
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible-deactivation anionic alternating ring-opening copolymerization of epoxides and cyclic anhydrides: access to orthogonally functionalizable multiblock aliphatic polyesters†
Maria J.
Sanford
 ,
Nathan J.
Van Zee
and
Geoffrey W.
Coates
*
,
Nathan J.
Van Zee
and
Geoffrey W.
Coates
*
Dept. of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, NY 14853-1301, USA. E-mail: coates@cornell.edu
First published on 15th November 2017
Abstract
The alternating copolymerization of epoxides and cyclic anhydrides is an increasingly popular route to aliphatic polyesters that are of interest as biodegradable replacements for petroleum-based polymers and for use in the biomedical field. However, broad and bimodal molecular weight distributions in these polymerizations continues to be an issue, limiting synthesis of multiblock copolymers. By use of a bifunctional catalytic system, the reversible-deactivation anionic alternating ring-opening copolymerization of epoxides and cyclic anhydrides gives unimodal polymers with Đ values generally less than 1.07. This allowed for the formation of well-defined triblock copolymers. Additionally, by incorporating both aldehyde and alkene functionalities into the polymer, orthogonal post-polymerization modification was achieved, giving access to well-defined highly modifiable aliphatic polyesters.
Introduction
Aliphatic polyesters have attracted interest as replacements for petroleum based polymers and for use in biomedical applications due to their biodegradability1–6 and biocompatibility.7 Currently there are only a few examples of commercially successful aliphatic polyesters used in applications ranging from packaging to biomedical devices.8–13 Applications have been limited as aliphatic polyesters are typically moderately semi-crystalline, hydrophobic materials with few opportunities for incorporation of functionalities. Incorporation of additional functionalities into these materials could modulate polymer properties, leading to potentially new applications, and provide additional opportunities for post-polymerization modification. However, the most common route to aliphatic polyesters—ring-opening polymerization of lactones or lactide7—offers few ways to introduce functionality into these monomers and the resulting polymers and those strategies that have been reported involve difficult synthetic routes and/or limited scope of functionality.14–16A promising alternative route to aliphatic polyesters is the alternating copolymerization of epoxides and cyclic anhydrides.17,18 Use of two distinct monomer sets allows for a wider range of polymer properties. Extensive catalyst development by our group19–27 and others including Williams,28–33 Darensbourg,34 and Duchateau,35–38 has resulted in the ability to successfully polymerize a large number of monomers.17,18 Furthermore, many of these readily available monomers contain functionalities that would be difficult to access via the ring-opening polymerization of lactones.17,18 Despite numerous advances in catalyst design a persistent problem spanning essentially all reported catalyst systems is a bimodal distribution of molecular weights. This bimodality is known to occur due to the presence of both monofunctional and bifunctional initiating species. Most catalyst systems use a halide (most commonly chloride), nitrate, or acetate as the initiating species,17,18 all of which are monofunctional and give rise to the lower molecular weight distribution. However, trace amounts of diacid in the anhydride or adventitious water can also initiate chains. These unwanted, bifunctional initiators give rise to a distribution with approximately twice the Mn value of the chains initiated by the intentionally added monofunctional initiator.23,26,28,30–32,39,40 This bimodality limits access to well-defined multiblock copolymers which could be used in a variety of applications. We sought to design a catalyst system that allows for unimodal polymer distributions giving access to a truly well-controlled system and improved block copolymer synthesis. Additionally, we hoped to employ a reversible-deactivation anionic ring-opening alternating copolymerization system as described below, and then apply it to incorporate new functionalities and post-polymerization modifications into aliphatic polyesters.
Reversible-deactivation anionic alternating ring-opening copolymerizations share many of the characteristics of living polymerizations including the ability to control Mn by the ratio of monomer to initiator, low Đ values, and the possibility of synthesizing block copolymers.41 In a reversible-deactivation anionic alternating ring-opening copolymerization—colloquially known as an immortal polymerization—reversible protic chain transfer allows chains to cycle between an active state that can propagate and a dormant state that can be reactivated by transferring a proton to an active chain (Scheme 1).41 In practical terms, an immortal polymerization differs from a living polymerization due to the addition of a protic chain transfer agent (CTA). Each CTA molecule initiates a polymer chain and also serves as a proton source facilitating the reversible protic transfer. Use of a CTA in the alternating copolymerization of epoxides and cyclic anhydrides has been previously reported with Cr based catalysts.27,37 A major advantage of an immortal polymerization over a living polymerization is that it allows for the generation of numerous chains per metal center while retaining the control of a living polymerization. Use of a CTA also provides an additional handle for tuning molecular weight as Mn is dependent upon the ratio of monomer to total initiators, including CTAs.41 Herein we report the development of a bifunctional catalytic system for the immortal polymerization of epoxides and cyclic anhydrides and its application to both the synthesis of multiblock copolymers and the orthogonal functionalization of these polymers.
 | ||
| Scheme 1 Reversible-deactivation anionic alternating ring-opening copolymerization, or immortal polymerization, of epoxides and cyclic anhydrides. | ||
Results and discussion
We previously found that use of (salph)AlCl complexes with electron-withdrawing substituents on the ligand allowed for exquisite control of side reactions including transesterification and epimerization.23,25,26 Given that control of side reactions is necessary for synthesis of well-defined materials and especially important for block copolymer synthesis, we sought to retain that general ligand framework. We hypothesized that due to the issue of unavoidable contamination by diacid or adventitious water it would be necessary to use a bifunctional system in order to achieve a unimodal distribution. Such a system could be achieved by using a non-initiating metal complex with a bifunctional cocatalyst. We found that displacing the chloride ligand of (Fsalph)AlCl with triflate (−OTf) was a facile and clean reaction leading to the formation of [(Fsalph)Al(THF)][OTf] (1a, Fig. 1). The −OTf anion is not very nucleophilic making it unlikely that it is capable of ring-opening an epoxide to initiate the polymerization. Control reactions confirmed that 1a was incapable of ring-opening an epoxide to initiate the polymerization.42 Previous work had shown that [PPN][1-adamantate] could be cleanly synthesized,22 leading us to believe that the related bifunctional [PPN]2[1,3-adamantane dicarboxylate] salt ([PPN]2[ADC], 1b, Fig. 1) would be readily accessible. Gratifyingly, we found that we could easily and cleanly synthesize 1b, giving rise to a bifunctional system when employed with 1a. | ||
| Fig. 1 Non-initiating metal complex and bifunctional cocatalyst employed in this study. | ||
We decided to use this system in an immortal polymerization (Scheme 1) with 1,3-adamantane dicarboxylic acid (AdA) as a bifunctional CTA. The metal complex/bifunctional cocatalyst (1a/1b) system was screened using propylene oxide (PO) and a citraconic anhydride based tricyclic anhydride (CPCA) with 2–10 equivalents of AdA as the CTA (Table 1, entries 1–3). Gratifyingly, these polymerizations yielded unimodal polymers (Fig. 2) with narrow dispersities (Đ = 1.04–1.07) and Mn values that were dependent upon the equivalents of AdA added (Table 1, entries 1–3). Both diacids and diols worked as bifunctional CTAs giving comparable results to AdA (Table 1, entries 4–8). A diacid terminated polymer (PEG600-diacid, Table 1, entry 6) was employed as a CTA and found to be comparable to the small molecule CTAs. The variety of CTAs that can be successfully used in this system suggests that this system could be employed to create diverse polymer structures that could be hard to synthesize via other routes.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | CTA | Equiv. CTAb | Conv.c (%) | M n theo (kDa) | M n (kDa) | Đ |
a [PO]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CPCA]:[1a]:[1b] = 2000:400:1:0.9 VPO = 0.9 mL VMeCN = 0.6 mL. MeCN was added as the solvent in stock solutions of 1a and 1b.
b Equivalents of CTA relative to 1a.
c Conversion of anhydride determined by 1H NMR spectroscopy.
d Determined by GPC in THF at 30 °C relative to polystyrene standards.
e When a diol is used as the CTA it should be noted that there is an anhydride prior to the repeat unit. :[CPCA]:[1a]:[1b] = 2000:400:1:0.9 VPO = 0.9 mL VMeCN = 0.6 mL. MeCN was added as the solvent in stock solutions of 1a and 1b.
b Equivalents of CTA relative to 1a.
c Conversion of anhydride determined by 1H NMR spectroscopy.
d Determined by GPC in THF at 30 °C relative to polystyrene standards.
e When a diol is used as the CTA it should be noted that there is an anhydride prior to the repeat unit.
|
||||||
| 1 | AdA | 2 | >99 | 32.6 | 12.2 | 1.04 |
| 2 | AdA | 5 | >99 | 16.1 | 7.1 | 1.04 |
| 3 | AdA | 10 | >99 | 8.7 | 4.2 | 1.07 |
| 4 | AA | 5 | >99 | 16.1 | 7.9 | 1.04 |
| 5 | TA | 5 | >99 | 16.1 | 6.9 | 1.06 |
| 6 | PEG600-diacid | 5 | 99 | 16.1 | 6.9 | 1.06 |
| 7e | 1,6-HD | 5 | 97 | 15.7 | 7.6 | 1.04 |
| 8e | BD | 5 | >99 | 16.1 | 7.3 | 1.04 |

 | ||
| Fig. 2 GPC traces of polymers described in Table 1, entries 1–3. | ||
While the low Đ values and dependence of Mn on the equivalents of CTA used suggested that the polymerization behaves in an immortal manner, we monitored the polymerization with respect to time and found Mn to be linearly related to monomer conversion (Scheme 2a). We also confirmed that Mn was linearly related to the ratio of monomer to initiating groups—in this case the sum of the moles of 1b and moles of CTA (Scheme 2b). Đ values remained narrow (<1.07) at all conversions (Scheme 2a) and ratios of monomer to initiators (Scheme 2b).
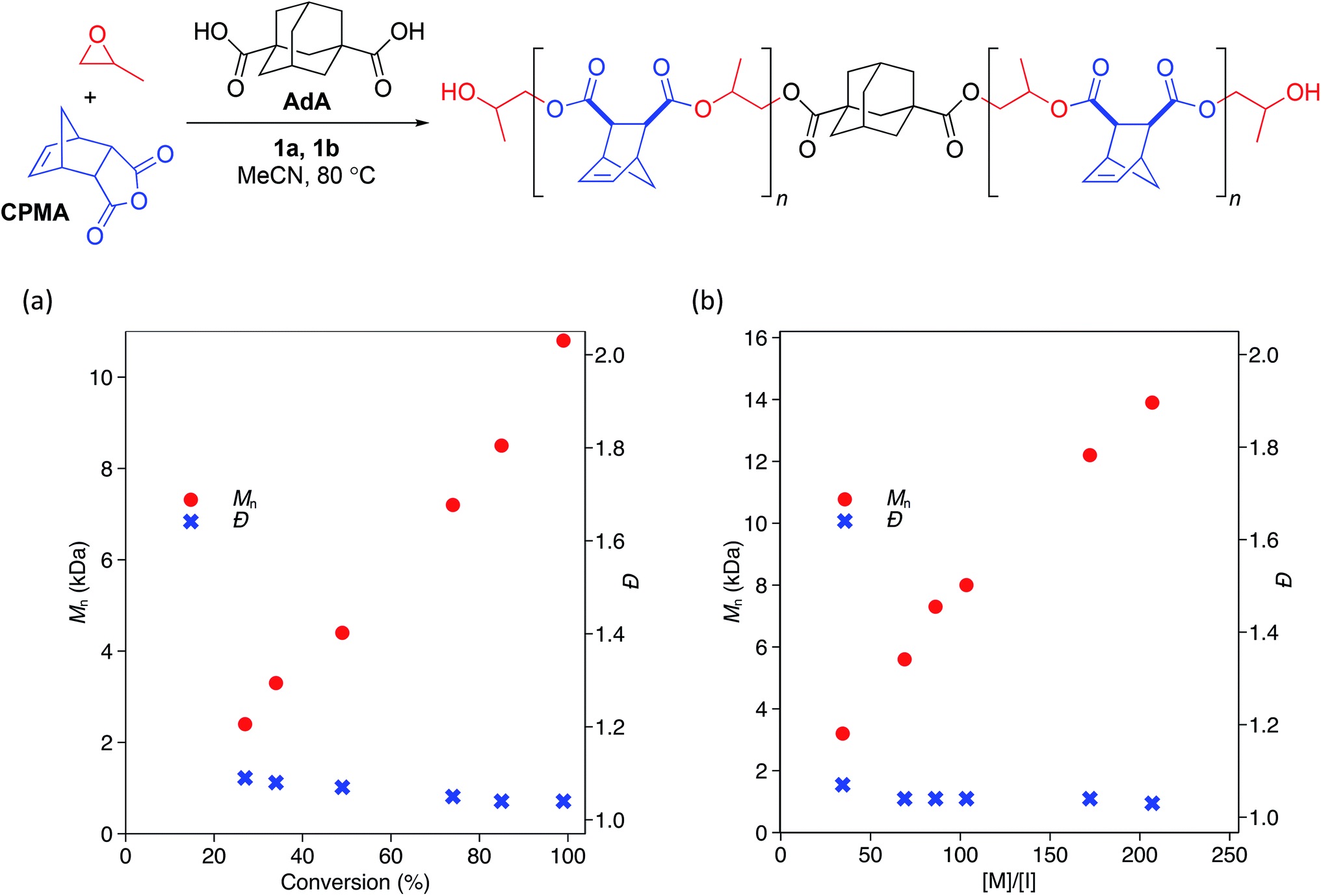 | ||
| Scheme 2 Change in Mn and Đ with respect to (a) conversion of monomera and (b) ratio of monomer to initiating species.b a[PO]:[CPMA]:[1a]:[1b]:[AdA] = 2000:400:1:0.9:2. Conversion of anhydride determined by 1H NMR analysis. Mn and Đ determined by GPC analysis in THF at 30 °C relative to polystyrene standards. b[PO]:[1a]:[1b]:[AdA] = 2000:1:0.9:2; ratio of [M] to [I] was changed by varying amount of anhydride. [M]/[I] = (mol anhyd.)/(mol 1b + mol AdA). All polymerizations were run to >99% conversion of anhydride as determined by 1H NMR analysis. Mn and Đ determined by GPC analysis in THF at 30 °C relative to polystyrene standards. | ||
We next screened a variety of epoxides and tricyclic anhydrides with this immortal polymerization system (Table 2). Mn values were well-controlled and Đ values remained under 1.10. Increasing the ratio of monomer to initiator produced higher molecular weight polymer (Mn = 20.0 kDa, Đ = 1.04; Table 2, entry 4). Perhaps the most interesting monomer employed was a vanillin-based epoxide, VGE, which contained a pendant aldehyde. The aldehyde did not interfere with the polymerization and we were able to incorporate aldehyde functionality into an aliphatic polyester (Table 2, entry 8). Our system is unique in that the aldehyde is directly incorporated into the polymer as itself and not as a masked structure, unlike in other systems containing a polyester or polycarbonate backbone.43,44 Because VGE is a solid epoxide, a large quantity of solvent had to be added if it was used as the only epoxide in the polymerization, significantly reducing the rate. In order to avoid this loss of rate, we used a combination of VGE and PO, allowing access to an aldehyde containing aliphatic polyester in a reasonable amount of time. Combining PO and VGE also allows the mol% incorporation of pendant aldehyde to be easily tuned.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Epoxide | Anhyd. | t rxn (h) | Conv.b (%) | M n (kDa) | Đ |
|
a [Epox.]:[Anhyd.]:[1a]:[1b]:[AdA] = 2000:400:1:0.9:5 VPO = 0.9 mL VMeCN = 0.6 mL MeCN was added solely as a the solvent in the stock solutions of 1a and 1b that were used.
b Conversion of anhydride determined by 1H NMR spectroscopy.
c Determined by GPC in THF relative to polystyrene standards.
d [PO]:[TMA]:[1a]:[1b]:[AdA] = 4000:1200:1:0.9:5 VPO = 1.8 mL VMeCN = 0.6 mL MeCN was added solely as the solvent in the stock solutions of 1a and 1b that were used.
e 100 equiv. VGE and 2000 equiv. PO. 1H NMR analysis showed that the polymer was 12% poly(VGE-alt-CPCA) and 88% poly(PO-alt-CPCA).
|
||||||
| 1 | PO | CPCA | 5.0 | >99 | 7.1 | 1.04 |
| 2 | PO | PMA | 4.5 | >99 | 9.3 | 1.04 |
| 3 | PO | TMA | 5.3 | >99 | 6.9 | 1.04 |
| 4d | PO | TMA | 11.0 | 95 | 20.0 | 1.04 |
| 5 | PO | PCA | 8.0 | >99 | 7.5 | 1.05 |
| 6 | PO | CHCA | 5.5 | >99 | 6.7 | 1.04 |
| 7 | DO | CPCA | 24.5 | >99 | 9.2 | 1.08 |
| 8e | VGE\PO | CPCA | 5.0 | >99 | 7.9 | 1.06 |
| 9 | AGE | PMA | 7.5 | >99 | 8.5 | 1.10 |
| 10 | TFEP | CPCA | 23.0 | >99 | 8.8 | 1.07 |
Since this immortal polymerization system yielded unimodal polymers with extremely narrow dispersities, we applied this system to the synthesis of a triblock copolymer. We used CPCA as the first anhydride with PO as the epoxide to generate the first block, resulting in a unimodal polymer (Mn = 6.6 kDa, Đ = 1.05, Scheme 3). After CPCA was fully consumed, an aliquot was removed for 1H NMR and GPC analysis followed by addition of PMA. Because the polymerization was initiated solely by bifunctional species, addition of a second monomer set resulted in growth of a triblock copolymer. We were pleased to see the expected increase in Mn (13.8 kDa) while Đ remained well-controlled (Đ = 1.04) and unimodal (Fig. 3).
 | ||
| Scheme 3 Synthesis of a triblock copolymer. [PO]:[Anhyd.]:[1a]:[1b]:[AdA] = 2000:200:1:0.9:2.5. Each block was run to >99% conversion of anhydride. Mn and Đ determined by GPC analysis in THF at 30 °C relative to polystyrene standards. | ||
 | ||
| Fig. 3 GPC traces of triblock copolymer described in Scheme 3. | ||
With a system in hand to synthesize a variety of aliphatic polyesters, including multiblock copolymers and some with both alkene and aldehyde functionalities that are potentially reactive for post-polymerization modification (PPM), we explored the possibility of orthogonally functionalizing these well-defined materials. PPM of aliphatic polyesters gives access to polymer properties and functionalities that may otherwise be difficult to incorporate into the polymer. Thiol–ene chemistry on norbornene-type moieties in the polymer backbone has been previously employed on polyesters synthesized from the alternating copolymerization of epoxides and cyclic anhydrides,45–48 as has cross-metathesis modification.49 Intriguingly incorporation of a pendant aldehyde into polymers containing reactive norbornene type moieties enables the possibility of orthogonal PPM, which is a much sought after functionalization.50
To this end, we first screened the thiol–ene post-polymerization modification on polymer samples that did not contain VGE. To our surprise, we found that the identity of the anhydride was important in allowing us to achieve this PPM. Polymers synthesized from monomers such as CPMA and CPCA with a [2.2.1] bicyclic structure were extremely reactive for the AIBN/heat catalyzed thiol–ene reaction with 1-octanethiol and reached full conversion of alkene within 2 hours (Table S1†). The thiol–ene reaction proceeded cleanly with Mn values increasing while Đ values and the unimodal distribution were maintained (Table S1, Fig. S1†). However, alkenes in [2.2.2] bicyclic structures, such as those arising from use of TMA, PMA, or PCA, were completely unreactive towards thiol–ene functionalization (Table S1†). No degradation of these unreactive polymers was observed and the GPC traces before and after the reaction were identical (Fig. S1†).
A sample of a triblock copolymer (Scheme 3) containing both [2.2.1] and [2.2.2] norbornene structures was submitted to these thiol–ene conditions. Gratifyingly, the reaction selectively occurred in the mid-block that was derived from CPCA while leaving the two end blocks derived from PMA untouched (Scheme 4). Derivatization with 1-octanethiol reduced the Tg of the polymer by approximately 50 °C, suggesting that with proper choice of midblock, endblocks, and thiol–ene post-polymerization modification this selective functionalization could be used to synthesize aliphatic polyester thermoplastic elastomers.
 | ||
| Scheme 4 Selective thiol–ene functionalization of triblock copolymer. | ||
Since aldehydes are reactive groups which have been previously used as handles for PPM,43,44,51 we next examined PPM reactions on polymers containing VGE. We hypothesized that the aldehyde should rapidly react with an amine to form an imine linkage. Imines have garnered interest for use in biomedical applications, particularly for use in drug delivery systems due to the pH sensitive nature of the imine bond.52–54 A polymer with 8 mol% VGE (Mn = 6.4 kDa, Đ = 1.06) was heated to 80 °C for 2.75 h in the presence of isopropylamine (Scheme 5).55 By 1H NMR analysis, the aldehyde was cleanly converted to an imine (Scheme 5). GPC analysis showed a slight increase in molecular weight (Mn = 6.6 kDa) while a narrow, unimodal distribution was maintained (Đ = 1.04; Scheme 5). Because imines are often in equilibrium with the aldehyde and amine, we used a large excess of amine to drive the reaction to completion. Given that the Mn value changed only slightly as would be expected from the reaction, and that the low Đ value was retained, it seems unlikely that the polyester backbone was cleaved by use of a large excess of amine.
 | ||
| Scheme 5 (a) Post-polymerization modification of aldehyde to imine and (b) 1H NMR spectra of polymer before and (c) after functionalization. | ||
In order to perform double post-polymerization modifications, it was necessary for the two PPM reactions to be orthogonal. We submitted a sample of poly(VGE-alt-CPMA) to typical thiol–ene conditions and saw full consumption of the alkene while the aldehyde remained unreacted (Scheme S1†). Similarly, in all imine PPM reactions performed in the absence of AIBN and thiol the alkene was untouched. This suggested that the two reactions are orthogonal to each other and could be performed on a polymer containing VGE and a [2.2.1] structure in the backbone either sequentially or in tandem. Given the similarities in reaction conditions between the imine functionalization and thiol–ene reaction, we performed the reactions in tandem. A sample of poly(PO-alt-CPMA)-ran-poly(VGE-alt-CPMA) that was 12 mol% VGE (Mn = 5.5 kDa; Đ = 1.07) was reacted with 1-octanethiol and n-propylamine in the presence of AIBN (Scheme 6). After 1.5 h, there was complete consumption of both the alkene and aldehyde, the latter of which was cleanly converted to an imine by 1H NMR. As anticipated, the molecular weight increased (Mn = 9.0 kDa) while the dispersity remained low (Đ = 1.07; Scheme 6). To the best of our knowledge this is the first example of a one-pot, orthogonal, double post-polymerization modification on an aliphatic polyester.
 | ||
| Scheme 6 Orthogonal, one-pot double post-polymerization modification reaction. | ||
Conclusions
In conclusion, we report the reversible-deactivation anionic alternating ring-opening copolymerization, or immortal polymerization, of epoxides with tricyclic anhydrides resulting in extremely well-controlled polymerization behavior and unimodal molecular weight distributions by use of a non-initiating metal complex/bifunctional cocatalyst catalytic system. This system exhibited immortal behavior; a CTA could be used to control Mn and to increase the number of chains of polymer produced per catalyst. Đ values were unimodal and generally less than 1.07 and Mn values were linearly related to conversion of monomer and to the ratio of monomer to initiator, allowing for the synthesis of a triblock copolymer. Due to differences in reactivity between [2.2.1] and [2.2.2] bicyclic structures in the polymer backbone it was possible to selectively functionalize one of the blocks in the triblock copolymer. By incorporating an epoxide with a pendant aldehyde (VGE), we were able to introduce an additional functionality that has orthogonal PPM behavior to the alkenes also present in the polymer. Both thiol–ene and aldehyde to imine PPM reactions proceeded cleanly and orthogonally towards each other allowing us to do a one-pot double PPM reaction. Because this catalyst system produced extremely well-defined polymers that can be further modified by two orthogonal reactions, we believe it will be highly useful for the synthesis of designer aliphatic polyesters that can be used for a variety of applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this project was provided by the Center for Sustainable Polymers, an NSF Center for Chemical Innovation (CHE-1413862). This work made use of the NMR facility at Cornell University which is supported, in part, by the NSF under award number CHE-1531632. We thank Dr Kyle O'Connor for numerous helpful discussions.Notes and references
- W. Amass, A. Amass and B. Tighe, Polym. Int., 1998, 47, 89–144 CrossRef CAS.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
- M. Vert, Biomacromolecules, 2005, 6, 538–546 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- D. A. Olson, S. E. A. Gratton, J. M. DeSimone and V. V. Sheares, J. Am. Chem. Soc., 2006, 128, 13625–13633 CrossRef CAS PubMed.
- P. Lecomte and C. Jérôme, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and A. T. Zevaco, Springer, Berlin, 2012, pp. 173–217 Search PubMed.
- Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef CAS.
- E. T. H. Vink, K. R. Rábago, D. A. Glassner and P. R. Gruber, Polym. Degrad. Stab., 2003, 80, 403–419 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- R. Datta and M. Henry, J. Chem. Technol. Biotechnol., 2006, 81, 1119–1129 CrossRef CAS.
- A. P. Gupta and V. Kumar, Eur. Polym. J., 2007, 43, 4053–4074 CrossRef CAS.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- S. A. Cairns, A. Schultheiss and M. P. Shaver, Polym. Chem., 2017, 8, 2990–2996 RSC.
- A. Buchard, D. R. Carbery, M. G. Davidson, P. K. Ivanova, B. J. Jeffery, G. I. Kociok-Köhn and J. P. Lowe, Angew. Chem., Int. Ed., 2014, 53, 13858–13861 CrossRef CAS PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed.
- J. M. Longo, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 15897–15900 CrossRef CAS PubMed.
- N. J. Van Zee and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 2665–2668 CrossRef CAS PubMed.
- A. M. DiCiccio, J. M. Longo, G. G. Rodriguez-Calero and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 7107–7113 CrossRef CAS PubMed.
- N. J. Van Zee, M. J. Sanford and G. W. Coates, J. Am. Chem. Soc., 2016, 138, 2755–2761 CrossRef CAS PubMed.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed.
- A. Thevenon, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS PubMed.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS PubMed.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- S. Huijser, E. HosseiniNejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- E. Hosseini Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef.
- E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS.
- E. Hosseini Nejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308–1313 RSC.
- J. Y. Jeon, S. E. Chan, J. K. Varghese and B. Y. Lee, Beilstein J. Org. Chem., 2014, 10, 1787–1795 CrossRef PubMed.
- R. Mundil, Z. Hošt'álek, I. Šeděnková and J. Merna, Macromol. Res., 2015, 23, 161–166 CrossRef CAS.
- S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861–2871 CrossRef CAS.
- See ESI† for control experiments supporting the inability of the triflate anion to ring-open an epoxide and initiate the polymerization.
- G. Ma, D. Li, J. Wang, X. Zhang and H. Tang, Aust. J. Chem., 2013, 66, 1576–1583 CrossRef CAS.
- G. S. Heo, S. Cho and K. L. Wooley, Polym. Chem., 2014, 5, 3555–3558 RSC.
- Z. Duan, X. Wang, Q. Gao, L. Zhang, B. Liu and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 789–795 CrossRef CAS.
- R. Baumgartner, Z. Song, Y. Zhang and J. Cheng, Polym. Chem., 2015, 6, 3586–3590 RSC.
- B. Han, L. Zhang, B. Liu, X. Dong, I. Kim, Z. Duan and P. Theato, Macromolecules, 2015, 48, 3431–3437 CrossRef CAS.
- G. Si, L. Zhang, B. Han, Z. Duan, B. Li, J. Dong, X. Li and B. Liu, Polym. Chem., 2015, 6, 6372–6377 RSC.
- L. Fournier, C. Robert, S. Pourchet, A. Gonzalez, L. Williams, J. Prunet and C. M. Thomas, Polym. Chem., 2016, 7, 3700–3704 RSC.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS.
- J. C. Foster and J. B. Matson, Macromolecules, 2014, 47, 5089–5095 CrossRef CAS.
- X. Cai, C. Dong, H. Dong, G. Wang, G. M. Pauletti, X. Pan, H. Wen, I. Mehl, Y. Li and D. Shi, Biomacromolecules, 2012, 13, 1024–1034 CrossRef CAS PubMed.
- L. Qiu, C.-Y. Hong and C.-Y. Pan, Int. J. Nanomed., 2015, 10, 3623–3640 CAS.
- X. Qu and Z. Yang, Chem.–Asian J., 2016, 11, 2633–2641 CrossRef CAS PubMed.
- This reaction works with other amines as well. See Table S2† for the reaction of an aldehyde containing polymer with an array of amines.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, polymerization, and functionalization procedures, and characterization data. See DOI: 10.1039/c7sc03643d |
| This journal is © The Royal Society of Chemistry 2018 |