
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCaged circular siRNAs for photomodulation of gene expression in cells and mice†
Liangliang
Zhang‡
a,
Duanwei
Liang‡
a,
Yuan
Wang‡
a,
Dong
Li‡
a,
Jinhao
Zhang
a,
Li
Wu
a,
Mengke
Feng
a,
Fan
Yi
a,
Luzheng
Xu
b,
Liandi
Lei
b,
Quan
Du
a and
XinJing
Tang
 *a
*a
aState Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, No. 38, Xueyuan Rd, Beijing 100191, China. E-mail: xinjingt@hsc.pku.edu.cn
bMedical and Health Analytical Center, Peking University, No. 38, Xueyuan Rd, Beijing 100191, China
First published on 19th October 2017
Abstract
By means of RNA interference (RNAi), small interfering RNAs (siRNAs) play important roles in gene function study and drug development. Recently, photolabile siRNAs were developed to elucidate the process of gene silencing in terms of space, time and degree through chemical modification of siRNAs. We report herein a novel type of photolabile siRNA that was synthesized through cyclizing two ends of a single stranded RNA with a photocleavable linker. These circular siRNAs became more resistant to serum degradation. Using reporter assays of firefly/Renilla luciferase and GFP/RFP, the gene silencing activities of caged circular siRNAs for both genes were evaluated in HEK293 cells. The results indicated that the target genes were successfully photomodulated using these caged circular siRNAs that were formed by caged circular antisense guide RNAs and their linear complementary sense RNAs. Using the caged circular siRNA targeting GFP, we also successfully achieved photomodulation of GFP expression in mice. Upon further optimization, this new type of caged circular siRNA is expected to be a promising tool for studying gene therapy.
Introduction
RNA interference is a potent and specific gene silencing approach1,2 which usually requires short double-stranded RNA and results in RISC formation and Dicer processing.3,4 Generally, two strategies are used to produce double-stranded RNA: (1) precursor shRNAs are transcribed endogenously from plasmids into shRNAs by RNA polymerases, followed by Dicer processing to produce active siRNAs within cells; (2) chemically synthetic siRNAs are formed by two complementary single-stranded RNAs. Although shRNA is more biologically relevant, synthetic siRNAs are currently still the most widely used for both laboratory and clinical applications.5–9Despite the potency and specificity of siRNAs in gene silencing, spatial and/or temporal regulation of siRNA activity is still difficult due to the constitutive transcription of shRNAs or delivery obstacles of siRNAs. Similar to other reported photolabile oligonucleotides,10–29 photolabile siRNAs have been developed to achieve dedicated regulation of gene expression.30–43 To date several approaches have been developed based on the unique structural properties of siRNAs and the processing of RNAi (Fig. 1). Disturbing a siRNA duplex conformation might interfere with the formation of siRNA/RISC complexes and/or further siRNA processing. Heckel et al. incorporated a 2-(2-nitrophenyl)propyl (NPP)-caged deoxyguanosine nucleotide in a siRNA antisense guide strand at the 9th to the 11th position. This modification approach did interfere with the formation of the siRNA/RISC complex, leading to compromised siRNA activity.37 In another study, Deiters et al. synthesized 6-nitropiperonyloxymethyl (NPOM)-caged guanosine and uridine phosphoramidites and site-specifically incorporated these caged nucleosides into the antisense guide strand of siRNAs at the cutting site and/or seed region.30 Stochastic labelling of the phosphate backbone of siRNA duplexes is another approach for the synthesis of photolabile siRNAs through coupling chemistry of the photolabile diazo moiety with phosphate groups.
 | ||
| Fig. 1 Summary of different approaches for caging siRNAs. (A) Caging moieties on nucleobases, the phosphate backbone and four terminal phosphates; (B) caged hairpin siRNA linked via a photocleavable linker; (C) caged dumbbell shaped siRNA with two ends linked via two photocleavable linkers; (D) caged circular siRNA with two ends of sense or antisense RNA linked via a photocleavable linker for this work. | ||
Friedman et al. reported DNMPE-caged double-stranded siRNAs to photomodulate the silencing of GFP expression without interrupting RFP expression in the cells.31–33 Using a similar approach, 2′-F substituted siRNAs were caged and their RNAi activities on transient GFP repression were photomodulated in zebrafish embryos with spatial resolution.41 The number of caging groups per siRNA increased with higher DMNPE diazo concentration. Heavily caged siRNA led to the elimination of its gene silencing activity, but such repression could not be fully recovered with cell-affordable light irradiation.33,37,41 By attaching a large caging group (cyclododecyl 4,5-dimethoxy-2-nitrophenyethyl moiety) at all four terminal phosphate groups, caged sdRNAs were developed for preventing RNA processing and further gene silencing activity.34 Site-specific labelling of the terminal phosphate group was also achieved through the attachment of DMNPE or a biotinylated photocleavable linker.36 However, this modification approach is partially tolerated due to the intact 5′-phosphoester moiety. Previously in our lab, we caged each phosphate group of siRNA through site-specific incorporation of a photolabile nucleotide phosphoramidite and screened all possible caging phosphate positions for efficient photomodulation of siRNA activity.44 We recently reported caged siRNA modified with a single vitamin E at the 5′ terminal phosphate of antisense RNA and successfully achieved the photomodulation of RNAi-induced gene silencing, possibly due to the binding of vitamin E receptor protein.45 Another kind of caged siRNA was developed by linking an antisense guide strand with a complementary sense strand RNA via a photocleavable linker. Unfortunately, no improvement of the photomodulation of siRNA activity was observed.36
Based on our and others’ previous achievements on caged circular antisense oligonucleotides,19–21,46 we further intended to develop a new generation of caged siRNAs with a circular structure. In the literature, usually sense and antisense strands are cyclized to form siRNAs or shRNAs with a dumbbell structure.40,47,48 Xi et al. reported circular dumbbell sdRNAs with an alkyl linkage. They found that this was more potent in RNAi gene silencing than their open-ended counterpart, likely due to their enhanced stability.48 Dmochowski et al. also reported a caged circular siRNA duplex with the dumbbell structure, where two photocleavable linkers were used to link the 5′ and 3′ ends of the sense and antisense siRNA strands (Fig. 1C).40 Different from previously reported circular RNAs, we proposed to develop a novel type of caged circular siRNA (Fig. 1D). Cyclization of the 5′ and 3′ ends of the sense or antisense single-stranded RNA was achieved to form a 21-nucleobase ring through both chemical and enzymatic cyclization methods, then a complementary RNA strand was hybridized to the caged circular RNA strand to form a caged circular siRNA duplex (Table 1). Upon exposure to light, the circular siRNAs were restored to the corresponding linear ones with a 5′ terminal phosphate group of RNA oligonucleotides. Using a dual luciferase assay49 and GFP expression assay, photoregulation of firefly luciferase reporter gene expression and GFP expression was achieved by this new type of caged circular siRNA. Further in vivo studies confirmed that it was possible to achieve photomodulation of siRNA activity in a mouse tumor model using the caged circular siRNA.

|
Experimental
General experimental procedures
All chemical reactions in the synthesis of the photocleavable linker were performed under an inert atmosphere using dry reagents and solvents. Column chromatography was performed with silica gel 60 (200–300 mesh). 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were taken on Bruker AVANCE III-400 spectrometers and standardized to the solvent NMR peak. Mass spectra of small molecules were obtained on a Waters Xevo TQD Mass Spectrometer using electrospray ionization (ESI). The reactions were conducted in a dark room when necessary.Purification of caged circular RNAs
Crude caged circular RNAs obtained from chemical synthesis or enzymatic synthesis were dissolved in PBS buffer and mixed with RNA loading buffer (0.25% bromophenol blue, 30% glycerol in DEPC-treated water). The solutions (6 μL per well) were loaded to 20% native PAGE (8.6 × 6.8 cm and 1 mm thick) gels. The gels were then electrophoresed at 80 V for 3 h, using Tris–borate–EDTA (TBE) buffer (pH 8.2). For each preparative gel, the two side lanes of the gel were cut and stained with SYBR Gold (Invitrogen) for gel imaging. The images were printed out according to the size of the gel in order to mark the location of the circular RNA without SYBR Gold stain. The gel at the marked location was cut, crumbled into tiny particles and immersed into TBE buffer at 37 °C overnight. After removing the solid gel particles through filtration, RNA solutions were desalted and concentrated using Millipore-amicon ultra-0.5 mL centrifugal filters (cutoff = 3000). The collected products were freeze-dried to the final caged circular single-stranded RNA.The caged circular single-stranded RNA was dissolved in PBS buffer to make a 20 μmol L−1 stock solution and was then mixed with an equal amount of the linear complementary RNA to form the caged circular siRNA duplex. This duplex was annealed by heating to 80 °C for 5 minutes and subsequent cooling to room temperature for at least 1 hour for further use. Tms of linear and circular siRNAs were measured with a DU800 UV-Vis spectrometer with 1 μM concentration of the corresponding duplexes.
Characterization of RNA oligonucleotides
Single-stranded oligonucleotides (∼0.2 nmol) were dissolved in water/acetonitrile (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, 20 μL) containing 1% triethylamine to make a final concentration of 10 μM. The solutions were then analyzed with a Waters Xevo G2 Q-Tof spectrometer with electrospray ionization (ESI) in the negative ion mode.
:50, 20 μL) containing 1% triethylamine to make a final concentration of 10 μM. The solutions were then analyzed with a Waters Xevo G2 Q-Tof spectrometer with electrospray ionization (ESI) in the negative ion mode.
The double-stranded siRNA was characterized with MALDI-TOF-MS. The matrix solution was made by dissolving 2,5-dihydroxybenzoic acid (DHB, 10 mg) in 50% acetonitrile/water solution (1 mL). The sample solution (10 μM, 0.8 μL) and the matrix solution (0.8 μL) were mixed and spotted on the surface of the sample plate. After the sample was dried, siRNA was then analysed with MALDI-TOF-MS according to the manufacturer’s protocol.
Photocleavage ability of caged circular single-stranded RNA
Caged circular single-stranded RNA was diluted with PBS buffer (pH 7.2) to make a final concentration of 2 μM (30 μL). The solution (5 μL per well) was irradiated using a UV-LED lamp (365 nm, 7 mW cm−2) from the bottom of the plate for 0, 1, 2, 3, 4, 5 and 6 minutes. After mixing with 1 μL RNA loading buffer, the samples were subject to gel analysis for photocleavage study using 20% native PAGE under similar gel running conditions of RNA purification.Serum stability of a caged circular siRNA
Caged circular siRNA or control linear siRNA (10 μM, 2 μL) was incubated at 37 °C in enzyme buffer solution to make a final concentration of 2 μM (20 μL). 30% fetal bovine serum (FBS) was used for studying siRNA stability. 5 μL of solution was aliquoted at different time points, and immediately frozen in liquid nitrogen and then stored at −80 °C until assaying. 1 μL of 6× RNA loading buffer was added to the aliquots. The samples were run on 20% native polyacrylamide gels in 1× TBE buffer according to the procedure mentioned before.Cell culture and RNAi assay (luciferase)
HEK293 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 units per mL penicillin and 100 μg mL−1 streptomycin (Life Technologies, Gibco). The cells were seeded into 24-well plates at ∼1 × 105 cells per well one day before transfection (lipofectamine 2000). siQuant vectors (100 ng per well) carrying the target site of the siRNA were transfected into HEK293 cells at ∼50% confluence, together with the pRL-TK control vector (50 ng per well), with or without the control siRNAs or caged circular siRNAs (1 or 5 nM). The activities of both luciferases were determined by a Synergy HT fluorometer (BioTek, USA) and then firefly luciferase activity was normalized to Renilla luciferase for each well. The silencing efficacy of each siRNA was calculated by comparison with samples transfected with only vectors without siRNA treatment. For light-irradiated cellular experiments, after transfection, the cells were cultured for another 4 h, washed with PBS, and then irradiated using a UV-LED lamp (365 nm, 7 mW cm−2) from the bottom of the plate for 3 minutes.Cell culture and RNAi assay (GFP expression assay)
HEK293 cells were trypsinized and seeded in 12 well plates with 1 mL 2 × 105 cells mL−1 for each well. According to the standard transfection protocol, the reporter plasmids (pEGFP-N1, pDsReds-N2) and corresponding siRNAs were cotransfected to cells using Lipofectamine 2000 in optiMEM. After 4 h, cells in one set of experiments were irradiated to induce uncaging of photolabile circular siRNAs. Then all the cells were subject to their mediums being replaced with 1 mL fresh DMEM containing 10% FBS for each well. After another 20 h culture in the incubator, the images of each well were collected by automatic inverted fluorescence microscopy (Olympus, IX83) under the same condition. The excitation and emission wavelengths are 488 nm and 509 nm for GFP and 560 nm and 585 nm for RFP, respectively.After plasmid and siRNA transfection and incubation under the same condition, the transfected cells were washed using 1× PBS buffer (pH 7.2) and trypsinized, and their fluorescence intensity was further quantified by flow cytometry (BD FACSAria II) with RFP gene expression as an internal control. All experiments were repeated at least three times.
Half-well patterning experiment
HEK293 cells were trypsinized and then seeded in 6 well plates with 2 mL 2 × 105 cells mL−1 for each well. After the cells were cotransfected with GFP (400 ng per well) and RFP (400 ng per well) plasmids as well as SG/c-AG (10 nM) for 4 h, black adhesive tape was aligned to cover half of each well to avoid light irradiation. Then the wells were irradiated for 3 minutes and the medium was replaced with 2 mL fresh DMEM containing 10% FBS. After incubation for another 20 h, the images were taken by automatic inverted fluorescence microscopy and were analyzed using Volocity Demo 6.1.1.Animal experiment
Six-week-old BALB/c nude mice (Department of Laboratory Animal Science of the Peking University Health Science, Beijing, China) were subcutaneously injected in the inner left and right thighs with U87-GFP cells (6 × 105 cells per site in a volume of 60 μL). When the tumors grew to around 0.2 cm in diameter (about 4 days), the mice were randomly divided into two groups for intratumoral injection. One group was injected with PBS on the left and with linear siRNAs (SG/AG) on the right side, respectively. Another group was injected with caged circular siRNAs (SG/c-AG) on both sides. For each injection, 3 nmol linear siRNAs (SG/AG) or caged circular siRNAs (SG/c-AG) was mixed with 20 μL transfection reagent (Entranster-in vivo; Engreen, Beijing, China) in vivo and then incubated for 15 minutes at room temperature. The volume of each injection was 60 μL. All injections were in accordance with the manufacturer’s instructions. 4 h after injection, the mice injected with the caged circular siRNA were fixed on a table and their left tumors were irradiated using a UV-LED lamp (365 nm, 7 mW cm−2) for 3 minutes, whereas other parts of the body were covered by tinfoil to avoid light exposure. The irradiated mice were then marked and returned to live with other mice in the group. These mice were then imaged at different time points (0 h, 12 h, 24 h and 48 h) using Maestro Automated In Vivo Imaging system. Then tumor fluorescence was quantified under the same parameters, and the fluorescence intensity of tumors at different time points was normalized to that of each mouse at just before the siRNA injection. The facility is in keeping with national standard “Laboratory Animal—Requirements of Environment and Housing Facilities” (GB 14925—2001). The care of laboratory animals and animal experimental operation conform to “Beijing Administration Rule of Laboratory Animal” and was approved by the Animal Care and Use Committee of Peking University.Results and discussion
Synthesis and purification of caged circular siRNAs
In our previous study, caged antisense oligonucleotides were synthesized by first coupling with photolabile o-nitrophenyl glycol phosphoramidite, followed by coupling of MMT–NH–C6 phosphoramidite.21,30 The MMT group was then removed to release free amine for the coupling reaction with succinic anhydride to introduce carboxylic acid for the cyclization of oligonucleotides. Here we simplified the procedure through a single step coupling reaction, in which a new photolabile phosphoramidite linker (PL, Table 1) was designed to introduce carboxylic acid at the 5′ terminal of single-stranded RNA for cyclization. A 5′ phosphate group was generated in the activated siRNA after PL was removed upon light activation. As shown in Scheme S1,†PL was readily synthesized in five steps with 37.6% yield through Grignard reagent exchange and coupling,50,51o-Cl-trityl protection of carboxylic acid, ketone reduction and phosphoramidition.To introduce an amine group at the 3′ terminal of the RNA strand, 5′-DMT-T (C6 amino)-succinyl-CPG was used as the first nucleotide of the sequence, which contains a 3′ terminal amine with no effect on the 3′ terminal hydroxyl group. After RNA synthesis, the photocleavable phosphoramidite linker (PL) was coupled to the RNA sequence as the last coupling monomer according to the standard RNA synthesis procedure in DMT-off mode. Raw oligonucleotides containing the 3′ amine and 5′ acid functional groups were obtained after cleavage and deprotection with concentrated ammonium hydroxide, followed by removal of TBDMS on RNA nucleotide monomers with TEA.3HF solution and RP-HPLC purification. The obtained single-stranded RNA was then cyclized with the formation of an amide bond (Fig. S1†) and the yield of isolated circular RNA is around 20%.
The caged circular RNA oligonucleotides were purified by RP-HPLC, as shown in Fig. S2.† However, when the collected peaks were further analyzed by PAGE, the presence of a tiny amount of single stranded starting RNA was still observed. Due to the fact that the pathway of siRNA is a catalytic process, the existence of even a small amount of linear siRNA may result in an obvious RNAi effect. We then chose to purify these caged circular RNAs through PAGE gels as shown in Fig. S3.† Compared to linear RNAs, the circular RNAs ran faster in PAGE gel, therefore facilitating further purification of the caged circular RNAs. The caged circular single-stranded RNA was then collected and analyzed by ESI-MS in negative mode [1% TEA in H2O/CH3CN (50:50)] as listed in Tables S1 and S2.† With the addition of an equal amount of complementary RNA, the caged circular siRNA was annealed and evaluated in this study.
Photocleavage of caged circular single-stranded RNAs
In order to find an appropriate irradiation time to efficiently cleave the photolabile linker, the single stranded caged circular RNA strand was treated by light irradiation to recover the corresponding linear RNA (Scheme S2†). Upon light irradiation (0, 1, 2, 3, 4, 5 and 6 min), the irradiated samples were then subject to gel analysis. Fig. 2 shows that the band of caged circular RNA decreased gradually and almost completely disappeared in 5 min. Simultaneously, the recovered linear RNA increased gradually and reached a maximum at around 5 minutes under our photolysis conditions, and no further improvement was observed even with a longer irradiation time. Under the same light irradiation condition, cell viability was also evaluated using MTT assay, revealing no significant toxicity within the 3 min light irradiation interval (Fig. S5†).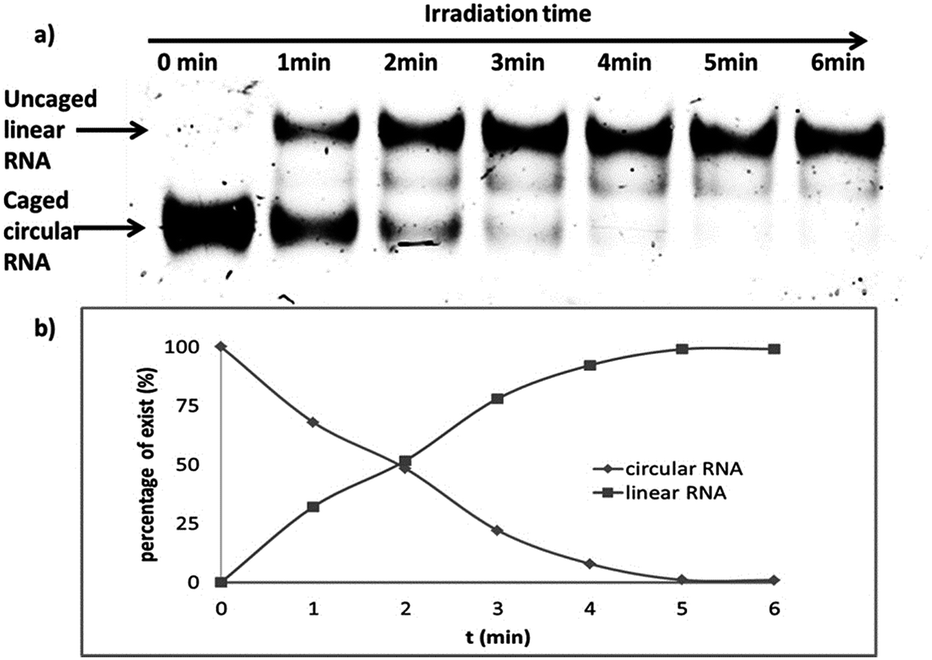 | ||
| Fig. 2 PAGE (20%) analysis of the photocleavage ability of a caged circular single-stranded RNA (2 μM, c-AL) in 1× PBS buffer (pH 7.2). The caged RNA was irradiated using a UV-LED lamp (365 nm, 7 mW cm−2) for 0, 1, 2, 3, 4, 5 and 6 minutes. The charts are percentages of the circular and cleaved RNAs upon light irradiation at different time intervals after gel was quantified with ImageQuant. | ||
Serum stability of the caged circular siRNA duplex
To evaluate the improvement of the serum stability of the caged circular siRNAs, the caged circular siRNA and linear siRNA duplexes were treated with fetal bovine serum (FBS). During 2 h treatment, aliquots were sampled at regular intervals, quickly frozen and stored for gel-shift analysis. When incubated in 30% FBS at 37 °C, the control linear siRNA (PC) started to be degraded in the first half hour with an obvious degradation band, as shown in Fig. 3. However, for the caged circular siRNA duplex (SL/c-AL), only a tiny amount of siRNA digestion occured in 4 hours, which indicated that the caged circular RNA was more resistant to FBS treatment. Therefore, we concluded that caged siRNA duplexes are stabilized by their circular structure in a biological environment. | ||
| Fig. 3 Serum stability of the caged circular siRNA (SL/c-AL) and control linear siRNA (PC) was analyzed with 30% fetal bovine serum (FBS). Aliquots were sampled at regular intervals, quickly frozen and stored for native PAGE gel-shift analysis. | ||
Gene silencing potency of caged circular siRNAs
The gene silencing potency of the caged circular siRNAs was evaluated together with their linear counterpart (PC) using a well-established dual-luciferase assay.49 Caged circular siRNA duplexes (c-SL/AL and SL/c-AL, Table 1) and control siRNA (PC) were transfected to cultured HEK293 cells at a final concentration of 1 nM, together with a fusion firefly luciferase vector carrying its target site and a Renilla luciferase vector serving as an internal control. Two sets of experiments were carried out under the same conditions except for light activation. After 4 hours of transfection, one set of experiments was subject to light irradiation for 3 minutes to activate the caged circular siRNA, while the other set was untreated and kept in dark conditions, serving as a control. As expected, no siRNAs induced gene silencing of Renilla luciferase under both irradiation and non-irradiation conditions, which indicated that light had no obvious off-target effect on gene expression under the current assay conditions (Table S3†). Instead, PC siRNA (positive control for firefly luciferase) showed efficient inhibition of firefly luciferase activity (Fig. 4). The caged siRNA duplex (c-SL/AL) with a circular sense strand RNA has significant basal activity for silencing firefly luciferase with or without light activation. For the caged siRNA with a circular antisense guide strand (SL/c-AL), RNAi activity was effectively inhibited before light irradiation. Upon light irradiation, the ring of caged circular siRNA was opened and siRNA was activated, inducing a 2.4 fold enhancement of the gene silencing potency. However, full recovery of siRNA activity was not restored. This is probably due to the blocking effect of the large residue of the photolabile moiety and long linker. | ||
| Fig. 4 Photomodulation of firefly luciferase expression in HEK293 cells with caged circular siRNAs using a dual-luciferase assay. “PC”, positive control; “c-SL/AL”, caged sense stranded siRNA; “SL/c-AL”, caged antisense stranded siRNA. The data were averaged in triplicate and all the experiments were repeated three times. | ||
In this study, we observed quite a strong inhibition of firefly luciferase activity with the caged siRNA duplex c-SL/AL even before light activation. According to our design, both the c-SL/AL and c-AL/SL have the same sequence. When they paired with their complementary RNA strand, they should form a similar circular structure and function similarly in RNAi-based gene silencing. However, different gene silencing abilities were found. We then tried to analyze the caged circular siRNA duplex. Equal amounts of caged circular RNA and its complementary strand were annealed and the duplex was run in a native PAGE gel at 37 °C. As shown in Fig. S4,† we did observe two bands on the gel. RNAs from both bands were recollected and were then subject to another round of PAGE analysis. Surprisingly, both fragments were divided into the same two bands again on the gel. These results indicated that these two fragments were two interchangeable conformers of the caged circular siRNA, similar to the previously predicted circular DNA duplex.52 This phenomenon also exhibited similarity with our previous observation, where multiple bands showed up when a 40 mer RNA binds to a circular 20 mer DNA on native PAGE gel.26 In addition, for the circular siRNA duplex, each strand of the siRNA duplex has another two additional unpaired dangling thymidines, which may cause a very crowded region and charge repulsion between phosphate groups of the two ends and can further decrease the thermostability of the caged circular siRNA duplex. This observation is confirmed by the Tms of the linear siRNA and circular siRNA duplexes (Fig. S5†). The Tm of linear siRNA (SL/AL) was 70 °C with the standard melting curve of a complementary duplex. However, the melting curve of circular siRNA (c-SL/AL) displayed a gradual increase of UV absorption at 260 nm from 30 to 85 °C, indicating that dissociation of two RNA strands of the circular siRNA happened at around 30 °C. So if the noncircular RNA strand is the antisense guide strand of the siRNA duplex (c-SL/AL), it is possible that the antisense guide strand will leave its circular partner and form a more stable RNA duplex with its target mRNA which triggers RNA interference even though no light irradiation is applied.
To confirm the generality of gene silencing with the caged circular siRNAs and overcome the blocking effect of the large residue of terminal modified thymidine, circular siRNA for GFP was redesigned using another photolabile linker (1-nitrobenzyl 1,2-glycol, PL2) and was synthesized through enzymatic cyclization. The phosphoramidite of this PL2 linker could be inserted in the middle of the RNA sequence in solid phase synthesis. The obtained modified RNA with a 5′ terminal phosphate group was then enzymatically cyclized to form circular RNA. After light activation, linear RNA was recovered with only a small residue at the 3′ terminal of RNA, which had little effect on siRNA activity (Scheme S2†). A caged circular antisense RNA strand of GFP-targeting siRNA was then prepared and hybridized with its complementary sense RNA, which was used for GFP gene photoregulation in cells. HEK293 cells were cotransfected for 4 hours with pEGFP-N1 and pDsReds-N2, as well as SG/AG or SG/c-AG (Table S2†). Two sets of experiments were performed with or without brief light activation. After another 20 hours of incubation, the cells were imaged and GFP/RFP expression was quantified by flow cytometry with RFP gene expression as an internal control. As expected, GFP and RFP expression were not affected by light irradiation in both negative and positive control experiments (Fig. 5A) which proved that the regulation of GFP and RFP expression was genetically specific. Fluorescence images showed that there were tremendous contrasts on the GFP expression in the set of caged circular siRNAs (SG/c-AG) with or without light irradiation. It is clear that the fluorescence intensity of cells with SG/c-AG transfection was similar to that of the negative control cells before light irradiation. However, after exposure to UV light, the cellular fluorescence intensity turned to the level of the positive control cells. As shown in Fig. 5B, the GFP expression level in cells transfected with GFP siRNA (SG/AG) at a 2.5 nM concentration was reduced to 34% of negative control cells (Fig. 5B). In the presence of caged circular siRNAs (SG/c-AG), almost no effect could be observed on GFP expression in the absence of light irradiation. This displays the inactivity of the caged circular siRNA in the cells. However, its gene silencing activity was recovered to the level of the corresponding linear siRNA after brief light activation. The dose dependency of uncaged siRNA on GFP gene silencing was also observed with more than 3 fold photomodulation of GFP gene silencing using SG/c-AG siRNA (10 nM), which indicated that our caged circular siRNA strategy is a new way to photomodulate gene expression in cells.
 | ||
| Fig. 5 Photomodulation of GFP expression in HEK293 cells cotransfected with pEGFP-N1, pDsRed-N1, and caged circular siRNAs (SG/c-AG). Cells were irradiated for 3 min (365 m, 7 W cm−2) or kept in the dark. (A) Cells were imaged using automatic inverted fluorescence microscopy with GFP and RFP channels and the scale bars represent 100 μm; (B) dose effect on photomodulation of GFP expression quantified through cell flow cytometry. The concentration of PC siRNA (SG/AG) was fixed at 2.5 nM. All the experiments were repeated three times; (C) the spatial regulation of GFP expression with patterned irradiation (left side view) and the scale bar represents 100 μm. | ||
Furthermore, the spatial control of GFP expression using caged circular siRNA (SG/c-AG) was tested. GFP/RFP plasmids and SG/c-AG were cotransfected to cells and then half of the cultured plate was irradiated to activate the caged circular siRNA. The cells were further incubated for another 20 h and were then imaged by automatic inverted fluorescence microscopy. As shown in Fig. 5C, GFP expression in the irradiated region was virtually silenced. Nevertheless the non-irradiated cells retained normal GFP expression activity. This result confirmed that our caged circular siRNA strategy has great effectivity on the spatial control of gene expression.
Photoregulation of GFP expression in vivo using caged circular siRNA
Based on the promising results of cell studies, we further applied the caged circular siRNA for photomodulation of GFP gene expression in mice (Fig. 6). An in vivo xenograft tumor model with U87-GFP cells was established with two tumors in each mouse. When the tumors grew to around 0.2 cm in diameter, PBS or linear or caged circular siRNAs (SG/c-AG or C-siRNA) were injected into the tumors. For the group injected with SG/c-AG, the left tumor was then tested with brief light exposure. The mice were imaged using Maestro Automated In Vivo Imaging system and the fluorescence intensity of the tumors was quantified at different time points. As expected, GFP expression in tumors gradually increased in the PBS injected mice as time went on, even with the same light exposure (Fig. S6†). For the tumors injected with linear siRNAs (SG/AG), an expected fluorescence decrease was observed after siRNA injection, but the tumor fluorescence intensities gradually returned to their original level over 48 h (Fig. 6B). For the tumors injected with caged circular siRNA (SG/c-AG or C-siRNA) shown in Fig. 6, fluorescence images indicated that there was a significant difference in the GFP expression with or without light irradiation (Fig. 6A). | ||
| Fig. 6 Photomodulation of GFP expression in vivo for the xenograft tumor model with U87-GFP cells using caged circular siRNA (SG/c-AG). (A) Typical in vivo real-time fluorescence imaging at the indicated time points after intratumor injection of caged circular siRNAs on both sides, and the left side tumor was exposed to light for 3 min at 4 h after injection. (B) Normalized GFP intensity bar chart of tumor GFP fluorescence after intratumor injection of PBS (1 mM), linear siRNA or caged circular siRNA with or without light irradiation. Mean and SD values are from experiments performed in four mice. | ||
In the absence of light irradiation, the GFP fluorescence intensity of the tumor decreased only slightly, then quickly recovered and surpassed the original GFP level. In comparison, upon exposure to UV light, the caged circular siRNA was uncaged and further down-regulated GFP expression efficiently with the same potency as that of linear siRNA after a single intratumor injection of siRNA. These tumors were then isolated after 48 hours and further imaged. The data indicated the more obvious observation of GFP silencing using caged circular siRNA for photomodulation of gene expression in vivo (Fig. S7†).
Conclusions
We designed and synthesized a new type of caged siRNA with a circular structure. These caged circular siRNAs showed increased stability to serum. Upon light exposure, these caged circular siRNAs were converted to the linear version of the siRNAs containing a 5′ phosphate group, with no need for further enzymatic phosphorylation. Different RNA induced gene silencing activity was observed for caged siRNAs with circular sense RNA or circular antisense RNA. A caged siRNA duplex with a caged circular sense RNA strand showed gene silencing activity even though no light was applied to open the circle of RNA. However, target gene expression (both firefly luciferase and GFP) was efficiently photomodulated for caged siRNAs with circular antisense RNAs. Further investigation on the formation of caged circular RNA with its complimentary RNA strand indicated that two interchangeable conformations of these circular siRNA duplexes existed. The circular siRNA duplex was thermodynamically less stable than the corresponding linear duplex and target RNA may compete to bind to the antisense RNA strand, which triggers RNAi activity. Further results indicated that the caged circular siRNA could be used to photomodulate siRNA induced gene silencing in a mouse tumor model. We expect that the optimized caged circular siRNA as a new addition to the caged siRNA family will be promising for practical applications in gene therapy in the future.Funding
The work was supported by the National Science Foundation of China (Grant No. 21422201 and 21372018), the National Basic Research Program of China (‘973’ Program; Grant No. 2012CB720600), the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (Grant No. 2017ZX09303013), the National High-tech R&D Program of China (2012AA022501) and the Beijing Natural Science Foundation (5132015).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Medical and Health Analytical Center, Peking University Health Science Center for the mouse studies.Notes and references
- A. P. McCaffrey, L. Meuse, T. T. Pham, D. S. Conklin, G. J. Hannon and M. A. Kay, Nature, 2002, 418, 38–39 CrossRef CAS PubMed.
- A. Kelly and A. F. Hurlstone, Briefings Funct. Genomics, 2011, 10, 189–196 CrossRef CAS PubMed.
- Y. Wang, G. Sheng, S. Juranek, T. Tuschl and D. J. Patel, Nature, 2008, 456, 209–213 CrossRef CAS PubMed.
- W. F. Lima, H. Wu, J. G. Nichols, H. Sun, H. M. Murray and S. T. Crooke, J. Biol. Chem., 2009, 284, 26017–26028 CrossRef CAS PubMed.
- M. Manoharan, Curr. Opin. Chem. Biol., 2004, 8, 570–579 CrossRef CAS PubMed.
- H. Peacock, A. Kannan, P. A. Beal and C. J. Burrows, J. Org. Chem., 2011, 76, 7295–7300 CrossRef CAS PubMed.
- W. F. Lima, T. P. Prakash, H. M. Murray, G. A. Kinberger, W. Li, A. E. Chappell, C. S. Li, S. F. Murray, H. Gaus, P. P. Seth, E. E. Swayze and S. T. Crooke, Cell, 2012, 150, 883–894 CrossRef CAS PubMed.
- D. Yu, H. Pendergraff, J. Liu, H. B. Kordasiewicz, D. W. Cleveland, E. E. Swayze, W. F. Lima, S. T. Crooke, T. P. Prakash and D. R. Corey, Cell, 2012, 150, 895–908 CrossRef CAS PubMed.
- D. Cejka, D. Losert and V. Wacheck, Clin. Sci., 2006, 110, 47–58 CrossRef CAS PubMed.
- D. D. Young, H. Lusic, M. O. Lively, J. A. Yoder and A. Deiters, ChemBioChem, 2008, 9, 2937–2940 CrossRef CAS PubMed.
- A. Deiters, Curr. Opin. Chem. Biol., 2009, 13, 678–686 CrossRef CAS PubMed.
- X. Ouyang, I. A. Shestopalov, S. Sinha, G. Zheng, C. L. Pitt, W. H. Li, A. J. Olson and J. K. Chen, J. Am. Chem. Soc., 2009, 131, 13255–13269 CrossRef CAS PubMed.
- A. Deiters, ChemBioChem, 2010, 11, 47–53 CrossRef CAS PubMed.
- A. Deiters, R. A. Garne, H. Lusic, J. M. Govan, M. Dush, N. M. Nascone-Yoder and J. A. Yoder, J. Am. Chem. Soc., 2010, 132, 15644–15650 CrossRef CAS PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- J. M. Govan, R. Uprety, J. Hemphill, M. O. Lively and A. Deiters, ACS Chem. Biol., 2012, 7, 1247–1256 CrossRef CAS PubMed.
- I. Ahmed and L. Fruk, Mol. BioSyst., 2013, 9, 565–570 RSC.
- C. M. Connelly, R. Uprety, J. Hemphill and A. Deiters, Mol. BioSyst., 2012, 8, 2987–2993 RSC.
- J. L. Richards, G. K. Seward, Y. H. Wang and I. J. Dmochowski, ChemBioChem, 2010, 11, 320–324 CrossRef CAS PubMed.
- L. Wu, Y. Wang, J. Wu, C. Lv, J. Wang and X. Tang, Nucleic Acids Res., 2013, 41, 677–686 CrossRef CAS PubMed.
- Y. Wang, L. Wu, P. Wang, C. Lv, Z. Yang and X. Tang, Nucleic Acids Res., 2012, 40, 11155–11162 CrossRef CAS PubMed.
- X. Tang, Photochemistry, ed. A. Albini and E. Fasani, 2013, vol. 41, pp. 319–341 Search PubMed.
- M. Su, F. Yang, C. Lv, L. Yu, X. Gu, J. Wang, Z. Li and X. Tang, J. Chin. Pharm. Sci., 2010, 19, 5–14 CAS.
- X. Tang, S. Maegawa, E. S. Weinberg and I. J. Dmochowski, J. Am. Chem. Soc., 2007, 129, 11000–11001 CrossRef CAS PubMed.
- X. Tang and I. J. Dmochowski, Nat. Protoc., 2007, 1, 3041–3048 CrossRef PubMed.
- X. Tang and I. J. Dmochowski, Angew. Chem., Int. Ed., 2006, 45, 3523–3526 CrossRef CAS PubMed.
- G. Zheng, L. Cochella, J. Liu, O. Hobert and W.-h. Li, ACS Chem. Biol., 2011, 6, 1332–1338 CrossRef CAS PubMed.
- P. K. Jain, V. Ramanan, A. G. Schepers, N. S. Dalvie, A. Panda, H. E. Fleming and S. N. Bhatia, Angew. Chem., Int. Ed., 2016, 55, 12628–12632 CrossRef.
- W. Zhou and A. Deiters, Angew. Chem., Int. Ed., 2016, 55, 5394–5399 CrossRef CAS PubMed.
- J. M. Govan, D. D. Young, H. Lusic, Q. Liu, M. O. Lively and A. Deiters, Nucleic Acids Res., 2013, 41, 10518–10528 CrossRef CAS PubMed.
- A. Kala and S. H. Friedman, Pharm. Res., 2011, 28, 3050–3057 CrossRef CAS PubMed.
- S. Shah, P. K. Jain, A. Kala, D. Karunakaran and S. H. Friedman, Nucleic Acids Res., 2009, 37, 4508–4517 CrossRef CAS PubMed.
- S. Shah, S. Rangarajan and S. H. Friedman, Angew. Chem., Int. Ed., 2005, 44, 1328–1332 CrossRef CAS PubMed.
- P. K. Jain, S. Shah and S. H. Friedman, J. Am. Chem. Soc., 2010, 133, 440–446 CrossRef PubMed.
- J. P. Casey, R. A. Blidner and W. T. Monroe, Mol. Pharm., 2009, 6, 669–685 CrossRef CAS PubMed.
- S. Shah and S. H. Friedman, Oligonucleotides, 2007, 17, 35–43 CrossRef CAS PubMed.
- V. Mikat and A. Heckel, RNA, 2007, 13, 2341–2347 CrossRef CAS PubMed.
- Q. N. Nguyen, R. V. Chavli, J. T. Marques, P. G. Conrad II, D. Wang, W. He, B. E. Belisle, A. Zhang, L. M. Pastor and F. R. Witney, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 394–403 CrossRef CAS PubMed.
- Y. Matsushita-Ishiodori and T. Ohtsuki, Acc. Chem. Res., 2012, 45, 1039–1047 CrossRef CAS PubMed.
- J. C. Griepenburg, B. K. Ruble and I. J. Dmochowski, Bioorg. Med. Chem., 2013, 21, 6198–6204 CrossRef CAS PubMed.
- R. A. Blidner, K. R. Svoboda, R. P. Hammer and W. T. Monroe, Mol. BioSyst., 2008, 4, 431–440 RSC.
- J. Hemphill, E. K. Borchardt, K. Brown, A. Asokan and A. Deiters, J. Am. Chem. Soc., 2015, 137, 5642–5645 CrossRef CAS PubMed.
- T. Lucas, F. Schafer, P. Muller, S. A. Eming, A. Heckel and S. Dimmeler, Nat. Commun., 2017, 8, 15162 CrossRef PubMed.
- L. Wu, F. Pei, J. Zhang, J. Wu, M. Feng, Y. Wang, H. Jin, L. Zhang and X. Tang, Chem. –Eur. J., 2014, 20, 12114–12122 CrossRef CAS PubMed.
- Y. Ji, J. Yang, L. Wu, L. Yu and X. Tang, Angew. Chem., Int. Ed., 2016, 55, 2152–2156 CrossRef CAS PubMed.
- S. Yamazoe, I. A. Shestopalov, E. Provost, S. D. Leach and J. K. Chen, Angew. Chem., Int. Ed., 2012, 51, 6908–6911 CrossRef CAS PubMed.
- N. Abe, H. Abe and Y. Ito, J. Am. Chem. Soc., 2007, 129, 15108–15109 CrossRef CAS PubMed.
- L. Wei, L. Cao and Z. Xi, Angew. Chem., Int. Ed., 2013, 52, 6501–6503 CrossRef CAS PubMed.
- M. Chen, L. Zhang, H.-Y. Zhang, X. Xiong, B. Wang, Q. Du, B. Lu, C. Wahlestedt and Z. Liang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2356–2361 CrossRef CAS PubMed.
- L. F. Fieser and W. H. Daudt, J. Am. Chem. Soc., 1941, 63, 782–788 CrossRef CAS.
- I. Sapountzis and P. Knochel, Angew. Chem., Int. Ed., 2002, 41, 1610–1611 CrossRef CAS PubMed.
- C. Kim, O. C. Lee, J. Y. Kim, W. Sung and N. K. Lee, Angew. Chem., Int. Ed., 2015, 54, 8943–8947 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc03842a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |