living cells (Fig. 6a). Without the cooperation of DBB-NO and TP-irradiation, there was no obvious red fluorescence for dead cells in the activated macrophage. This result again proves the low dark cytotoxicity of DBB-NO and negligible cytotoxicity caused by TP-irradiation. In the presence of DBB-NO and TP-irradiation, a remarkable red fluorescence in the activated macrophages reveals the significant cellular death. Moreover, the obvious cell shrinkage and the formation of numerous blebs of activated macrophage in the bright field of the activated macrophages also indicate the endogenous NO-activatable TP-PDT with high efficiency. Such a highly efficient TP-PDT toward the activated macrophage should be attributed to the simultaneous enhancement of δ and ΦΔ. In addition, we also compared the cellular phototoxicity of DBB-NO toward the activated macrophage and normal macrophage. Clearly, DBB-NO can only exhibit a highly efficient NO-activatable TP-PDT in the activated macrophage. This is because the normal macrophage is unable to generate sufficient NO to transform the benign DBB-NO into phototoxic DBB. Furthermore, we performed a quantitative evaluation for the TP-PDT of DBB-NO using a MTT (Fig. S16†) assay. The viability of HeLa cells and the normal macrophage incubated with DBB-NO remained nearly 100%, while the viability of the activated macrophages incubated with DBB-NO was increasingly reduced to 19.1%. In addition, a flow cytometric assay (Fig. S17†) also revealed an efficient TP-PDT only in the activated macrophage. From these results, we demonstrated that DBB-NO has an NO-activatable feature for potential TP-PDT in activated macrophages.
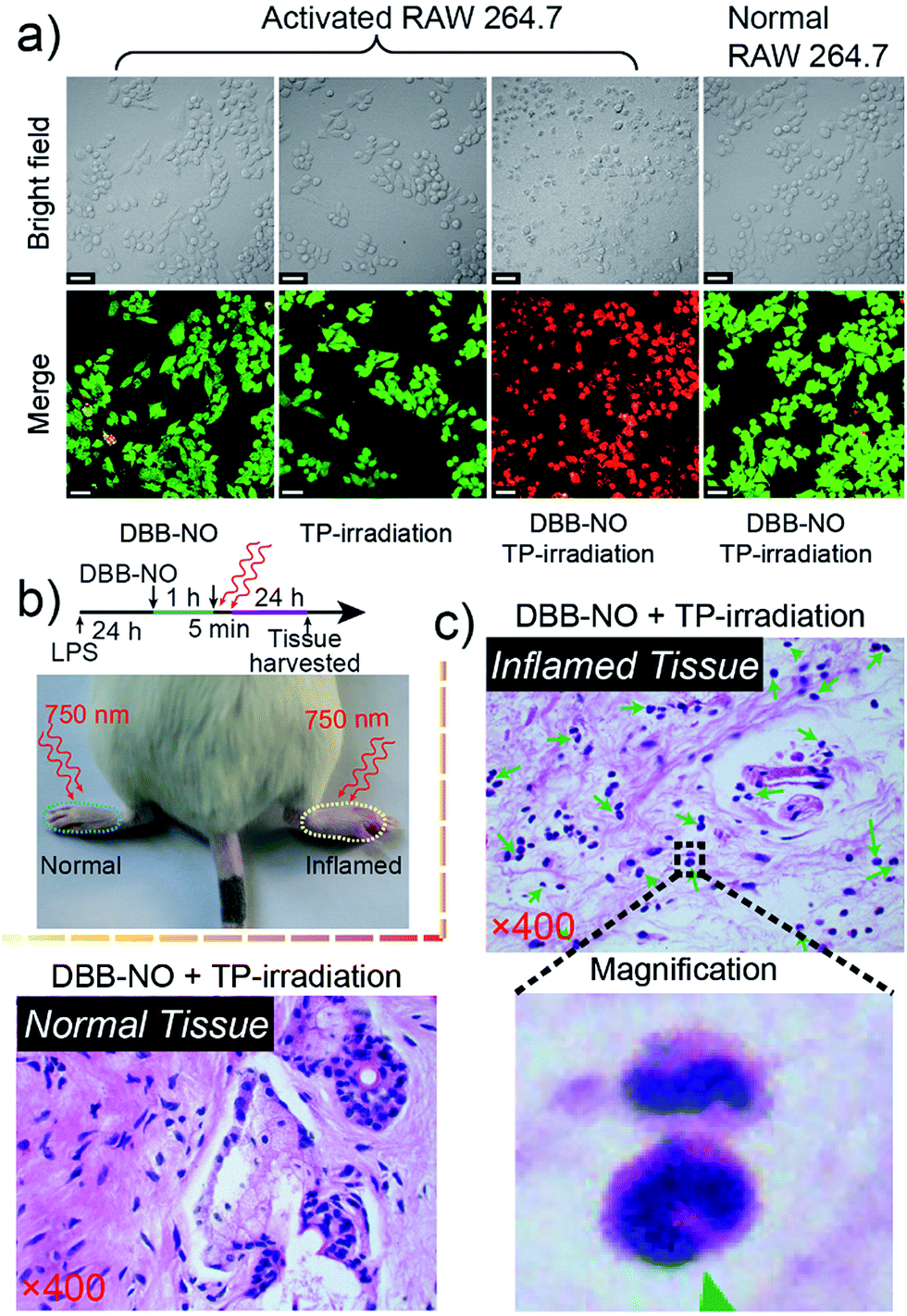 | ||
| Fig. 6 (a) Live/dead assay of the activated RAW 264.7 macrophage after 5 min TP-PDT. The green color represents live cells, and the red color represents dead cells. (b) Illustration for in vivo TP-PDT of the inflamed mouse model. (c) Histological H & E assay for the tissues at 24 h after the treatment with DBB-NO under TP-irradiation at 750 nm for 5 min. Green arrowheads point to crescent-shaped nuclei and the magnification region gives a representative structure of the crescent-shaped nuclei. The mass emergence of crescent-shaped nuclei represents the macrophage death. | ||
With the abovementioned merits, we performed an in vivo TP-PDT of DBB-NO using the LPS-induced inflamed mouse model. After 5 min TP-irradiation and subsequent housing for another 24 h (Fig. 6b), the skin of the inflamed (right paw) and normal (left paw) tissues were harvested and sectioned for hematoxylin–eosin (H & E) staining assay. Compared to the normal tissue (Fig. 6c) and the control groups treated with DBB-NO or TP-irradiation alone (Fig. S18†), the mass emergence of crescent-shaped nuclei (green arrowheads) in inflamed tissue after TP-PDT implied a prominent macrophage apoptosis,25 thus illustrating an endogenous NO-activatable phototoxicity of DBB-NO to the inflamed tissue. This result preliminarily demonstrated that DBB-NO can serve as a smart theranostic agent to selectively destroy activated macrophages and not the normal ones for precision therapy.
Conclusions
In summary, for the first time we reported a NO-activatable TP-FPS for efficient TP-imaging and TP-PDT in activated macrophages. By coupling NO-responsive OPD with zwitterionic DBB, a NO-activatable DBB-NO was constructed. Upon responding to NO, DBB-NO simultaneously exhibited a remarkably enhanced ΦΔ and ΦF as well as an extremely NO-elevated δ. These properties make DBB-NO bright and phototoxic only in the NO-upregulated activated macrophage, thus allowing DBB-NO to effectively distinguish activated macrophages from normal ones and subsequently kill them for precision therapy. More importantly, by simply replacing the stimulus-responsive moiety or linker, the zwitterionic structure such as DBB with an ultralarge δ can act as an active molecular template to construct other pathological condition-activatable materials with the TPA feature for precise theranostics.Conflicts of interest
All authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Basic Research Program of China (No. 2012CB933301), the National Natural Science Foundation of China (No. 21674048, 61378081, 11404219 and 21574064), the Synergetic Innovation Center for Organic Electronics and Information Displays and the Natural Science Foundation of Jiangsu Province of China (No. BK20171020).Notes and references
- (a) J. N. Sharma, A. Al-Omran and S. S. Parvathy, Inflammopharmacology, 2007, 15, 252 CrossRef CAS PubMed; (b) W. P. Fan, N. Lu, P. Huang, Y. Liu, Z. Yang, S. Wang, G. C. Yu, Y. J. Liu, J. K. Hu, Q. J. He, J. L. Qu, T. F. Wang and X. Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 1229 CrossRef CAS PubMed; (c) C. Bogdan, Nat. Immunol., 2001, 2, 907 CrossRef CAS PubMed.
- (a) G. C. Chiou, J. Ocul. Pharmacol. Ther., 2001, 17, 189 CrossRef CAS PubMed; (b) S. Snyder, Science, 1992, 257, 494 CAS.
- M. Forte, V. Conti, A. Damato, M. Ambrosio, A. A. Puca, S. Sciarretta, G. Frati, C. Vecchione and A. Carrizzo, Oxid. Med. Cell. Longevity, 2016, 2016, 7364138 Search PubMed.
- (a) H. Li, D. Zhang, M. Gao, L. Huang, L. Tang, Z. Li, X. Chen and X. Zhang, Chem. Sci., 2017, 8, 2199 RSC; (b) Z. Mao, H. Jiang, Z. Li, C. Zhong, W. Zhang and Z. Liu, Chem. Sci., 2017, 8, 4533 RSC; (c) L. Yuan, W. Lin, Y. Xie, B. Chen and S. Zhu, J. Am. Chem. Soc., 2012, 134, 1305 CrossRef CAS PubMed; (d) H. Yu, Y. Xiao and L. Jin, J. Am. Chem. Soc., 2012, 134, 17486 CrossRef CAS PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889 CrossRef CAS PubMed.
- (a) J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795 CrossRef CAS PubMed; (b) H. Yuan, B. Wang, F. Lv, L. Liu and S. Wang, Adv. Mater., 2014, 26, 6978 CrossRef CAS PubMed; (c) F. Hu, Y. Huang, G. Zhang, R. Zhao, H. Yang and D. Zhang, Anal. Chem., 2014, 86, 7987 CrossRef CAS PubMed; (d) X. Q. Shen, L. Li, A. C. M. Chan, N. Y. Gao, S. Q. Yao and Q. H. Xu, Adv. Opt. Mater., 2013, 1, 92 CrossRef.
- (a) X. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 1604053 CrossRef; (b) J. F. Lovell, T. W. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839 CrossRef CAS PubMed.
- Y. Gabe, Y. Urano, K. Kikuchi, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2004, 126, 3357 CrossRef CAS PubMed.
- (a) A. V. Kachynski, A. Pliss, A. N. Kuzmin, T. Y. Ohulchanskyy, A. Baev, J. Qu and P. N. Prasad, Nat. Photonics, 2014, 8, 455 CrossRef CAS; (b) Y. Shen, A. J. Shuhendler, D. Ye, J. J. Xu and H. Y. Chen, Chem. Soc. Rev., 2016, 45, 6725 RSC.
- (a) K. Ogawa and Y. Kobuke, Anti-Cancer Agents Med. Chem., 2008, 8, 269 CrossRef CAS; (b) J. D. Bhawalkar, N. D. Kumar, C. F. Zhao and P. N. Prasad, J. Clin. Laser Med. Surg., 1997, 15, 201 CAS.
- (a) S. Brown, Nat. Photonics, 2008, 2, 394 CrossRef CAS; (b) H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049 CrossRef CAS PubMed.
- (a) Q. L. Fan, Y. Zhou, X. M. Lu, X. Y. Hou and W. Huang, Macromolecules, 2005, 38, 2927 CrossRef CAS; (b) Q.-L. Fan, S. Lu, Y.-H. Lai, X.-Y. Hou and W. Huang, Macromolecules, 2003, 36, 6976 CrossRef CAS.
- W. B. Hu, T. C. He, R. C. Jiang, J. Yin, L. Li, X. M. Lu, H. Zhao, L. Zhang, L. Huang, H. D. Sun, W. Huang and Q. L. Fan, Chem. Commun., 2017, 53, 1680 RSC.
- (a) T. Wang, E. F. Douglass, K. J. Fitzgerald and D. A. Spiegel, J. Am. Chem. Soc., 2013, 135, 12429 CrossRef CAS PubMed; (b) X. Ye, S. S. Rubakhin and J. V. Sweedler, J. Neurosci. Methods, 2008, 168, 373 CrossRef CAS PubMed.
- J. Hu, M. R. Whittaker, H. Duong, Y. Li, C. Boyer and T. P. Davis, Angew. Chem., Int. Ed., 2014, 53, 7779 CrossRef CAS PubMed.
- Y. Q. Sun, J. Liu, H. Zhang, Y. Huo, X. Lv, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 12520 CrossRef CAS PubMed.
- A. Katritzky, T. Ibrahim, S. Tala, N. Abo-Dya, Z. Abdel-Samii and S. El-Feky, Synthesis, 2011, 2011, 1494 CrossRef.
- (a) T. He, Z. B. Lim, L. Ma, H. Li, D. Rajwar, Y. Ying, Z. Di, A. C. Grimsdale and H. Sun, Chem.–Asian J., 2013, 8, 564 CrossRef CAS PubMed; (b) T. He, P. C. Too, R. Chen, S. Chiba and H. Sun, Chem.–Asian J., 2012, 7, 2090 CrossRef CAS PubMed.
- (a) L. Rong, C. Zhang, Q. Lei, S. Y. Qin, J. Feng and X. Z. Zhang, Adv. Sci., 2015, 3, 1500211 CrossRef PubMed; (b) H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014 CrossRef CAS PubMed; (c) K. Zheng, W. Lin, L. Tan, H. Chen and H. Cui, Chem. Sci., 2014, 5, 3439 RSC.
- M. Y. Kim, J. H. Park, J. S. Mo, E. J. Ann, S. O. Han, S. H. Baek, K. J. Kim, S. Y. Im, J. W. Park, E. J. Choi and H. S. Park, J. Cell Sci., 2008, 121, 1466 CrossRef CAS PubMed.
- K. Y. Lu, P. Y. Lin, E. Y. Chuang, C. M. Shih, T. M. Cheng, T. Y. Lin, H. W. Sung and F. L. Mi, ACS Appl. Mater. Interfaces, 2017, 9, 5158 CAS.
- L. Yuan, L. Wang, B. K. Agrawalla, S. J. Park, H. Zhu, B. Sivaraman, J. Peng, Q. H. Xu and Y. T. Chang, J. Am. Chem. Soc., 2015, 137, 5930 CrossRef CAS PubMed.
- W. B. Hu, H. H. Ma, B. Hou, H. Zhao, Y. Ji, R. C. Jiang, X. M. Hu, X. M. Lu, L. Zhang, Y. F. Tang, Q. L. Fan and W. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 12039 CAS.
- (a) W. G. Fisher, W. P. Partridge, C. Dees and E. A. Wachter, Photochem. Photobiol., 1997, 66, 141 CrossRef CAS PubMed; (b) M. Khurana, H. A. Collins, A. Karotki, H. L. Anderson, D. T. Cramb and B. C. Wilson, Photochem. Photobiol., 2007, 83, 1441 CrossRef CAS PubMed.
- (a) F. A. Oberhammer, K. Hochegger, G. Fröschl, R. Tiefenbacher and M. Pavelka, J. Cell Biol., 1994, 126, 827 CrossRef CAS PubMed; (b) U. Ziegler and P. Groscurth, Physiology, 2004, 19, 124 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details of synthesis, characterization and supplementary figures. See DOI: 10.1039/c7sc04044j |
| ‡ Present address: Key Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University (NanjingTech), Nanjing 211800, China. |
| This journal is © The Royal Society of Chemistry 2018 |