
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Lewis superacid Al[N(C6F5)2]3 and its higher homolog Ga[N(C6F5)2]3 – structural features, theoretical investigation and reactions of a metal amide with higher fluoride ion affinity than SbF5†
J. F.
Kögel‡
a,
D. A.
Sorokin
a,
A.
Khvorost
a,
M.
Scott
a,
K.
Harms
a,
D.
Himmel
 b,
I.
Krossing
b and
J.
Sundermeyer
*a
b,
I.
Krossing
b and
J.
Sundermeyer
*a
aFachbereich Chemie der Philipps-Universität, Hans-Meerwein Str., 35043 Marburg, Germany. E-mail: jsu@staff.uni-marburg.de
bInstitut für Anorganische und Analytische Chemie, Freiburger Materialforschungszentrum (FMF), Freiburg Institute for Advanced Studies (FRIAS), Section Soft Matter Science, Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany
First published on 23rd October 2017
Abstract
Herein we present the synthesis of the two Lewis acids Al[N(C6F5)2]3 (ALTA) and Ga[N(C6F5)2]3 (GATA) via salt elimination reactions. The metal complexes were characterized by NMR-spectroscopic methods and X-ray diffraction analysis revealing the stabilization of the highly Lewis acidic metal centers by secondary metal–fluorine contacts. The Lewis acidic properties of Al[N(C6F5)2]3 and Ga[N(C6F5)2]3 are demonstrated by reactions with Lewis bases resulting in the formation of metallates accompanied by crucial structural changes. The two metallates [Cs(Tol)3]+[FAl(N(C6F5)2)3]− and [AsPh4]+[ClGa(N(C6F5)2)3]− contain interesting weakly coordinating anions. The reaction of Al[N(C6F5)2]3 with trityl fluoride yielded [CPh3]+[FAl(N(C6F5)2)3]− which could find application in the activation of metallocene polymerization catalysts. The qualitative Lewis acidity of Al[N(C6F5)2]3 and Ga[N(C6F5)2]3 was investigated by means of competition experiments for chloride ions in solution. DFT calculations yielded fluoride ion affinities in the gas phase (FIA) of 555 kJ mol−1 for Al[N(C6F5)2]3 and 472 kJ mol−1 for Ga[N(C6F5)2]3. Thus, Al[N(C6F5)2]3 can be considered a Lewis superacid with a fluoride affinity higher than SbF5 (493 kJ mol−1) whereas the FIA of the corresponding gallium complex is slightly below the threshold to Lewis superacidity.
Introduction
Lewis acidic compounds play an important role in synthetic chemistry and have been successfully applied to Diels–Alder reactions,1 rearrangements,2 conjugate additions3 or Friedel–Crafts reactions4 to name only a few examples. Thus, Lewis acid catalysis has been the subject of various review articles5 and the scientific activity in the field of Lewis acids was additionally kindled by the development of frustrated Lewis pair chemistry by Stephan in 2006.6The importance of Lewis acids as valuable synthetic tools has evoked a fundamental interest in the phenomenon of Lewis acidity and its underlying principles. In this context, Haartz and McDaniel in 1973 introduced the fluoride ion affinity in the gas phase (FIA) as the benchmark for the quantification of Lewis acidity.7 Bartlett et al. took up on this and extended the scale.8,9 Christe and Dixon were the first to introduce a reliable isodesmic calculation recipe for the FIA.10 However, the first FIA value (without naming it as such) was presented for BF3 already in 1955.11 By definition, SbF5 as the strongest conventional molecular Lewis acid with a calculated FIA of 493 kJ mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 marks the threshold to Lewis superacidity. Krossing et al. reported on the preparation of the fluorobenzene adduct of the homoleptic aluminum complex Al[OC(CF3)3]3 (Chart 1, FIA: 505 kJ mol−1 in case of the PhF adduct8 and 543 kJ mol−1 for the corresponding adduct free form9) and highlighted important requirements for the design of Lewis superacids: The generation of an extremely electron-poor metal center can be achieved by ligands with weak donor properties that usually contain strongly electron withdrawing substituents such as perfluorinated alkyl groups. In case of Al[OC(CF3)3]3, the aluminum center is stabilized by the formation of two hemilabile aluminum–fluorine interactions masking the high Lewis acidity of the metal center. These metal–fluorine contacts break up in the presence of a Lewis base. The incorporation of additional O- or N-donor atoms in the ligand backbone instead of the carbon-bonded fluorine atoms would allow the formation of stable chelates, which drastically reduce the Lewis acidic properties of the metal complex. Furthermore, sufficient bulkiness of the ligand moieties should prevent oligomerization which would have a reducing effect on the Lewis acidity and complicate the theoretical determination of the FIA. Such decrease in the Lewis acidity due to aggregation is observed for aluminum triiodide and aluminum tribromide that reach the demanded FIA for Lewis superacidity in their monomeric forms in the gas phase (AlI3: 535 kJ mol−1 AlBr3: 510 kJ mol−1),9 but show dramatically lower values in the solid state (AlI3: 429 kJ mol−1 AlBr3: 408 kJ mol−1) because of their high monomerisation enthalpies of 106 kJ mol−1 and 102 kJ mol−1, respectively.13 Eventually, the ligand regime has to provide inertness towards intramolecular or intermolecular degradation processes like the abstraction of fluorine atoms from the ligand backbone. Beside Al[OC(CF3)3]3, the related Al[O(C6F10(C6F5))]3 (530 kJ mol−1)14 also meets the criterion for Lewis superacidity. Lately Wiesner et al. revealed the enormous Lewis acidity of Al(OTeF5)3 which could be isolated as an acetonitrile adduct.15
12 marks the threshold to Lewis superacidity. Krossing et al. reported on the preparation of the fluorobenzene adduct of the homoleptic aluminum complex Al[OC(CF3)3]3 (Chart 1, FIA: 505 kJ mol−1 in case of the PhF adduct8 and 543 kJ mol−1 for the corresponding adduct free form9) and highlighted important requirements for the design of Lewis superacids: The generation of an extremely electron-poor metal center can be achieved by ligands with weak donor properties that usually contain strongly electron withdrawing substituents such as perfluorinated alkyl groups. In case of Al[OC(CF3)3]3, the aluminum center is stabilized by the formation of two hemilabile aluminum–fluorine interactions masking the high Lewis acidity of the metal center. These metal–fluorine contacts break up in the presence of a Lewis base. The incorporation of additional O- or N-donor atoms in the ligand backbone instead of the carbon-bonded fluorine atoms would allow the formation of stable chelates, which drastically reduce the Lewis acidic properties of the metal complex. Furthermore, sufficient bulkiness of the ligand moieties should prevent oligomerization which would have a reducing effect on the Lewis acidity and complicate the theoretical determination of the FIA. Such decrease in the Lewis acidity due to aggregation is observed for aluminum triiodide and aluminum tribromide that reach the demanded FIA for Lewis superacidity in their monomeric forms in the gas phase (AlI3: 535 kJ mol−1 AlBr3: 510 kJ mol−1),9 but show dramatically lower values in the solid state (AlI3: 429 kJ mol−1 AlBr3: 408 kJ mol−1) because of their high monomerisation enthalpies of 106 kJ mol−1 and 102 kJ mol−1, respectively.13 Eventually, the ligand regime has to provide inertness towards intramolecular or intermolecular degradation processes like the abstraction of fluorine atoms from the ligand backbone. Beside Al[OC(CF3)3]3, the related Al[O(C6F10(C6F5))]3 (530 kJ mol−1)14 also meets the criterion for Lewis superacidity. Lately Wiesner et al. revealed the enormous Lewis acidity of Al(OTeF5)3 which could be isolated as an acetonitrile adduct.15
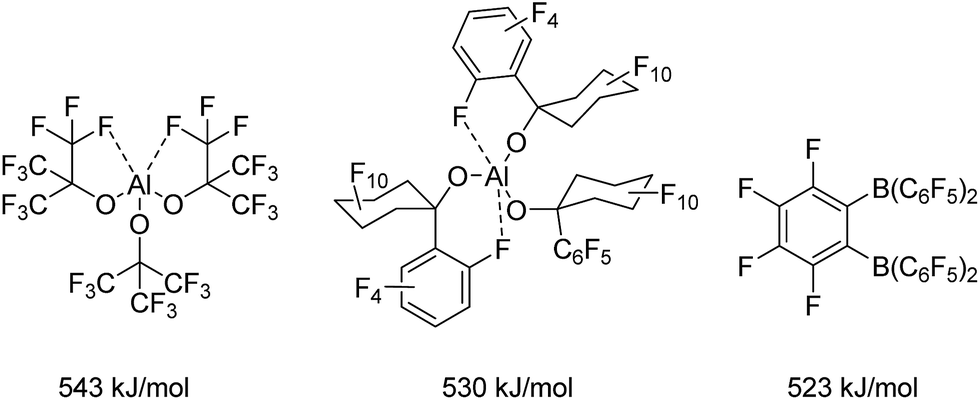 | ||
| Chart 1 Examples for Lewis superacids and their calculated FIAs. | ||
Among perfluorinated aluminum aryl Lewis acids, Al(C6F5)3 is the most prominent exhibiting an FIA of 530 kJ mol−1.8,16 It has been tested in metallocene17 and alkyne activation reactions18 as well as a component of weakly coordinating anions (WCAs).19 Only recently Chen and Chen reported a [Si–H⋯Al] interaction in a crystal structure of [Et3Si-H-Al(C6F5)3]20 in analogy to works by Piers and Tuononen21 and Stephan22 who demonstrated [Si–H⋯B] interactions. However, despite its considerably higher Lewis acidity, the explosive Al(C6F5)3 has received less attention than the corresponding boron compound.13 Whereas common boranes like the widely used B(C6F5)3 (452 kJ mol−1) show FIAs below the threshold to Lewis superacidity, only the chelating 1,2-[(C6F5)2B]2C6F4 (523 kJ mol−1) exhibits an FIA higher than that of SbF5.9,23 Very recently the group of Mitzel published tris(perfluorotolyl)boran which turned out to be more Lewis acidic than its parent compound B(C6F5)3.24,25
The design of highly Lewis acidic metal complexes has also been the subject of theoretical works.26 Frenking et al. reported on the enhancement of the Lewis acidity of B, Al and Ga compounds with adamantyl substituents by pyramidalization of the coordination geometry.27
The research in the field of strong Lewis acids goes hand in hand with the investigation of the corresponding weakly coordinating anions derived from the reaction of a Lewis acid with a Lewis basic anion. The metal center is shielded by the hydrophobic and sterically demanding perfluorinated ligand regime granting delocalization of the negative charge. Thus, WCAs allow the stabilization of highly reactive cationic species28 like the carbocations [CCl3]+ and [CBr3]+,29 the tritylium cation,30 a radical cation of benzidine,31 a stable [AsBr4]+ cation32 or a [Ag2Se12]2+ cage.33 In this context, especially Ag+[Al(OC(CF3)3)4]− has emerged as a versatile reagent for the abstraction of chloride ions from neutral precursors to generate reactive cations stabilized by the WCA [Al(OC(CF3)3)4]−.34 Such reactions yielded stabilized amido-substituted germanium(II) and tin(II) monocations,35 homoleptic ethylene complexes of the coinage metals,36 the tBu3Si+ source [tBu3Si–Ga–SitBu3]+,37 gallium(I) arene complexes38 or univalent gallium and indium phosphane complexes.39
Aluminum and gallium are proper metals for the generation of strong Lewis acids because of the small size and the high charge of their M3+ cations. The principle for the preparation of Lewis acidic aluminum or gallium compounds is to find a negatively charged ligand with weak donor character leaving a high positive partial charge on the metal center. This can be achieved by delocalizing the ligand's negative charge over perfluorinated electron withdrawing groups. As described above, it has been demonstrated that perfluorinated alkoxo ligands are able to form Lewis superacidic aluminum complexes. This article is concerned with the question: can certain perfluorinated metal amides be Lewis superacids and display a higher fluoride affinity than SbF5? Representative amido ligands of intrinsically weak donor capability, [N(C6F5)(C(CF3)3)]−40 and [N(C6F5)(SO2RF)]−41 were introduced by us only recently. In this context, we also turned our attention to bis(pentafluorophenyl)amide [N(C6F5)2]− as a promising ligand for the preparation of strong Lewis acids.42 HN(C6F5)2 can be easily prepared in large scale43 and its two strongly electron withdrawing pentafluorophenyl substituents should provide complexes with good solubility in nonpolar solvents and sufficient sterical shielding of the metal center. Furthermore, the NH-acid is known for its ability to form hemilabile metal–fluorine contacts via its ortho-fluorine atoms stabilizing the metal center and leading to interesting coordination modes. It has already been incorporated into complexes of lithium,44 the f-block metals neodymium,45 cerium, lanthanum46 and uranium47 and the d-block metals titanium, zirconium, vanadium, iron, cobalt48 and tungsten.49
Results and discussion
Preparation
Al[N(C6F5)2]3 and Ga[N(C6F5)2]3 were both prepared via reactions of LiN(C6F5)2 with the corresponding metal trichlorides in toluene at 90 °C leading to the precipitation of lithium chloride (Scheme 1). Al[N(C6F5)2]3 was first isolated from an alkane elimination reaction between trimethylaluminum and HN(C6F5)2 in toluene at 105 °C, but this route only yielded traces of the desired product. The 19F NMR spectra of Al[N(C6F5)2]3 (δ = −153.1, −158.6 and −161.1 ppm) and Ga[N(C6F5)2]3 (δ = −151.8, −158.1 and −161.4 ppm) in [D6]benzene reveal three signals with similar chemical shifts in a 2:1:2 ratio for the three aromatic fluorine atoms. As expected, especially the aluminum compound turned out to be extremely moisture-sensitive.
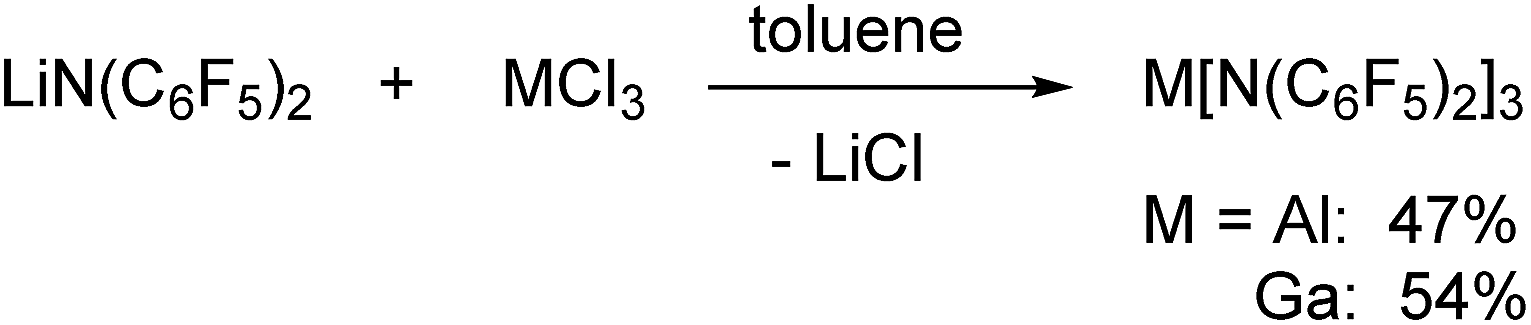 | ||
| Scheme 1 Preparation of Al[N(C6F5)2]3 and Ga[N(C6F5)2]3via salt elimination reactions. | ||
Structural features of the free Lewis acids
A trigonal planar AlN3 coordination geometry is found for Al[N(C6F5)2]3 with Al–N distances of 1.843(2) Å, 1.840(2) Å and 1.805(2) Å (Chart 2). The metal center is further stabilized by two axial aluminum–fluorine contacts with Al⋯F distances of 2.084(1) and 2.060(1) Å and an F12–Al–F24 angle of 164.93(6)°. The incorporation of the two ortho-fluorine atoms in Al⋯F contacts leads to an elongation of the corresponding C–F bonds (1.3897(2) and 1.3867(1) Å compared to 1.3459(2) and 1.3480(2) Å found for the two other C–Fortho bond lengths in the corresponding C6F5 rings). Similar to the κ-N3F2 configuration experimentally verified for this aluminum trisamide, a κ-O3F2 configuration with longer Al⋯F contacts (2.143 and 2.155 Å) was proposed for the alkoxido superacid Al[OC(CF3)3]3 on the basis of DFT calculations.8 | ||
| Chart 2 Molecular structure of Al[N(C6F5)2]3 (ellipsoids with 30% probability). Selected bond lengths/Å and angles/°: Al–N1 1.843(2), Al–N2 1.840(2), Al–N3 1.805(2), Al⋯F12 2.084(1), Al⋯F24 2.060(1), N1–Al–N2 123.7(1), N2–Al–N3 116.2(1), N3–Al–N1 120.09(9), F24–Al–F12 164.93(6). | ||
As observed for the corresponding aluminum complex, the molecular structure of Ga[N(C6F5)2]3 (Chart 3) reveals a trigonal planar GaN3 coordination geometry with Ga–N bond lengths of 1.826(5), 1.798(5) and 1.848(5) Å. These values are shorter than the M–F distances found in the molecular structure of Al[N(C6F5)2]3. In addition to the three nitrogen donors, the gallium atom is coordinated by six ortho-fluorine atoms with gallium–fluorine distances ranging from 2.914(4) to 3.102(4) Å. The complex is further stabilized by π-stacking interactions between the pentafluorophenyl rings of neighboring N(C6F5)2 moieties (distances of neighboring rings' centroids: 3.5192(3), 3.7848(4) and 3.5463(3) Å). Similar coordination modes were observed for the homoleptic lanthanum and cerium complexes of HN(C6F5)2 recently reported by Yin et al.46 La[N(C6F5)2]3 and Ce[N(C6F5)2]3 show longer N–M bond lengths (La–N between 2.410(2) and 2.512(2) Å, Ce–N between 2.406(3) and 2.430(2) Å), but shorter M⋯F contacts (La⋯F between 2.6695(17) and 2.8942(16) Å, Ce⋯F between 2.6764(16) and 2.7064(17) Å) compared to Ga[N(C6F5)2]3. According to a review article on interactions between metal atoms and organically bound fluorine atoms by Plenio, this kind of coordination mode is one of the two recurrent structure motives for gallium complexes with fluorinated ligands.50 The coordination of a gallium center by six fluorine atoms incorporated in the organic ligand backbone was also observed in case of tris(2,4,6-tris(trifluoromethyl)phenyl)gallium exhibiting Ga⋯F distances between 2.683 and 2.821 Å.51
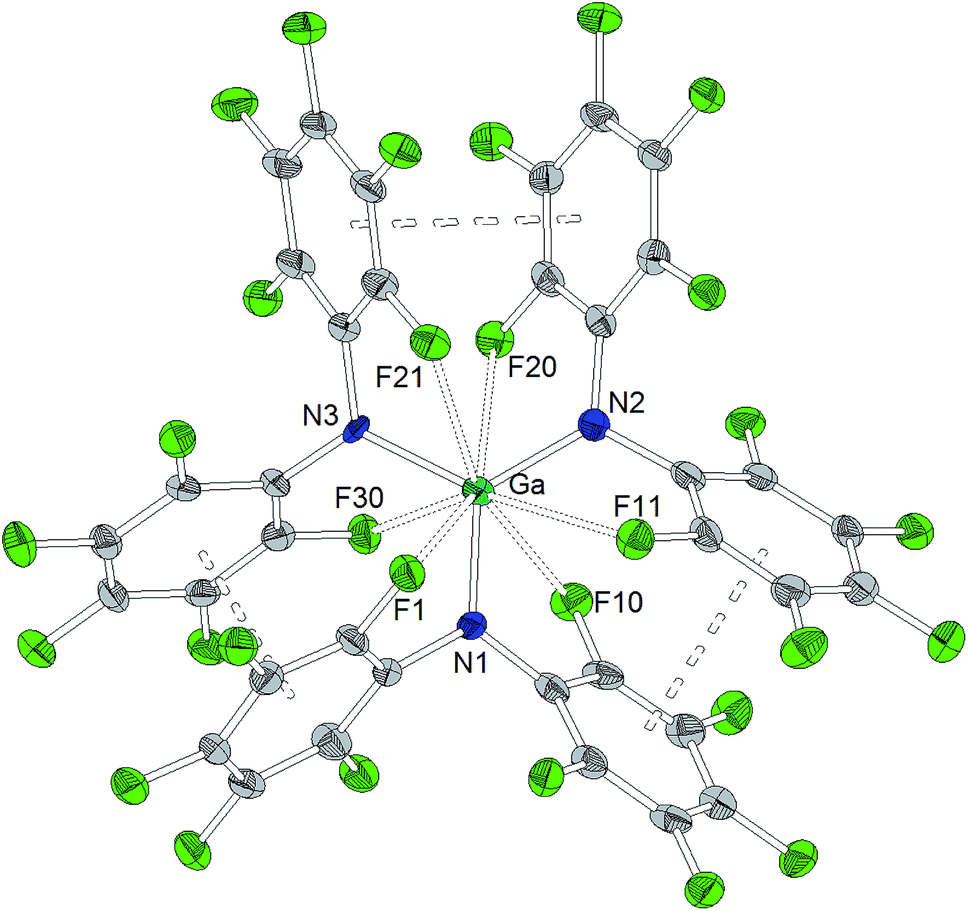 | ||
| Chart 3 Molecular structure of Ga[N(C6F5)2]3 (ellipsoids with 30% probability). Selected bond lengths/Å and angles/°: Ga–N1 1.826(5), Ga–N2 1.798(5), Ga–N3 1.848(5), Ga⋯F1 3.032(4), Ga⋯F10 2.981(4), Ga⋯F11 3.096(4), Ga⋯F20 2.994(4), Ga⋯F21 3.102(4), Ga⋯F30 2.914(4), N1–Ga–N2 122.8(2), N2–Ga–N3 123.0(2), N3–Ga–N1 114.3(2). | ||
Reactivity and experimental fluoride ion affinity of Al[N(C6F5)2]3
The enormous fluoride ion affinity of Al[N(C6F5)2]3 could also be demonstrated experimentally. The reaction of Al[N(C6F5)2]3 with [PPh4]+[SbF6]− in toluene at 100 °C resulted in the precipitation of a mixture of [PPh4]+[FAl(N(C6F5)2)3]− and the formation of HN(C6F5)2 (Scheme 2(a)). The 19F NMR spectrum of [PPh4]+[FAl(N(C6F5)2)3]− exhibits signals with chemical shifts of −149.6, −166.2, −167.5 and −172.3 ppm in a 12:6:12:1 ratio. The formation of HN(C6F5)2 is plausible as it is known, that SbF5 reacts with toluene to give (p-Tol)3SbF2 and three equivalents of HF in moderate yields.52 The lewis acid Al[N(C6F5)2]3 as well as the corresponding anion [FAl(N(C6F5)2)3]− can be protolyzed by HF under formation of HN(C6F5)2. Further proofs for the high fluoride affinity of Al[N(C6F5)2]3 could be obtained when dissolving the Lewis acid in hexafluorobenzene or F2ClCCF2Cl. Both solutions slowly turn dark and the 19F NMR spectrum of both reaction mixtures reveal unselective reactions which can be referred to the abstraction of F− from the solvent leading to further reactions of the highly reactive carbocations.
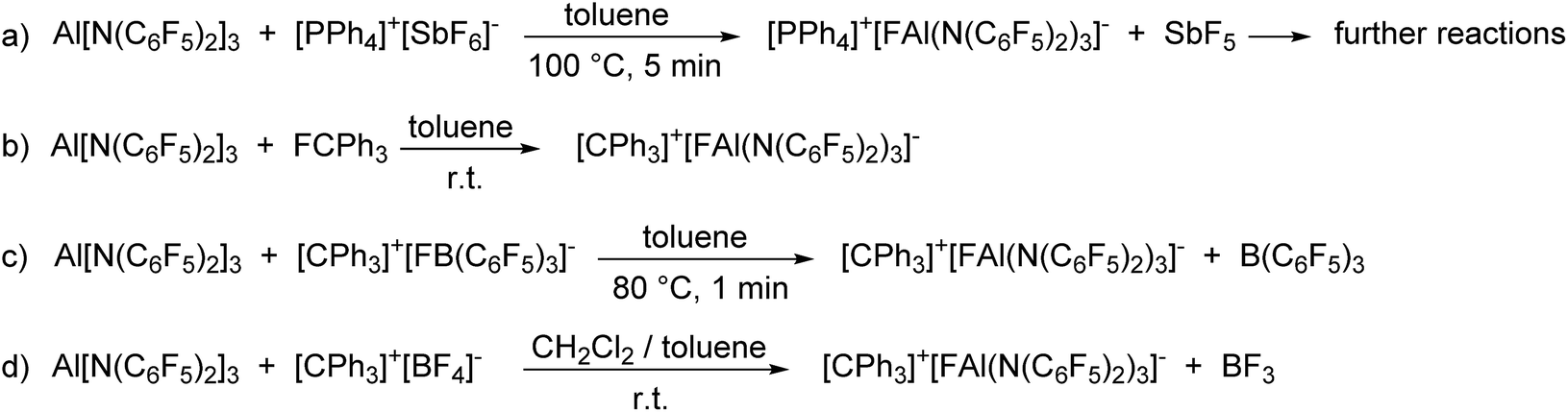 | ||
| Scheme 2 Reactions proving the high Lewis acidity of Al[N(C6F5)2]3. | ||
The reaction of Al[N(C6F5)2]3 with trityl fluoride in toluene yields [CPh3]+[FAl(N(C6F5)2)3]− which is stable in solution for at least three days (Scheme 2(b)). Attempts to isolate the yellow compound resulted in its decomposition after 24 h at room temperature. In agreement with the theoretically predicted trend of the FIA in the gas phase, [CPh3]+[FAl(N(C6F5)2)3]− is also formed when reacting Al[N(C6F5)2]3 with [CPh3]+[FB(C6F5)3]− in toluene and with [CPh3]+[BF4]− in a mixture of toluene and dichloromethane (Scheme 2(c) and (d)).
In situ generated [CPh3]+[FAl(N(C6F5)2)3]− was able to abstract a methyl group from dimethyl zirconocene to give [Cp2ZrMe]+[FAl(N(C6F5)2)3]− as a white solid (Scheme 3(a)). The 19F NMR spectrum shows four signals with chemical shifts of −148.1, −149.4, −160.6 and −163.5 ppm in a 1:12:6:12 ratio. The aluminum bound fluorine atom is shifted to lower field compared to [FAl(N(C6F5)2)3]− (δ = −170.8 ppm). This can be referred to the coordination of the fluorine atom to the zirconium center which was also observed in a low quality crystal structure of [Cp2ZrMe]+[FAl(N(C6F5)2)3]−. The strong Zr⋯F interaction explains why [Cp2ZrMe]+[FAl(N(C6F5)2)3]− turned out to be inactive in ethene polymerization reactions.
 | ||
| Scheme 3 Reactions of [CPh3]+[FAl(N(C6F5)2)3]− with Cp2ZrMe2 and n-butyllithium. | ||
Orange crystals suitable for X-ray diffraction analysis were grown directly from the solution of reaction (a) in Scheme 3 and revealed the formation of [CPh3]+[((C6F5)2N)3AlF–Li–FAl(N(C6F5)2)3]– (Chart 4). The presence of lithium in the crystal structure can be referred to traces of lithium chloride or Li+[ClAl(N(C6F5)2)3]− in the used Al[N(C6F5)2]3. The selective preparation of [CPh3]+[((C6F5)2N)3AlF–Li–FAl(N(C6F5)2)3]− could be achieved from the reaction of two equivalents of in situ generated [CPh3]+[FAl(N(C6F5)2)3]− with n-butyllithium (Scheme 3(b)). A quartet with a chemical shift of −179.3 ppm and a 1J(F,Li) coupling constant of 94 Hz is observed for the fluorine atoms bound to aluminum atoms in the 19F NMR spectrum. The crystal structure reveals Al–F distances of 1.714(3) and 1.710(3) Å. The lithium cation possesses a distorted octahedral coordination sphere and is coordinated by the two aluminum bound fluorine atoms and additional four ortho-fluorine atoms of C6F5 units. The Li⋯F distances to the aluminum-bound fluorine atoms are comparably short (1.793(9) and 1.809(9) Å) whereas the distances to the organically bound fluorine atoms range from 2.185(12) to 2.392(10) Å. The R3Al–F–Li–F–AlR3 structure motive has already been described in the literature for [Ag(PhCH3)3]+[{((SiMe3)3C)2Al2F5}2Li]− (Al–F 1.688(2) Å, Li–F 1.854(6) Å),53 [Li(Me3Si)3CAlF3(THF)]4 (Al–F 1.694(2) Å and 1.701(2) Å, Li–F 1.873(6) Å and 1.801(6) Å)54 and Li+[(Me3Si)3CAlF3]−·THF (mean Al–F 1.687(8) Å, mean Li–F 1.85(2) Å).55 In contrast to [CPh3]+[((C6F5)2N)3AlF–Li–FAl(N(C6F5)2)3]−, all lithium atoms in these references exhibit a tetrahedral coordination sphere. The three compounds described in the literature show slightly shorter Al–F distances and longer Li–F distances than found for [CPh3]+[((C6F5)2N)3AlF–Li–FAl(N(C6F5)2)3]−.
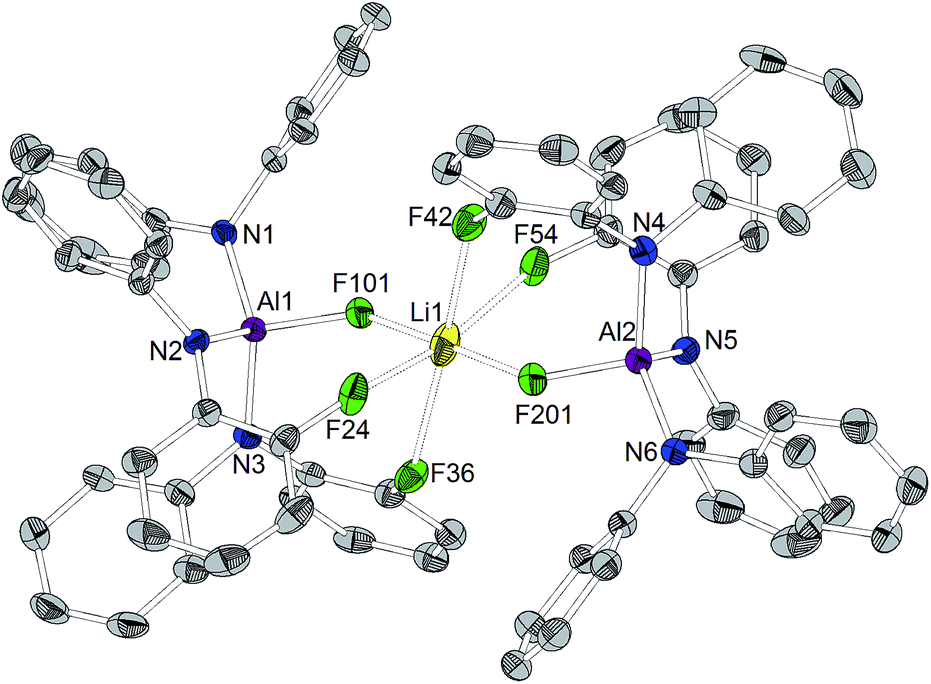 | ||
| Chart 4 Molecular structure of [CPh3]+[((C6F5)2N)3AlF–Li–FAl(N(C6F5)2)3]− (ellipsoids with 30% probability, non-coordinating fluorine atoms, the cation and two toluene molecules omitted for clarity). Selected bond lengths/Å and angles/°: Al1–N1 1.862(3), Al1–N2 1.859(4), Al1–N3 1.857(3), Al1–F101 1.714(3), Al2–N4 1.865(4), Al2–N5 1.859(4), Al2–N6 1.861(4), Al2–F201 1.710(3), Li1⋯F101 1.793(9), Li1⋯F201 1.809(9), Li1⋯F24 2.185(12), Li1⋯F36 2.392(10), Li1⋯F42 2.280(10), Li1⋯F54 2.287(12), N1–Al1–N2 113.51(15), N2–Al1–N3 102.24(16), N1–Al1–N3 123.30(16), N1–Al1–F101 99.83(15), N2–Al1–F101 114.48(15), N3–Al1–F101 103.57(14), N4–Al2–N5 102.41(17), N5–Al2–N6 115.61(17), N4–Al2–N6 122.56(17), N4–Al2–F201 102.66(15), N5–Al2–F201 112.48(16), N6–Al2–F201 100.55(16). | ||
Synthesis and structural features of the metallates
The reaction of Al[N(C6F5)2]3 with cesium fluoride in toluene and the reaction of Ga[N(C6F5)2]3 with tetraphenylarsonium chloride in dichloromethane yielded the two metallates [Cs(Tol)3]+[FAl(N(C6F5)2)3]− and [AsPh4]+[ClGa(N(C6F5)2)3]− (Scheme 4).56 The aluminum bound fluorine atom in the former compound exhibits a chemical shift of −157.4 ppm in the 19F NMR spectrum. Both metallates could be structurally characterized (Charts 5 and 6, Table 1). The two weakly coordinating anions show the pyramidalization of the coordination geometry around the metal atoms. The M–N bonds are elongated in comparison to the parent compounds whereas this is more pronounced in case of the gallium compound (Al–N between 1.860(3) and 1.865(3) Å, Ga–N between 1.912(2) and 1.931(2) Å). The molecular structure of [Cs(Tol)3]+[FAl(N(C6F5)2)3]− reveals η2-, η3- and η6-coordination of the cesium atom by three toluene molecules and the cesium atom also interacts with the fluorine atom bound to the aluminum center with a Cs⋯F distance of 2.878(2) Å. Furthermore, the crystal structure reveals four metal–fluorine contacts between the cesium atom and carbon bound fluorine atoms (Cs⋯F between 3.094(2) and 3.807(3) Å).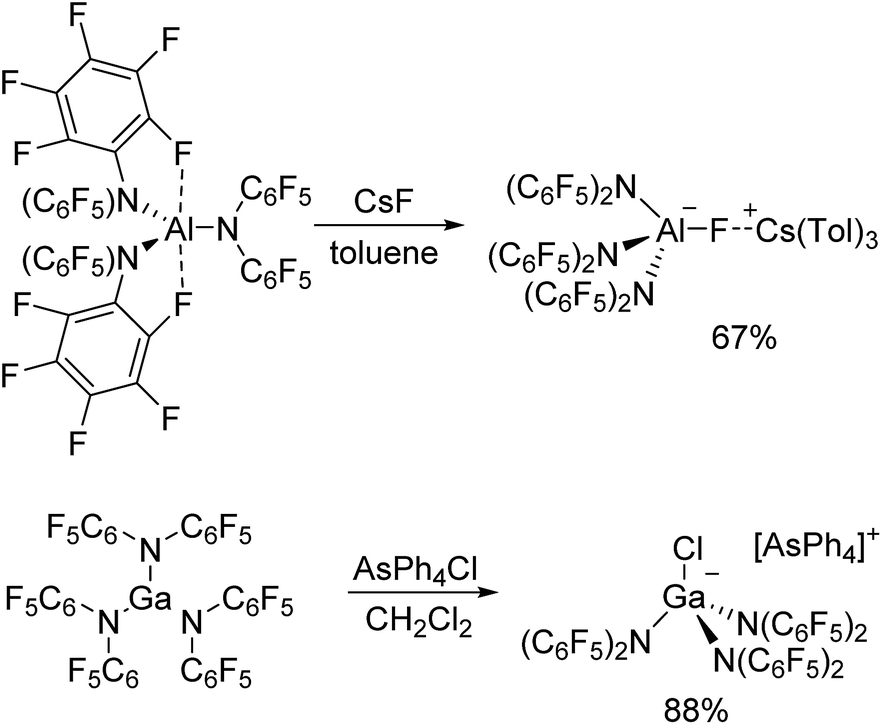 | ||
| Scheme 4 Preparation of [Cs(Tol)3]+[FAl(N(C6F5)2)3]− and [AsPh4]+[ClGa(N(C6F5)2)3]−. | ||
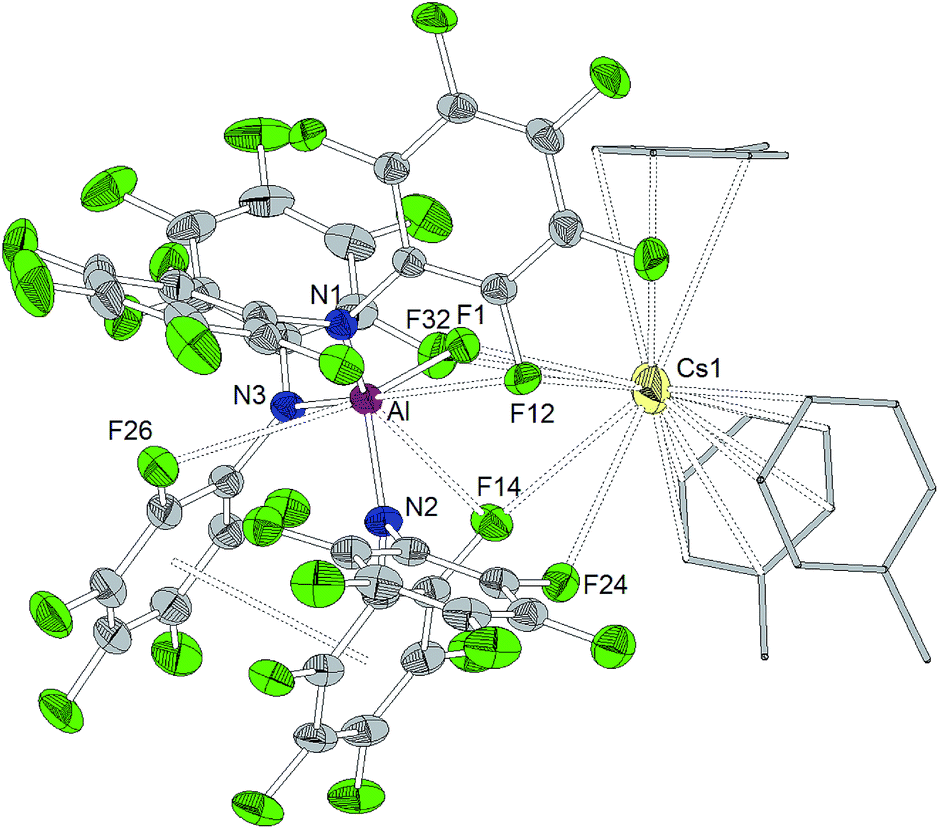 | ||
| Chart 5 Molecular structure of [Cs(Tol)3]+[FAl(N(C6F5)2)3]− (ellipsoids with 30% probability, the three toluene molecules displayed in wireframe design). Selected bond lengths/Å and angles/°: Al–N1 1.863(3), Al–N2 1.860(3), Al–N3 1.865(3), Al–F1 1.689(2), Al⋯F12 3.099(3), Al⋯F14 3.169(3), Al⋯F26 3.202(3), Cs1⋯F1 2.878(2), Cs1⋯F12 3.773(2), Cs1⋯F14 3.094(3), Cs1⋯F24 3.807(3), Cs1⋯F32 3.692(3), N1–Al–N2 112.31(14), N2–Al–N3 102.59(14), N3–Al–N1 123.22(15), N1–Al–F1 98.64(12), N2–Al1–F1 113.94(14), N3–Al1–F1 106.43(14). | ||
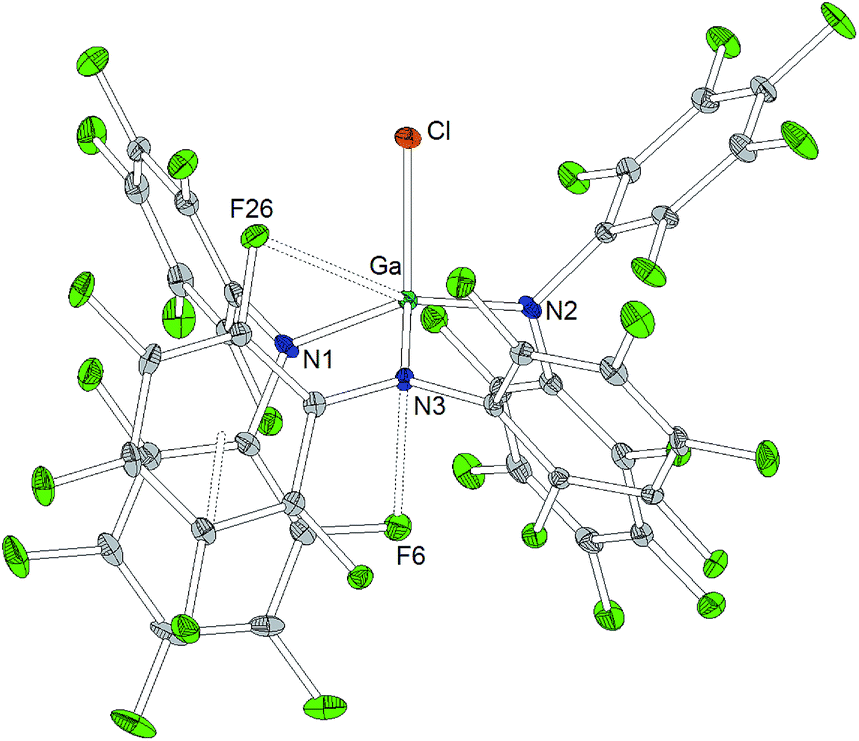 | ||
| Chart 6 Molecular structure of [AsPh4]+[ClGa(N(C6F5)2)3]− (ellipsoids with 30% probability, the [AsPh4]+ cation and one pentane molecule are omitted for clarity). Selected bond lengths/Å and angles/°: Ga–N1 1.912(2), Ga–N2 1.912(2), Ga–N3 1.931(2), Ga⋯F6 3.244(2), Ga⋯F26 3.205(2), Ga–Cl1 2.1890(8), N1–Ga–N2 117.4(1), N2–Ga–N3 115.7(1), N3–Ga–N1 99.9(0), N1–Ga–Cl1 113.90(8), N2–Ga–Cl1 98.83(7), N3–Ga–Cl1 111.85(7). | ||
| Al[N(C6F5)2]3 | Ga[N(C6F5)2]3 | [Cs(Tol)3]+[FAl(N(C6F5)2)3]− | [AsPh4]+[ClGa(N(C6F5)2)3]− | |
|---|---|---|---|---|
| M–N1 | 1.843(2) | 1.826(5) | 1.863(3) | 1.912(2) |
| M–N2 | 1.840(2) | 1.798(5) | 1.860(3) | 1.912(2) |
| M–N3 | 1.805(2) | 1.848(5) | 1.865(3) | 1.931(2) |
| M–Hal | — | — | 1.689(2) | 2.1890(8) |
Qualitative chloride ion affinity in dichloromethane
To evaluate the Lewis acidity of Al[N(C6F5)2]3 and Ga[N(C6F5)2]3 in solution, their chloride ion affinity in dichloromethane was studied by means of qualitative competition experiments via19F NMR spectroscopy. Both compounds turned out to be able to abstract a chloride ion from [AsPh4]+[ClB(C6F5)3]−. Thus, the borane B(C6F5)3 is a weaker Lewis acid towards a chloride ion in solution than both of the two metal amido title compounds. The addition of one equivalent of tetraphenylarsonium chloride to a 1:1 mixture of Al[N(C6F5)2]3 and Ga[N(C6F5)2]3 resulted in the formation of [AsPh4]+[ClAl(N(C6F5)2)3]− identifying the aluminum complex the stronger Lewis acid under the applied conditions (Scheme 5). Hence, the experimental results for the chloride ion affinity follow the trend of the fluoride ion affinity predicted by theoretical calculations (vide infra).
 | ||
| Scheme 5 Qualitative competition experiments for the determination of the chloride ion affinity of Al[N(C6F5)2]3 and Ga[N(C6F5)2]3. | ||
Theoretical section
As the M[N(C6F5)2]3 (M = Al, Ga) Lewis acids are too large for high level ab initio calculations, quantum chemical calculations on the Lewis acidities (i.e. ion affinities) of M[N(C6F5)2]3 (M = Al, Ga) were split into two parts to increase the accuracy. Ligand exchange reactions with their MF3 counterparts were calculated at the BP8657–59-D360/def-TZVP61 (including Grimme's 2010 dispersion correction) level of theory: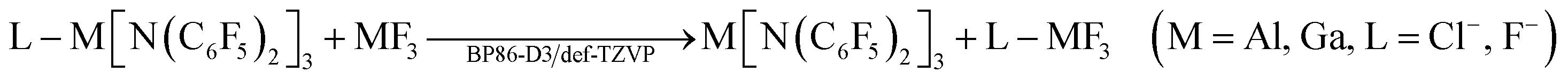
As these reactions are isodesmic, the error in the metal–ligand bond strengths should largely cancel out retaining high accuracy despite of the formally rather low level of theory.
The ligand dissociation energies of the L–MF3 complexes were calculated with a CCSD(T)–MP2 compound method based on single point calculations on MP2/def2-QZVPP62 structures. Due to the very similar basis set dependency of CCSD(T) and MP2, CCSD(T)/A′VQZ accuracy can be approximated by calculating
| ΔECCSD(T)/A′VQZ ≈ ΔEcompound = ΔECCSD(T)/A′VDZ + ΔEMP2/A′VQZ − ΔEMP2/A′VDZ |
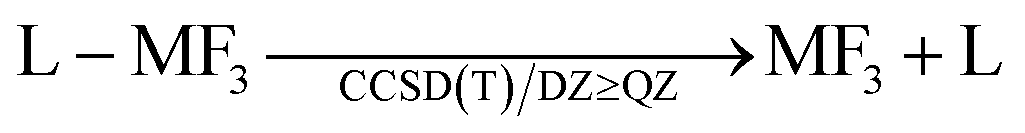
Addition of both reaction energetics gives the M[N(C6F5)2]3 (M = Al, Ga) dissociation energies. Thermal corrections to enthalpies and Gibbs energies were done at the BP86-D3/def-TZVP level of theory.
Theoretical calculations reveal an outstanding fluoride ion affinity in the gas phase of 555 kJ mol−1 for Al[N(C6F5)2]3 (Table 2). This value is practically identical to the FIAs of B(CF3)3 (556 kJ mol−1)9 and AuF5 (556 kJ mol−1).8 Thus, Al[N(C6F5)2]3 is not only lot more Lewis acidic than the strongest conventional Lewis acid antimony pentafluoride (495 kJ mol−1),9 but even outnumbers the aluminum-based Lewis acids Al[OC(C5F10)C6F5]3 (530 kJ mol−1)11 and Al[OC(CF3)3]3 (543 kJ mol−1).9 As discussed above, the Lewis acidity of Al[N(C6F5)2]3 is reduced by two weak dative bonds from ortho-fluorine atoms at the phenyl rings. To evaluate this effect, we calculated the FIA of Al[N(C6H2F3)2]3 with all ortho-fluorine atoms replaced by hydrogen, obtaining an even higher FIA of 598 kJ mol−1.
| FIA/kJ mol−1 | Chloride ion affinity/kJ mol−1 | |
|---|---|---|
| Al[N(C6H2F3)2]3 | 598 | — |
| AuF5 | 5569 | — |
| Al[N(C 6 F 5 ) 2 ] 3 | 555 | 362 |
| B(CF3)3 | 55612 | 35812 |
| Al[OC(CF3)3]3 | 54312 | 35212 |
| SbF5 | 49312 | 34112 |
| Ga[N(C 6 F 5 ) 2 ] 3 | 472 | 324 |
| AlF3 | 47112 | 30612 |
| B(C6F5)3 | 45212 | 23612 |
| BF3 | 34212 | 14612 |
Ga[N(C6F5)2]3 exhibits a computational FIA of 472 kJ mol−1. Therefore, it is similarly Lewis acidic as for example monomeric AlF3 (482 kJ mol−1), but stronger than the widely used B(C6F5)3 (452 kJ mol−1).9 However, the Lewis acidity of Ga[N(C6F5)2]3 is located slightly below the threshold to Lewis superacidity. The lower FIA of Ga[N(C6F5)2]3 compared to the aluminum homolog can be referred to the stabilization of the Lewis acidic gallium center by six metal fluorine contacts (Chart 3). The formation of the gallate [FGa(N(C6F5)2)3]− after the uptake of a fluoride anion leads to an unfavorable situation due to repulsion of the C6F5 moieties. In case of isostructural aluminum and gallium centered Lewis acids, a decrease in Lewis acidity is also expected for the gallium compound due to gallium's higher electronegativity as a consequence of the d-block contraction.
The Lewis acids treated herein show chloride ion affinities in the gas phase of 362 kJ mol−1 for Al[N(C6F5)2]3 and 324 kJ mol−1 in case of the corresponding gallium complex. These values are in agreement with the experimental chloride affinities in dichloromethane. They are considerably lower than the corresponding FIA values and the comparison to the chloride affinities of other Lewis acids shows that Al[N(C6F5)2]3 and Ga[N(C6F5)2]3 are in the same positions as in the FIA ranking.67 However, it is striking that the difference between the chloride affinities of Al[N(C6F5)2]3 and Ga[N(C6F5)2]3 is much smaller than the deviation between the FIA values. This can be attributed to the softer character of chloride in comparison to the hard fluoride anion.
Conclusions
We presented the first metal amide with a higher fluoride ion affinity in the gas phase and in solution than the benchmark compound for Lewis superacidity SbF5. In this context, the synthesis and full characterization of two homoleptic group 13 metal decafluorodiphenylamides Al[N(C6F5)2]3 (ALTA) and Ga[N(C6F5)2]3 (GATA) are described. The origin for such high fluoride affinity of Al[N(C6F5)2]3 is originating from a weak amide donor capability and a trigonal planar AlN3 coordination motif with two hemilabile secondary ortho-CF → Al contacts. In contrast, the slightly weaker Lewis acid Ga[N(C6F5)2]3 has a trigonal pyramidal GaN3 configuration with six extra ortho-CF → Ga contacts in the solid state. In solution the secondary interactions of both compounds are involved in a fast dynamic exchange process. In contrast to the prominent Lewis superacidic perfluoroalkoxide [Al(OC(CF3)3)3] which has been crystallized as base adducts only, the amide Al[N(C6F5)2]3 can be isolated as crystalline Lewis acid. The fluoride affinity is experimentally substantiated by fluoride abstraction from F–X (X = BF3, B(C6F5)3, SbF5 and CPh3+) as well as by competition experiments between both title compounds. The prominent status of the Lewis superacid Al[N(C6F5)2]3 is emphasized by the calculated fluoride ion affinity (FIA) in the gas phase of 555 kJ mol−1versus 493 kJ mol−1 for SbF5.We believe that these and related Lewis acidic aluminum and gallium amides add new perspectives to the highly topical field of frustrated Lewis pairs that has mainly been dominated by boron-based Lewis acids so far. Furthermore, the weakly coordinating anions derived from our metal amides will be applied for stabilization of highly reactive cations in catalysis and fundamental chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Fabian G. Schröder and Lars H. Finger for their valuable support with crystal structure refinement. Financial support by the Fonds der Chemischen Industrie (doctoral scholarship for J. F. K.) is gratefully acknowledged.Notes and references
- (a) K. N. Houk and R. W. Strozier, J. Am. Chem. Soc., 1973, 95, 4094 CrossRef CAS; (b) S. Otto, F. Bertoncin and J. B. F. N. Engberts, J. Am. Chem. Soc., 1996, 118, 7702 CrossRef CAS; (c) F. Fringuelli, O. Piermatti, F. Pizzo and L. Vaccaro, Eur. J. Org. Chem., 2001, 3, 439 CrossRef.
- (a) L.-X. Shao, Y.-P. Zhang, M.-H. Qi and M. Shi, Org. Lett., 2007, 9, 117 CrossRef CAS PubMed; (b) A. Zicmanis, S. Katkevica and P. Mekss, Catal. Commun., 2009, 10, 614 CrossRef CAS.
- (a) M. P. Sibi, J. Ji, J. H. Wu, S. Guertler and N. A. Porter, J. Am. Chem. Soc., 1996, 118, 9200 CrossRef CAS; (b) M. P. Sibi, J. J. Shay, M. Liu and C. P. Jasperse, J. Am. Chem. Soc., 1998, 120, 6615 CrossRef CAS.
- K. Suyama, K. Matsumoto and T. Katsuki, Heterocycles, 2009, 77, 817 CrossRef CAS.
- (a) A. Corma and H. Garcia, Chem. Rev., 2003, 103, 4307 CrossRef CAS PubMed; (b) S. Kobayashi and K. Manabe, Pure Appl. Chem., 2000, 72, 1373 CrossRef CAS; (c) S. Antoniotti, V. Dalla and E. Dunach, Angew. Chem., 2010, 122, 8032 ( Angew. Chem., Int. Ed. , 2010 , 49 , 7860 ) CrossRef.
- (a) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed; (b) J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., 2007, 119, 5056 ( Angew. Chem., Int. Ed. , 2007 , 46 , 4968 ) CrossRef; (c) S. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535 RSC; (d) D. W. Stephan and G. Erker, Angew. Chem., 2010, 122, 50 ( Angew. Chem., Int. Ed. , 2010 , 49 , 46 ) CrossRef.
- J. C. Haartz and D. H. McDaniel, J. Am. Chem. Soc., 1973, 95, 8562 CrossRef CAS.
- T. E. Mallouk, G. L. Rosenthal, G. Müller, R. Brusasco and N. Bartlett, Inorg. Chem., 1984, 23, 3167 CrossRef CAS.
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., 2008, 120, 7772 ( Angew. Chem., Int. Ed. , 2008 , 47 , 7659 ) CrossRef.
- K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151 CrossRef CAS.
- A. P. Altshuller, J. Am. Chem. Soc., 1955, 77, 6187 CrossRef CAS.
- H. Böhrer, N. Trapp, D. Himmel, M. Schleep and I. Krossing, Dalton Trans., 2015, 44, 7489 RSC.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, 93rd edn, 2012 Search PubMed.
- K. A. Kraft, N. Trapp, D. Himmel, H. Böhrer, P. Schlüter, H. Scherer and I. Krossing, Chem.–Eur. J., 2012, 18, 9371 CrossRef PubMed.
- A. Wiesner, T. W. Gries, S. Steinhauer, H. Beckers and S. Riedel, Angew. Chem., 2017, 129, 8375 ( Angew. Chem., Int. Ed. , 2017 , 56 , 8263 ) CrossRef.
- (a) T. Belgardt, J. Storre, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Inorg. Chem., 1995, 34, 3821 CrossRef CAS; (b) J. Klosin, G. R. Roof and E. Y.-X. Chen, Organometallics, 2000, 19, 4684 CrossRef CAS.
- N. G. Stahl, M. R. Salata and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 10898 CrossRef CAS PubMed.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396 CrossRef CAS PubMed.
- R. E. LaPointe, G. R. Roof, K. A. Abboud and J. Klosin, J. Am. Chem. Soc., 2000, 122, 9560 CrossRef CAS.
- J. Chen and E. Y.-X. Chen, Angew.Chem., 2015, 127, 6946 ( Angew. Chem., Int. Ed. , 2015 , 54 , 6842 ) CrossRef.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamaki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983 CrossRef CAS PubMed.
- D. W. Stephan, Nat. Chem., 2014, 6, 952 CrossRef CAS PubMed.
- V. C. Williams, W. E. Piers, W. Clegg, M. R. J. Elsegood, S. Collins and T. B. Marder, J. Am. Chem. Soc., 1999, 121, 3244 CrossRef CAS.
- L. A. Körte, J. Schwabedissen, M. Soffner, S. Blomeyer, C. G. Reuter, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., 2017, 129, 8701 ( Angew. Chem., Int. Ed. , 2017 , 56 , 8578–8582 ) CrossRef.
- It should be noted that the discussion above is only concerned with neutral Lewis acids. For FIA values of strong cationic Lewis acids see ref. 12 as well as: (a) J. M. Slattery and S. Hussein, Dalton Trans., 2012, 41, 1808 RSC; (b) M. Schleep, C. Hettich, J. Velázquez Rojas, D. Kratzert, T. Ludwig, K. Lieberth and I. Krossing, Angew. Chem., 2017, 129, 2926 ( Angew. Chem., Int. Ed. , 2017 , 56 , 2880 ) CrossRef; (c) T. O. Petersen, D. Simone and I. Krossing, Chem.–Eur. J., 2016, 22, 15847 CrossRef CAS PubMed.
- A. Y. Timoshkin and K. Morokuma, Phys. Chem. Chem. Phys., 2012, 14, 14911 RSC.
- L. A. Mück, A. Y. Timoshkin and G. Frenking, Inorg. Chem., 2012, 51, 640 CrossRef PubMed.
- For a recent review see: T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789 RSC.
- A. J. Lehner, N. Trapp, H. Scherer and I. Krossing, Dalton Trans., 2011, 40, 1448 RSC.
- I. Krossing, H. Brands, R. Feuerhake and S. Koenig, J. Fluorine Chem., 2001, 112, 83 CrossRef CAS.
- X. Chen, B. Ma, X. Wang, S. Yao, L. Ni, Z. Zhou, Y. Li, W. Huang, J. Ma, J. Zuo and X. Wang, Chem.–Eur. J., 2012, 18, 11828 CrossRef CAS PubMed.
- M. Gonsior and I. Krossing, Dalton Trans., 2005, 7, 1203 RSC.
- T. Köchner, N. Trapp, T. A. Engesser, A. J. Lehner, C. Röhr, S. Riedel, C. Knapp, H. Scherer and I. Krossing, Angew. Chem., 2011, 124, 11253 ( Angew. Chem., Int. Ed. , 2011 , 50 , 11253 ) CrossRef PubMed.
- A. Bihlmeier, M. Gonsior, I. Raabe, N. Trapp and I. Krossing, Chem.–Eur. J., 2004, 10, 5041 CrossRef CAS PubMed.
- J. Li, C. Schenk, F. Winter, H. Scherer, N. Trapp, A. Higelin, S. Keller, R. Pöttgen, I. Krossing and C. Jones, Angew. Chem., 2012, 124, 9695 ( Angew. Chem., Int. Ed. , 2012 , 51 , 9557 ) CrossRef.
- (a) G. Santiso-Quiñones, A. Reisinger, J. Slattery and I. Krossing, Chem. Commun., 2007, 5046 RSC; (b) I. Krossing and A. Reisinger, Angew. Chem., 2003, 115, 5903 ( Angew. Chem., Int. Ed. , 2003 , 42 , 5725 ) CrossRef; (c) A. Reisinger, T. Trapp, C. Knapp, D. Himmel, F. Breher, H. Rüegger and I. Krossing, Chem.–Eur. J., 2009, 15, 9505 CrossRef CAS PubMed; (d) J. Schaefer, D. Himmel and I. Krossing, Eur. J. Inorg. Chem., 2013, 52, 2712 CrossRef.
- A. Budanow, T. Sinke, J. Tillmann, M. Bolte, M. Wagner and H.-W. Lerner, Organometallics, 2012, 31, 7298 CrossRef CAS.
- J. M. Slattery, A. Higelin, T. Bayer and I. Krossing, Angew. Chem., 2010, 122, 3297 ( Angew. Chem., Int. Ed. , 2010 , 49 , 3228 ) CrossRef.
- A. Higelin, U. Sachs, S. Keller and I. Krossing, Chem.–Eur. J., 2012, 18, 10029 CrossRef CAS PubMed.
- J. F. Kögel, L. H. Finger, N. Frank and J. Sundermeyer, Inorg. Chem., 2014, 53, 3839 CrossRef PubMed.
- (a) T. Linder and J. Sundermeyer, Chem. Commun., 2009, 29146 Search PubMed; (b) J. F. Kögel, T. Linder, F. G. Schröder, J. Sundermeyer, S. K. Goll, D. Himmel, I. Krossing, K. Kuett, J. Saame and I. Leito, Chem.–Eur. J., 2015, 21, 5769 CrossRef PubMed.
- A. Khvorost, Dissertation, Philipps-Universität Marburg, 2003 Search PubMed.
- (a) G. M. Brooke, J. Burdon, M. Stacey and J. C. Tatlow, J. Chem. Soc., 1960, 1768 RSC; (b) R. Koppang, Acta Chem. Scand., 1971, 25, 3067 CrossRef CAS.
- A. Khvorost, P. L. Shutov, K. Harms, J. Lorberth, J. Sundermeyer, S. S. Karlov and G. S. Zaitseva, Z. Anorg. Allg. Chem., 2004, 630, 885 CrossRef CAS.
- D. R. Click, B. L. Scott and J. G. Watkin, Chem. Commun., 1999, 633 RSC.
- H. Yin, A. J. Lewis, P. Carroll and E. J. Schelter, Inorg. Chem., 2013, 52, 8234 CrossRef CAS PubMed.
- H. Yin, A. J. Lewis, U. J. Williams, P. J. Carroll and E. J. Schelter, Chem. Sci., 2013, 4, 798 RSC.
- G. R. Giesbrecht, J. C. Gordon, D. L. Clark, C. A. Hijar, B. L. Scott and J. G. Watkin, Polyhedron, 2003, 22, 153 CrossRef CAS.
- D. V. Peryshkov and R. R. Schrock, Organometallics, 2012, 31, 7278 CrossRef CAS.
- H. Plenio, Chem. Rev., 1997, 97, 3363 CrossRef CAS PubMed.
- R. D. Schluter, H. S. Isom, A. H. Cowley, D. A. Atwood, R. A. Jones, F. Olbrich, S. Corbelin and R. J. Lagow, Organometallics, 1994, 13, 4058 CrossRef CAS.
- G. A. Olah, P. Schilling and I. M. Gross, J. Am. Chem. Soc., 1974, 96, 876 CrossRef CAS.
- H. Hatop, H. W. Roesky, T. Labahn, C. Röpken, G. M. Sheldrick and M. Bhattacharjee, Organometallics, 1998, 17, 4326 CrossRef CAS.
- H. Hatop, M. Schiefer, H. W. Roesky, R. Herbst-Irmer and T. Labahn, Organometallics, 2001, 20, 2643 CrossRef CAS.
- A. G. Avent, W.-Y. Chen, C. Eaborn, I. B. Gorrell, P. B. Hitchcock and J. D. Smith, Organometallics, 1996, 15, 4343 CrossRef CAS.
- The reaction of the metal amides with neutral donor molecules yielded Lewis acid-base complexes. Their structural characterization and theoretically determined binding affinity will be treated in a separate publication. Unlike in case of the aluminum compound, the reaction of Ga[N(C6F5)2]3 with CsF in toluene did not yield the corresponding metallate [FGa(N(C6F5)2)3]− in a highly selective manner. Thus, we only present the chloride ion adduct in this work. However, synthesis of [FGa(N(C6F5)2)3]− should of course be possible.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- T. H. Dunning, K. A. Peterson and A. K. Wilson, J. Chem. Phys., 2001, 114, 9244 CrossRef CAS.
- N. J. DeYonker, K. A. Peterson and A. K. Wilson, J. Phys. Chem. A, 2007, 111, 11383 CrossRef CAS PubMed.
- R. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS.
- A. Kraft, J. Beck and I. Krossing, Chem.–Eur. J., 2011, 17, 12975 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1557072–1557076. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03988c |
| ‡ Current address: FB Biologie/Chemie, Universität Bremen, Leobener Str. im NW2, 28359 Bremen, Germany. |
| This journal is © The Royal Society of Chemistry 2018 |