
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Challenges and opportunities for alkane functionalisation using molecular catalysts
Xinxin
Tang
,
Xiangqing
Jia
and
Zheng
Huang
 *
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: huangzh@sioc.ac.cn
First published on 9th November 2017
Abstract
The conversion of vast low-value saturated hydrocarbons into valuable chemicals is of great interest. Thanks to the progression of organometallic and coordination chemistry, transition metal catalysed C sp3–H bond functionalisation has now become a powerful tool for alkane transformations. Specifically, methods for alkane functionalisation include radical initiated C–H functionalisation, carbene/nitrene insertion, and transition metal catalysed C–H bond activation. This perspective provides a systematic and concise overview of each protocol, highlighting the factors that govern regioselectivity in these reactions. The challenges of the existing catalytic tactics and future directions for catalyst development in this field will be presented.
1. Introduction
Alkanes, or paraffins, with the general chemical formula of CnH2n+2, have the simplest structure among organic molecules, which range in complexity from the smallest hydrocarbon, methane, to macromolecules such as polyethylene. Alkanes are the major constituents of natural gas and crude oil and thus the direct conversion of abundant and low-cost hydrocarbon feedstocks into value-added products has the potential to be a revolutionary technology in the chemical industry. Selectively converting propane to propene, for instance, is a process with tremendous applicability due to the huge demand of propene for the production of polypropene and commodity chemicals, such as acrylic acid, acrylonitrile, and cumene.Alkanes are among the least reactive organic molecules as they consist only of strong C sp3–H and single C sp3–C sp3 bonds. The chemical inertness of alkanes is somewhat reflected in the forcing conditions that are required in the very few alkane transformations performed industrially: (hydro) cracking or reforming processes in oil refineries using heterogeneous catalysts operate at temperatures in the range of 400 to 600 °C. Another general concern for alkane functionalisation is the control of regioselectivity. In the absence of a directing or activating group, the reactivity of one C–H or C–C bond in an alkane is expected to be similar to that in the others.1 Taking C–H bonds as an example, the bond dissociation energies (BDEs) of the primary (1°), secondary (2°) and tertiary (3°) C sp3–H bonds of alkanes fall in the range of 96 to 101 kcal mol−1, with 1° C–H bonds being stronger than 2° and 3° bonds.2 The BDE values are close, which renders the discrimination of two C sp3–H bonds, in particular the same type of bonds, very difficult (Fig. 1). Finally, the product of alkane functionalisation is generally more reactive than the starting material. Hence, the transformation will be subject to an upper limit on the yield of the end product.
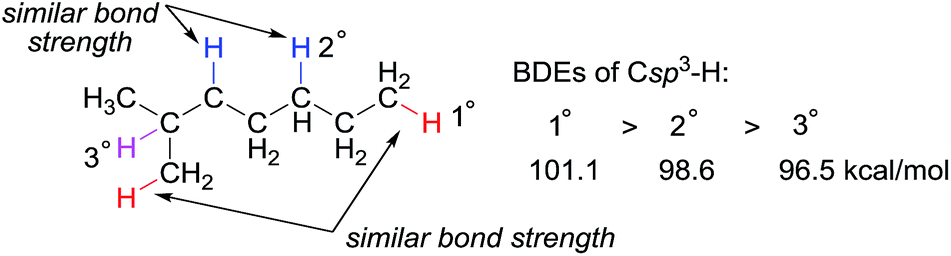 | ||
| Fig. 1 Bond dissociation energies (BDEs) of primary, secondary, and tertiary C sp3–H bonds in an alkane. | ||
Developing homogeneous catalysts for alkane functionalisations has garnered increasing attention during the past four decades due to the potential advantages of milder conditions, higher selectivity, and an enhanced diversity of transformation modes compared to heterogeneous catalysis. While many methods using small-molecule catalysts have been developed for the selective functionalisation of a specific C sp3–H bond, examples of C–C bond activation in simple alkanes are rare.3 Therefore, catalytic alkane transformations have greatly benefited from the spectacular progress in the field of C–H bond functionalisation seen over the last several decades. The reactions fall under three general categories: (i) stepwise radical pathways, (ii) carbene/nitrene insertion, and (iii) metal-mediated C–H bond activation. It is important to note that, depending on the pathways through which the C–H bond is cleaved, the relative reactivity of different types of C–H bond varies. Although 1° C–H bonds are less reactive towards a free radical or metallocarbene than 2° and 3° C–H bonds, many organometallic reagents and catalysts react preferentially with 1° C–H bonds over 2° and 3° C–H bonds. This perspective will provide representative examples for each class of alkane functionalisation with an emphasis on the selectivity for different types of C sp3–H bond, highlight the inherent challenges for these catalytic reactions and outline future prospects in this area. Although it is very important, methane functionalisation is beyond the scope of this perspective and heterogeneous and enzymatic systems will not be covered here either.
2. Radical-induced alkane functionalisation
Alkane functionalisation can occur through hydrogen abstraction from a C–H bond by either a high valent metal-based radical (e.g. porphyrin oxoiron(IV) cation radical) or an organic oxidant (e.g. alkyl peroxide) to form an alkyl radical, which is trapped by a metal complex to form the functionalised product or rebound to a metal centre to form a metal-alkyl species that is subject to further transformation (Fig. 2). The past 40 years have seen significant advances in the development of enzymatic catalysis for alkane oxidations and the reader is directed to several recent reviews for an in-depth coverage.4 Meanwhile, many synthetic metalloporphyrin and non-heme metal complexes have been developed for alkane hydroxylations, halogenations, and other transformations.5 The selectivity of these reactions in general correlates with the C–H bond strength.6 However, when a sterically demanding metal complex is involved in the recombination with an alkyl radical, the reaction tends to occur at the 2° and 1° C–H bonds, rather than at the 3° C–H bonds, as demonstrated by one example shown below.7,8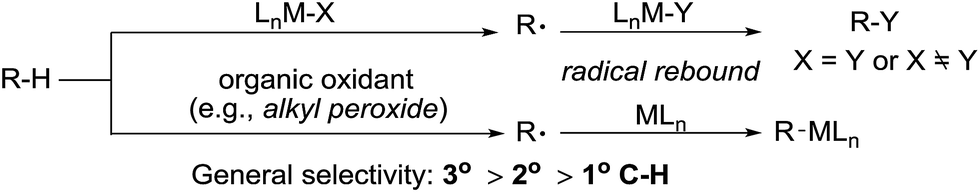 | ||
| Fig. 2 Alkane functionalisation through a radical intermediate. | ||
In 1979, Groves reported the first example of the porphyrin Fe-catalysed hydroxylation of C sp3–H bonds with iodosylbenzene as the oxidant. The oxidation of adamantane using [FeIII(TPP)Cl] (TPP = tetraphenylporphyrin) gives low yields of alcohols. The product distribution reveals the selective hydroxylation of 3° C–H over 2° C–H bonds (eqn (1)).9 Importantly, an oxoiron(IV) porphyrin cation radical was well-characterized by NMR, EXAFS and Mössbauer spectroscopy, thereby providing evidence in support of a radical mechanism.10
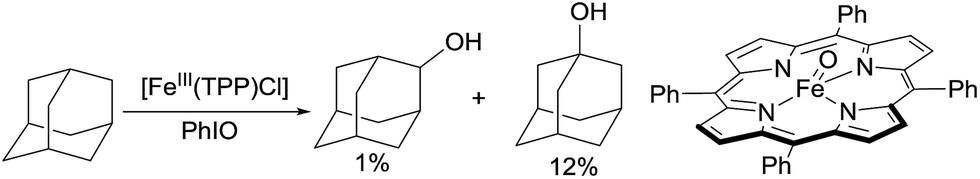 | (1) |
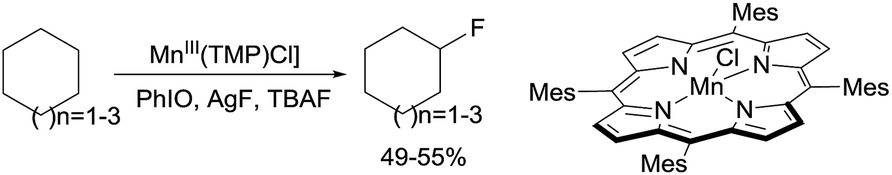 | (2) |
Following their early work on C–H bond hydroxylation, recently the same group reported the halogenation of alkanes catalysed by porphyrin Mn complexes through a so-called heteroatom-rebound catalysis strategy.5c Selective halogenation over common hydroxylation is due to the decelerated oxygen-rebound rate when the trans-axial ligand (L) bound to the Mn centre (L–MnV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) is a OH− or F− group. Brominations,11 chlorinations,11 and even fluorinations12 occur with moderate-to-high yields for cycloalkanes. For example, the fluorination of C6–C8 cycloalkanes with AgF/TBAF using Mn(TMP)Cl (TMP = tetramesitylporphyrin) affords the mono-fluorinated products in ∼50% yields (eqn (2)). More recently, the group extended the Mn catalysis to the azidation of 2° and 3° alkane C–H bonds using aqueous NaN3 as the azide source.13
O) is a OH− or F− group. Brominations,11 chlorinations,11 and even fluorinations12 occur with moderate-to-high yields for cycloalkanes. For example, the fluorination of C6–C8 cycloalkanes with AgF/TBAF using Mn(TMP)Cl (TMP = tetramesitylporphyrin) affords the mono-fluorinated products in ∼50% yields (eqn (2)). More recently, the group extended the Mn catalysis to the azidation of 2° and 3° alkane C–H bonds using aqueous NaN3 as the azide source.13
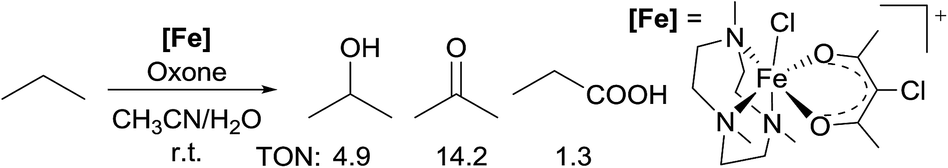
Most of the radical reactions use cycloalkanes as the substrates, in part because the regioselectivity issue does not arise in such cases. In 2014, Che reported the oxidation of acyclic light alkanes, ethane and propane, with oxone as the oxidant and a non-heme Fe complex [FeIII(Me3tacn)(Cl-acac)Cl]+ (Me3tacn = 1,4,7-trimethyl-1,4,7-triazacyclononane) as the precatalyst.14 The reaction of propane gives a mixture of iso-propanol/acetone/propanoic acid in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11:1 ratio, with a total turnover number (TON) of 20 (eqn (3)). The regioselectivity observed in the reactions is consistent with the selectivity of 2° > 1° for alkane oxidation by a radical pathway. Thorough mechanistic studies established an oxoiron(IV) radical intermediate, [FeIV(Me3tacn)(Cl-acac˙+)(O)]2+.
:11:1 ratio, with a total turnover number (TON) of 20 (eqn (3)). The regioselectivity observed in the reactions is consistent with the selectivity of 2° > 1° for alkane oxidation by a radical pathway. Thorough mechanistic studies established an oxoiron(IV) radical intermediate, [FeIV(Me3tacn)(Cl-acac˙+)(O)]2+.
 | (3) |
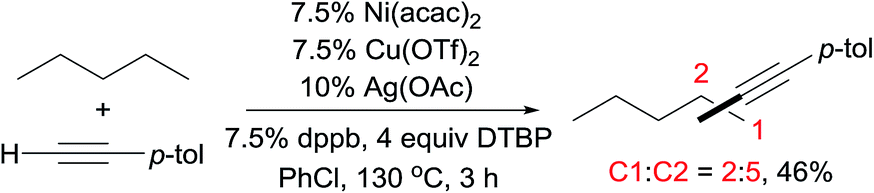
Alkane functionalisations with alkyl peroxides or hypervalent iodine compounds as the radical initiators have also been documented.7,15 In 2014, Hartwig reported Cu-catalysed amidations or imidations of alkanes with amides, sulfonamides, or imides in the presence of tBuOOtBu (DTBP).7 Intriguingly, the regioselectivity of 2° > 1° > 3°, as demonstrated by the reaction of 3-ethylpentane (eqn (4)), differs from the regiochemistry observed for most radical-induced C–H bond functionalisations. The low reactivity of the 3° C–H bond in this case is attributed to steric effects: the tertiary radical is too sterically encumbered to recombine with phen-ligated Cu amidate, a proposed catalytic intermediate. More recently, Lei published an interesting example of alkane alkynylation through a radical oxidative C sp3–H/C sp–H cross-coupling using a mixed Cu/Ni/Ag catalytic system. The product distributions shown in eqn (5) indicate that the 2° C–H bonds are more reactive than the 1° C–H bonds for the coupling.15c
 | (4) |
 | (5) |
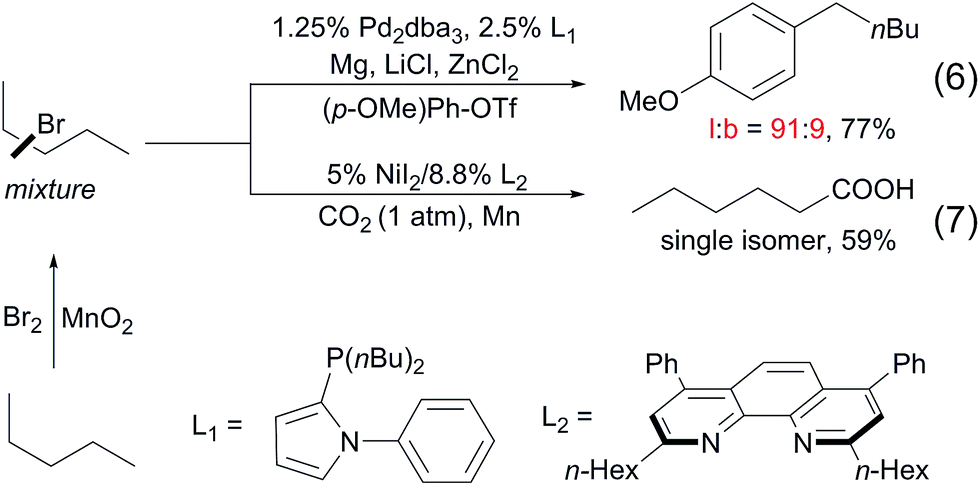
To date, a number of methods for alkane functionalisation via radical pathways have been reported15,16 but the substrate scopes are primarily limited to cycloalkanes. Catalytic functionalisations of linear alkanes invariably lead to mixtures of regioisomers, as exemplified above. In particular, the direct functionalisation of alkanes at the terminal positions by a radical mechanism remains unknown. Most recently, the Baudoin17 and Martin18 groups independently developed two-step protocols for the terminal functionalisation of alkanes. Such systems comprise a Mn-mediated alkane bromination with Br2, yielding a mixture of alkyl bromides and a subsequent Pd/Ni-catalysed tandem chain-walking and arylation/carboxylation sequence. These methods show high selectivity for linear products (eqn (6) and (7)).
 | (8) |
3. Alkane functionalisation by carbene or nitrene insertion
Alkane functionalisations by means of carbene insertion into C–H bonds occurs through a concerted pathway, involving a three-centered transition state formed by the overlap of an empty p-orbital of the carbene carbon with the C–H σ-orbital of the alkane (Fig. 3). Consequently, the product is formed with the retention of the configuration at the carbon centre.19 Like the radical mechanism shown above, the metal centre does not interact directly with the C–H bond, but the metal fragments have important effects on the stability, reactivity, and selectivity of the carbene moiety: (i) an appropriate transition metal can serve as a stabilizing entity, protecting the carbene from decomposition by forming a transient metallocarbene; (ii) a metal complex with an electron-withdrawing ligand can enhance the electrophilicity of the carbene, making it more reactive towards weak nucleophiles such as alkanes and (iii) the general regiochemical preference for carbene insertion into 3° over 2° C–H bonds and 2° over 1° C–H bonds is similar to that observed for the radical pathway, but the steric environment of the ligand may have a large impact on regiochemistry and even on stereochemistry.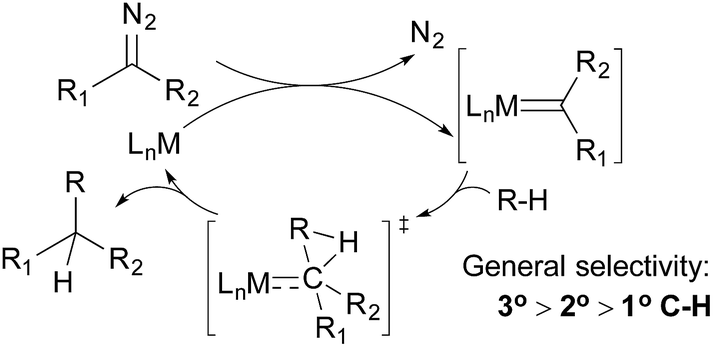 | ||
| Fig. 3 Alkane functionalisation via carbene insertion. | ||
In the 1970s, Scott and DeCicco disclosed the first example of carbene insertion into alkane C–H bonds using simple copper sulfate as the catalyst; the reaction of cyclohexane with ethyl diazoacetate (EDA) results in a low yield of the C–C bond-forming product.20 Years later, Noels and Teyssie reported an improved system using dirhodium catalysts with dicarboxylate ligands; the reactions of various cyclic and linear alkanes give moderate-to-good yields of C–H monoinsertion products.21 These seminal works set the stage for catalyst development in this field – most systems developed later use Rh- and coinage metal-based catalysts.
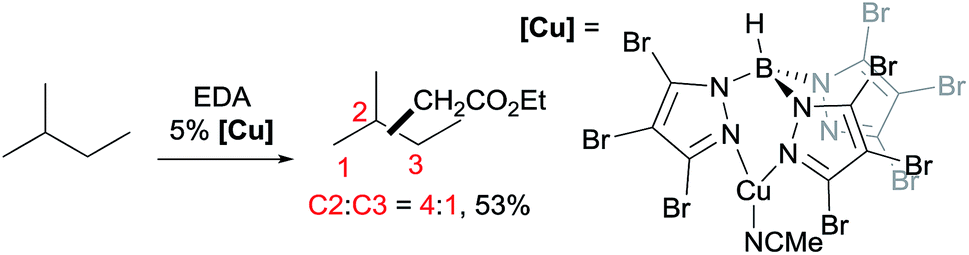
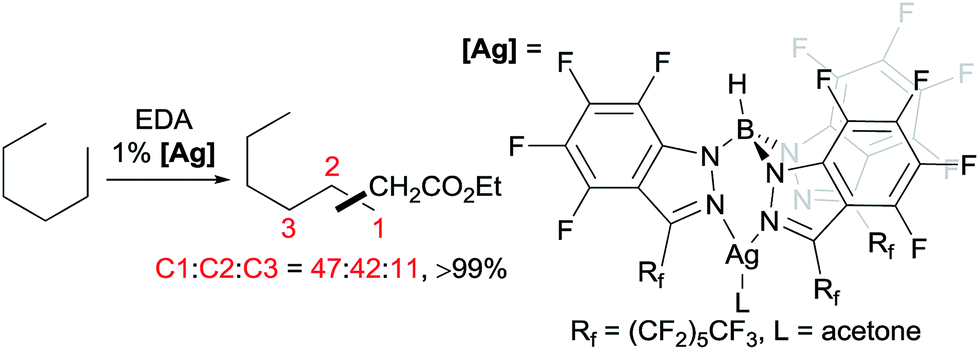
A series of contributions mainly from the Pérez group on coinage metal catalysis significantly advanced the area of alkane functionalisation via carbene insertion.22–25 A Cu complex of electron-withdrawing perbromo trispyrazolylborate catalyses carbene insertion into the C–H bonds of linear alkanes with moderate to good yields and a regioselectivity of 3° > 2° > 1° C–H bonds (eqn (8)).23 While the Cu complex is hardly active for 1° C–H bonds,23,24 Ag24b,25 and Au24a analogs show enhanced primary selectivity. Among them, a highly electrophilic Ag complex of perfluoro trisindazolylborate gives 47% primary selectivity in the reaction of n-hexane with EDA (eqn (9)).25b
 | (9) |
 | (10) |
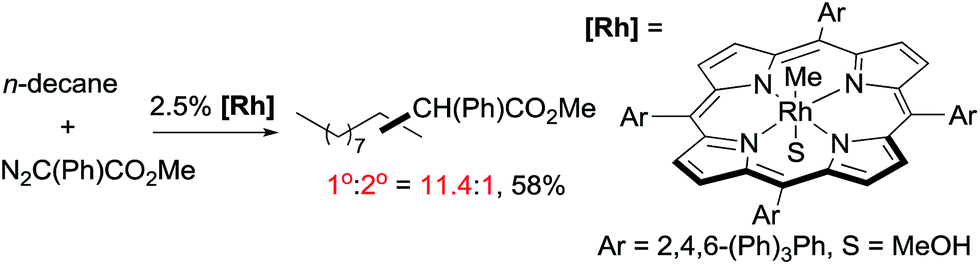
As mentioned above, the selective carbene insertion into a primary C–H bond is a challenge. Thus far the best terminal selectivity was achieved by Che using a Rh catalyst with a highly sterically demanding porphyrin ligand and a relatively bulky carbene source, N2C(Ph)CO2Me. For example, the reaction of n-decane offers ∼90% primary selectivity (eqn (10)).26 The steric contribution, evidently, prevails over the electronic effect in controlling the regioselectivity in this case.
 | (11) |
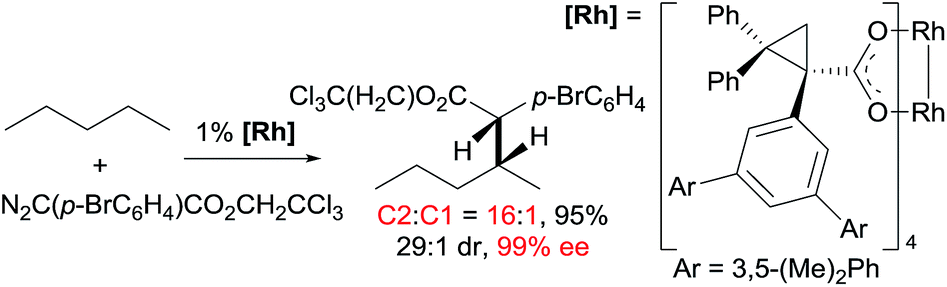
Asymmetric carbene insertion into alkane C–H bonds has also been achieved. The Davies group disclosed that dirhodium tetrakis (S-(N-dodecylbenzenesulfonyl)prolinate) could induce the asymmetric functionalisation of cycloalkanes with phenyldiazoacetate with modest-to-high enantioselectivity.27 Che extended the substrates to linear alkanes: the reaction of n-hexane with a sterically hindered chiral rhodium porphyrin is moderately selective for 1° C–H bonds (78% terminal selectivity), giving the linear product with 66% ee.26 A more recent example by Davies showed that the reaction of n-pentane and trichloroethyl 2-(4-bromophenyl)-2-diazoacetate using a dirhodium catalyst occurs selectively at the C2-position, accompanied with exceptionally high enantio- and diastereoselectivity (eqn (11)).28
 | (12) |
While carbene insertion reactions have been well documented, far less is known about nitrene insertions for alkane functionalisations. Early examples came from the Breslow29 and Barton30 groups using an Fe or Mn catalyst for aminations of cyclohexane and adamantane. Since then, several metal catalysts (Rh,31 Ag,31b,32 and Cu32b,33) have proven effective for the amination of cycloalkanes with sulfonylazide or hypervalent iodine as the nitrene source.
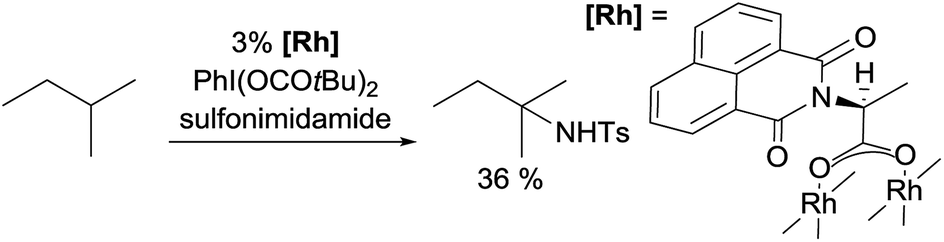
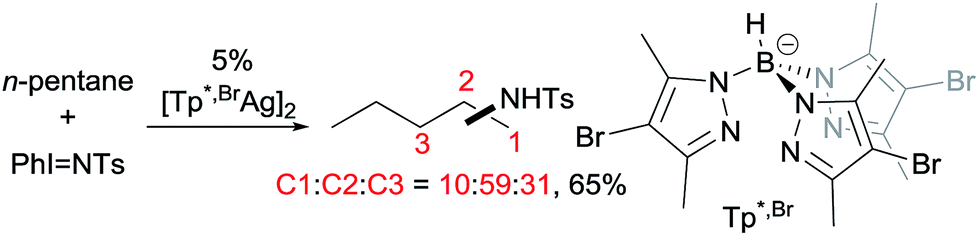
Examples of nitrene insertion reactions with acyclic alkanes are particularly scarce.32a The regioselectivity of 3° > 2° > 1° C–H bonds observed in the few reactions indicates that the product distributions are governed by the BDE of C–H bonds. For example, the Rh-catalysed reaction of 2-methybutane with sulfonimidamide is highly selective for tertiary C–H bonds, albeit in low yields (eqn (12)).31c The Ag-catalysed nitrene insertion into n-pentane gives a total yield of 65%, with branched amines as the major products (eqn (13)).32c Of note is that the asymmetric nitrene insertion into alkane C–H bonds has not been reported.
 | (13) |
 | (14) |
4. Alkane functionalisation via C–H bond activation at the transition metal centre
The term C–H bond activation refers to a type of reaction in which a transition metal reacts with a C–H bond to form a metal–carbon bond. Such transformations, traditionally, are classified into three categories: (i) oxidative addition with an electron-rich late transition metal;34 (ii) electrophilic activation with an electron-deficient late transition metal35 and (iii) σ-bond metathesis with an early transition metal via a four-membered transition state.36 Recently, the σ-bond metathesis mechanism has been extended to late transition metal systems involving σ complexes as intermediates, which was designated as σ complex-assisted metathesis (σ-CAM).37 Moreover, a variation of the electrophilic activation mechanism, termed concerted metalation–deprotonation (CMD), which often involves an electrophilic metal carboxylate, has been revealed recently (Scheme 1).38 Such routes by which metal–carboxylate complexes cleave C–H bonds, however, have been barely reported for the functionalisations of simple alkanes.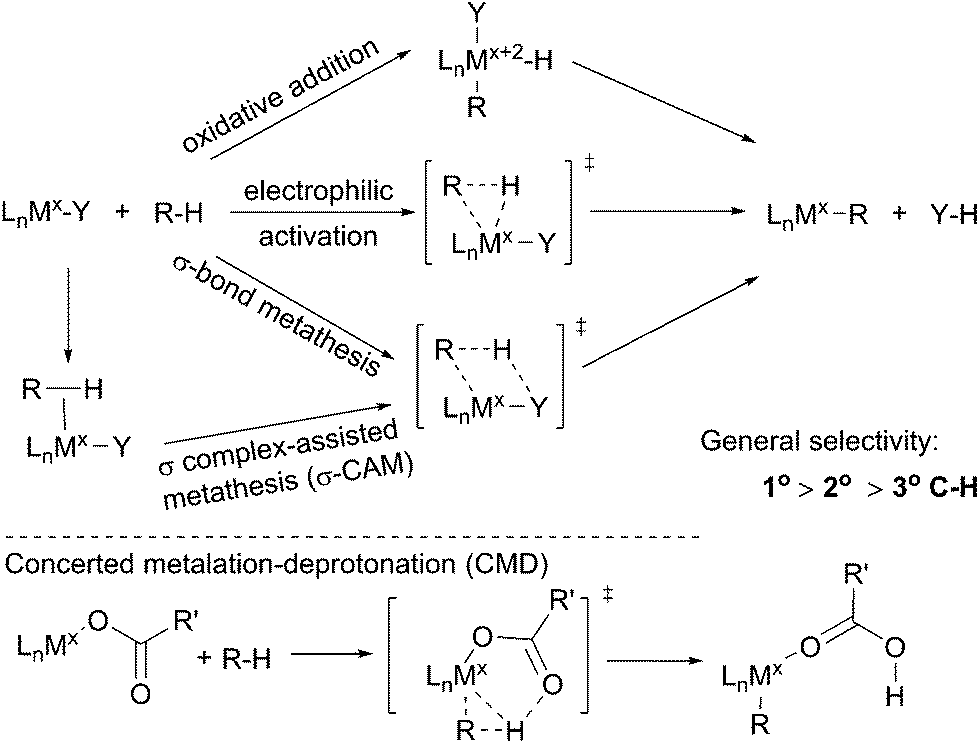 | ||
| Scheme 1 Pathways for metal-mediated C–H bond activation. | ||
One attractive feature of metal-mediated C–H bond activation is the selective cleavage of the most sterically accessible primary C–H bonds. Bergman39 and Jones40 reported that Cp*M(PMe3) (M = Ir or Rh), generated upon the UV irradiation of Cp*Rh(PMe3)H2, reacts with an alkane to form Cp*M(PMe3)(n-alkyl)(H) with exclusive terminal regioselectivity. These proof-of-concept studies demonstrated the potential of C–H activation for the terminally selective functionalisation of an alkane. The known catalytic C–H bond functionalisations encompass alkane oxidation, borylation, carbonylation, dehydrogenation, and dehydrogenation based reactions, which will be presented in the following sections.
4.1. Alkane oxidation
In early 1970s, Shilov reported the selective partial oxidation of an alkane to an alcohol or alkyl chloride catalysed by PtCl2 using H2PtCl6 as the oxidant.41 The rate-determining step involves the electrophilic activation of R–H with a Pt(II) centre to form a Pt(II)–R complex. Next the Pt(II)–R species is oxidized by [PtCl6]2− to generate a Pt(IV)–R complex, which undergoes nucleophilic attack by OH− or Cl− with the release of the product and regeneration of the Pt(II) catalyst.42In principle, the expensive Pt(IV) oxidant could be replaced by a cheaper oxidant. Indeed, Cu(II)/O2, Fe(III)/O2 and polyoxometallate [H3PMo9V3O40]/O2 can act as effective oxidants for alkane oxidations.43 Periana discovered that SO3 or H2SO4 is an active oxidant for the catalytic oxidation of methane to methyl bisulfate in high yields. Most systems developed by this group were applied for methane oxidization;44 the reactions of higher alkanes were plagued by the formation of multiple products.
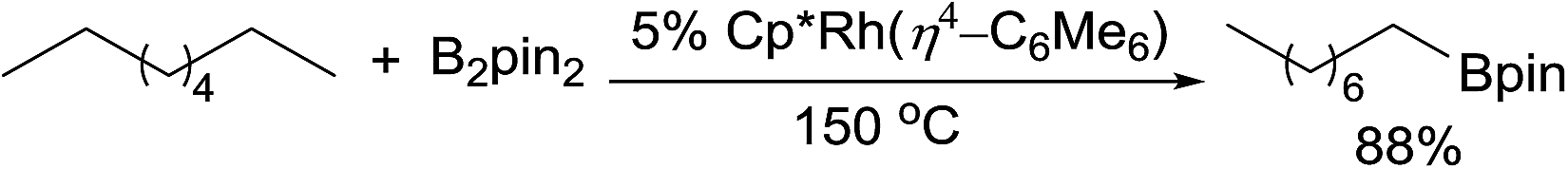 | (15) |
The selectivity for electrophilic C–H activation follows the trend of 1° > 2° > 3° C–H bonds, but the regioselectivity observed in the oxidation of an acyclic alkane (e.g. n-pentane in eqn (14)) is rather low.45 While the Pt-catalysed oxidations of alkyl sulfonates46 and ammonium salts47 offer moderate-to-good selectivity for primary C–H bond hydroxylation, to date, oxidations of simple alkanes with high terminal selectivity via electrophilic C–H activation have remained underexploited.43e,48
4.2. Alkane borylations
The catalytic alkane borylation developed by the Hartwig group is a unique example of the selective functionalisation of a primary C–H bond.49 The earlier systems with carbonyl complexes employed photochemical conditions. In 1997, Hartwig reported that a boryl W carbonyl complex, under irradiation, reacts with n-pentane to form a linear alkylboronate ester with excellent terminal selectivity.50 The stoichiometric reaction was then expanded to a catalytic reaction using a tricarbonyl Re catalyst.51 Under a CO atmosphere, the photocatalytic borylation of n-pentane with 1 equiv. of B2pin2 forms the linear product in high yield. Following the success of photocatalysis, this group developed methods for thermal alkane borylation. A Rh complex Cp*Rh(η4-C6Me6), upon thermally dissociating the dative ligand, hexamethylbenzene, proved effective for alkane borylation at 150 °C (eqn (15)).52 This catalytic system was also applied to the selective borylation of 1° C–H bonds in polyolefins.49,53 Subsequently, a simple Cp*Ru dichloride complex was found to be a suitable precatalyst for the borylation.54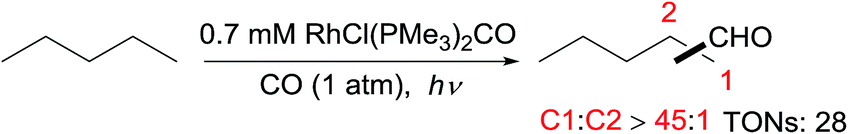 | (16) |
Calculations and experiments showed that the rate-limiting step in the Rh-catalysed borylation is the C–H bond activation,50–52 which very likely occurs through the σ-CAM route involving the interaction between a Rh–B bond and the coordinated C–H bond.55
4.3. Photocatalytic alkane carbonylation
Tanaka first reported alkane carbonylation with CO via C–H activation catalysed by RhCl(CO)(PMe3)2 under irradiation.56 The primary C–H bond of an acyclic alkane is selectively activated to form linear aldehydes, along with small amounts of branched products (eqn (16)). However, the resulting aldehyde could undergo a secondary Norrish Type II photoreaction to give a terminal olefin, acetaldehyde and ethanol as byproducts. The Goldman group revealed that this reaction occurs via at least two different mechanisms, one via a photochemical pathway and the other via a radical process. The former is responsible for the high linear selectivity, while the latter is less regioselective.57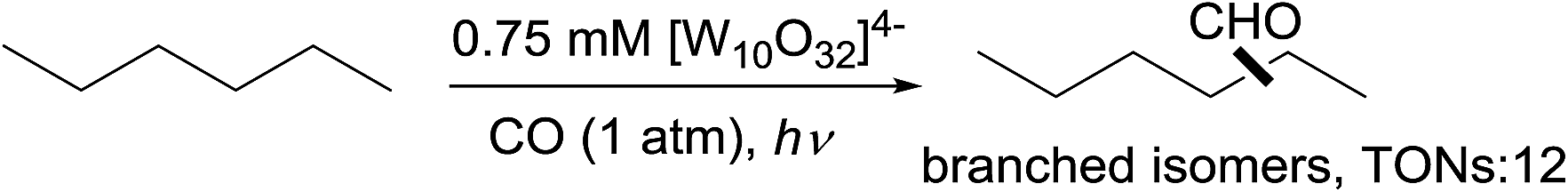 | (17) |
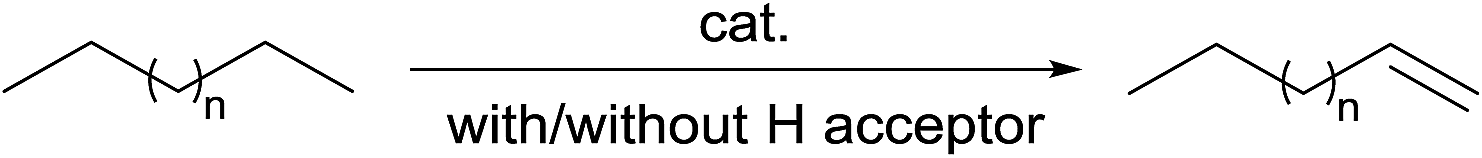 | (18) |
A pure radical process for alkane carbonylation was also reported – Hill showed that the polyoxotungstate-catalysed carbonylation of n-hexane with CO under irradiation produced a mixture of branched aldehydes (eqn (17)), in sharp contrast to the high linear selectivity gained in the rhodium catalysis.58 Note that catalytic thermal alkane carbonylation is an unknown reaction.
4.4. Alkane dehydrogenation
Alkane dehydrogenation (AD) is an atom-economic method for the synthesis of olefins, which are valuable synthetic intermediates for petrochemicals. Heterogeneous AD requires high reaction temperatures and suffers from low product selectivity.59 In this respect, the development of a mild and selective homogeneous AD system is desired. Pioneered by Crabtree and Felkin, significant advances have been achieved in the development of molecular catalysts for AD with or without a hydrogen acceptor (eqn (18)). In the majority of these systems Ir pincer complexes have been extensively studied, although the development of other metal systems has also received research interest.60 Most of these processes proceed via C–H oxidative addition and the formation of metal–carbon bonds tend to occur at 1° C–H bonds rather than 2° and 3° C–H bonds. Thus, organometallic AD is fundamentally promising for the production of α-olefins. | (19) |
In 1979, Crabtree reported the first stoichiometric AD reaction: [IrH2(acetone)2(PR3)]+ reacts with cylcooctene (COE) to form cyclooctadiene Ir complexes with tert-butylethylene (TBE) as the hydrogen acceptor.61 Soon after that, the first examples of the catalytic AD transfer dehydrogenation (TD) of cylcooctane (COA) with TBE were independently developed by Crabtree62 and Felkin63 using Ir- or Re-based catalysts, albeit with low TONs. The hydrogenation of TBE overcomes the thermodynamic challenge associated with hydrogen release during the dehydrogenation process.
A breakthrough in homogeneous AD was achieved in the mid-1990s by Jensen, Kaska, and Goldman, showing that bis(phosphine)-based (PCP)Ir pincer complexes are highly efficient for the TD of cyclic and linear alkanes. The success of this type of Ir system in AD is attributed in a large part to their high thermostability. The TD of COA/TBE using a tBu-substituted complex (tBu4PCP)IrH21a (Fig. 4) at 200 °C gives an initial rate of 12 turnovers per minute. TONs of up to 1000 could be achieved by the addition of portions of TBE as the catalyst is subject to TBE inhibition at high [TBE].64
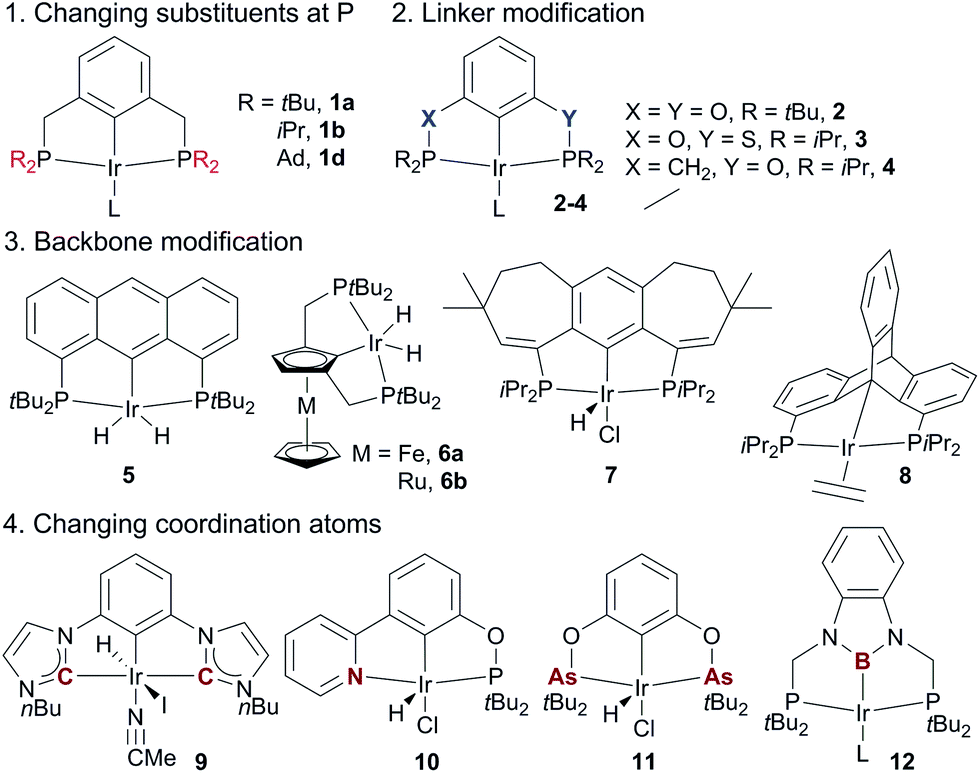 | ||
| Fig. 4 The design of various pincer Ir dehydrogenation catalysts. | ||
Catalyst 1a is even effective for acceptorless dehydrogenation, giving 360 TONs in a run with cyclodecane at 201 °C after a day.65 The efficient hydrogen removal under refluxing conditions is crucial to the catalytic turnover. A sterically less hindered iPr-substituted analog (iPr4PCP)IrH41b is even more effective than 1a, producing a TON of 1000 for the acceptorless dehydrogenation of cyclodecane.66
Remarkably, Goldman demonstrated that catalysts 1a and 1b exhibit high kinetic selectivity for valuable α-olefins in the early stages of the TD of linear alkanes with TBE, NBE or α-olefin as the hydrogen acceptor. Unfortunately, subsequent olefin isomerization quickly leads to thermodynamically more stable internal olefins.67
The superior performance of (PCP)Ir complexes 1a and 1b in AD has stimulated extensive research in developing new pincer Ir complexes with the goal of more effective catalysts. Four strategies have been used for ligand design (Fig. 4). (1) Variation of substituents on the P-atoms:68 the tBu-to-Me substitution, for example, leads to a more efficient AD catalyst (tBu3MePCP)IrH41c.68a (2) Variation of the linkers between the backbone and two P-atoms: (tBu4POCOP)Ir catalyst 2 containing two O-linkers69 and (iPr4PSCOP)Ir catalyst 3 with O and S as the linkers70 are less subject to TBE inhibition relative to the (tBu4PCP)Ir system, producing TONs of up to 6000 in the reaction of COA/TBE TD. (3) Backbone modifications: complexes of various aromatic skeletons,71 including anthraphos 5,71a ferrocene/ruthenocene 6,71b 7-6-7 fused-ring backbones 7,71c and triptycene 871d backbones, exhibit high thermostability and high activity for AD. (4) Substitution of coordination atom(s):67 bis(carbene) Ir (CCC, 9),72a,b (PBP)Ir 12,72c and pyridine–phosphinite (NCP, 10)72d complexes display low-to-moderate activity in the COA/TBE TD. The novel (AsCAs)Ir complex 11 shows good TD activity, but decomposes readily at >175 °C.72e
Non-iridium precious metal catalysts have also been prepared and studied for AD, but in general they are less efficient than the classic pincer Ir complexes (Fig. 5). The studies of Rh catalysis mainly focused on photocatalytic AD (see below). One recent exception was the study by Brookhart using a carbazolide-based PNP Rh complex 13, which affords moderate activity in the thermal TD of COA/TBE.73 Early investigations into Ru-catalysed dehydrogenation used Ru polyhydride63b and bis(phosphine) Ru(II) bis(allyl) complexes,74 which showed low catalytic activity. A π-accepting pincer-ligated [(CF3)4PCP]RuH(COD) 14 gives a limited TON due to facile catalyst decomposition,75 while the Os analog 15 has a significantly enhanced lifetime relative to 14, furnishing a TON of ∼600 in the COA/TBE TD.76 The author’s group developed a series of hydrido Ru(II) olefin complexes supported by iPr-substituted pincer ligands, which are moderately active for the COA/TBE TD. Although less efficient, pincer Ru catalysts are more tolerant of polar functional groups than the related pincer Ir catalysts.75,77 A lipophilic tBu2dpb-ligated hydrido Pt complex 16 shows low activity for the TD of cycloalkanes with TBE.78
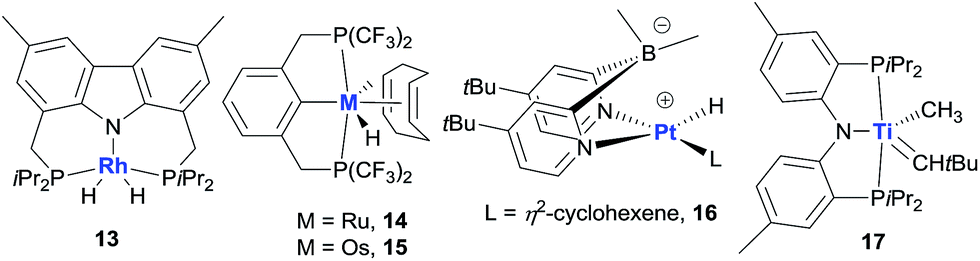 | ||
| Fig. 5 Non-iridium metal dehydrogenation catalysts. | ||
Apart from the extensively studied noble metal-based AD catalysts, very recently, Mindiola and Baik reported a rare base-metal catalyst, (PNP)Ti alkylidene (17, Fig. 5), for TD with a phosphorus ylide as a hydrogen acceptor under mild reaction conditions (75 °C). Significantly, α-olefins could be selectively formed in this system, albeit with low TONs (<4).79
Compared to TBE, the most commonly used hydrogen acceptor for TD reactions, ethylene and propylene, are more practical acceptors in terms of cost and accessibility. Goldman and Brookhart reported the use of propylene as a hydrogen acceptor for the pincer-Ir-catalysed dehydroaromatization of n-alkane (vide supra).80 Furthermore, Brookhart demonstrated that n-pentane could be effectively dehydrogenated by pincer-Ir using ethylene or propylene as the acceptor; the resulting diene product then undergoes a sequence of a Diels–Alder reaction with ethylene and heterogeneous AD by Pt/C to form toluene.81 More recently, Goldman found that the TD of linear alkanes with ethylene or propylene in a heterogeneous solid–gas system (i.e. the combination of a solid molecular pincer-Ir catalyst and gaseous substrates) gives a high yield of α-olefins. The isomerization of a terminal alkene to an internal alkene, to a large extent, could be inhibited. Calculations reveal that the gaseous acceptor effectively captures the Ir dihydride species, which is partly responsible for alkene isomerization.82
Catalytic AD reactions under photochemical conditions have also been explored. In general, the photochemical AD proceeds at lower temperatures and offers a higher selectivity for α-olefins in comparison to the thermochemical reactions. An additional advantage is that a hydrogen acceptor is not required in these photochemical reactions. Crabtree reported the first photochemical AD using IrH2(PCy3)2(η2-O2CCF3), giving a TON of seven for the acceptorless dehydrogenation of COA.83 Normura and Saito found that RhCl(PMe3)2CO is a more effective catalyst: the photochemical dehydrogenation of n-octane resulted in a turnover frequency (TOF) of 650 h−1 at 86 °C.84 Goldman applied this Rh catalyst to the photochemical dehydrogenation of cycloalkanes: TONs of up to 5000 with a TOF of 300 h−1 were achieved in the run with COA. Mechanistic studies established that the photoextrusion of CO from RhCl(PMe3)2CO is part of the catalytic cycle.85 Using the same catalyst, very recently, the Beller group observed that the added 4,4′-bipyridine could enhance the catalyst stability and light transmittance has a profound effect on the efficiency. Under the optimal light transmittance, the acceptorless dehydrogenation of n-octane gives TONs of ∼1500 with 35% selectivity for 1-octene.86
4.5. Dehydrogenation-based alkane transformation
Several catalytic methods of alkane transformation initiated by AD have been developed during the past decade. Goldman and Brookhart reported a tandem dual catalyst system for alkane metathesis (AM), in which the pincer Ir catalyst (1a) effects AD and Schrock catalyst effects olefin metathesis. The process enables the conversion of a linear alkane to linear products, but rapid olefin isomerization prior to metathesis results in the formation of a range of alkanes. For example, the metathesis of n-hexane at 125 °C yields C2–C5 and C7–C15 n-alkanes (150–200 equiv. relative to Ir) (Fig. 6a). Switching the olefin metathesis catalyst to Re2O7/γ-Al2O3, a heterogeneous catalyst, leads to a more robust system, giving TONs of ∼500 for n-decane metathesis at 175 °C.87,88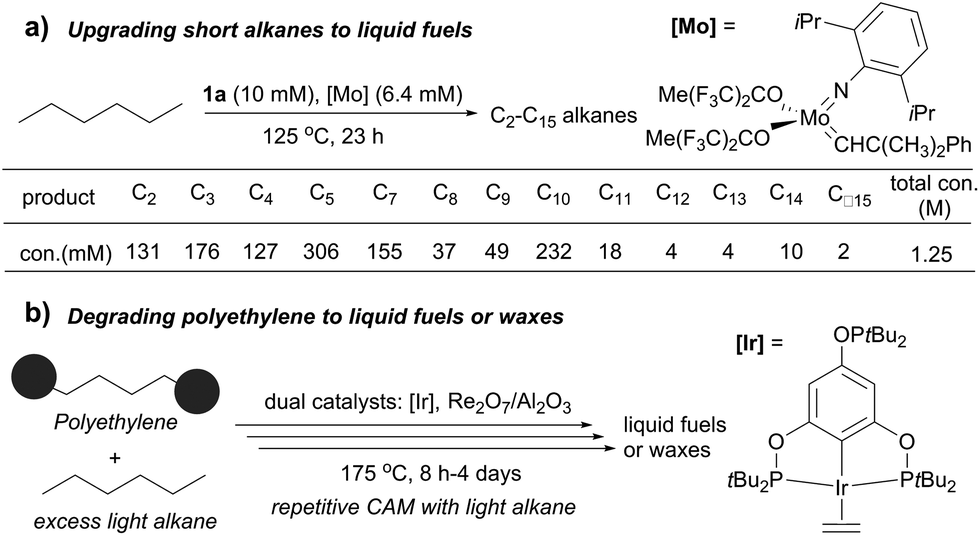 | ||
| Fig. 6 Applications of alkane metathesis. | ||
The Goldman–Brookhart AM system holds promise for upgrading low carbon number n-alkanes (e.g. C4–C8) to higher molecular weight fuel alkanes. Its reverse process, the degradation of polyethylene (PE) into fuel alkanes using a cross-AM strategy, was recently reported by Huang and Guan (Fig. 6b). Using the Goldman–Brookhart Ir/Re catalyst system, the cross-AM between PE and a light alkane results in the breakdown of a PE chain. Because a large excess of light alkanes is used as the reactant/solvent, the first cross-AM products react further with the light alkanes to give even shorter hydrocarbons. After multiple cycles of cross-AM with light alkanes, the authors demonstrated that HDPE (high density PE) with a molecular weight of up to 1.7 million could be fully degraded to oil and wax. The catalyst system can tolerate commercial HDPE, LDPE and LLDPE that contains phosphinite-based stabilizers and zinc stearate which allowed for the degradation of various post-consumer PE plastics waste.
Bercaw and Labinger devised a novel method, the net coupling of an alkane and a terminal alkene, to upgrade light hydrocarbons into heavier fuel alkanes (eqn (19)).89 This system comprises a catalyst for the TD of an alkane and a Ta catalyst (Cp*TaCl2(CH2CH2)) for the dimerization of the resulting alkene product with another alkene molecule. The subsequent Ir-mediated TD would yield the upgraded alkanes, together with a new alkene for the next cycle of dimerization. Because the Ta catalyst is inactive for the dimerization of internal olefins, the choice of the terminal-selective Ir catalyst (1b) is important for this transformation. The slow addition of the terminal alkene, which minimizes the degree of undesired isomerization, is also crucial to the success of the coupling. The authors showed that gradually adding 1-hexene via a syringe pump to a mixture of Ir/Ta catalysts in n-heptane forms branched C13 and C14 in a 40% total yield (relative to 1-hexene), corresponding to a TON of 60 for 1b. Despite the low efficiency, the alkane/alkene coupling yields products with controllable selectivity for a desired weight fraction, in contrast to a stochastic product distribution observed for AM.
 | (20) |
Aromatic molecules, especially the BTX family of benzene, toluene, and xylene, are valuable feedstocks in the chemical industry. Goldman and Brookhart reported the first example of the homogeneously catalyzed dehydroaromatization of n-alkanes to benzene, n-alkylarenes or o-dialkylarenes (Fig. 7).80,81,90 Using hybrid complex (iPr4PCOP)Ir(C2H4) 4 as the catalyst, the reaction of n-octane with four equiv. of TBE affords o-xylene as the dominant product, while the reaction of n-decane yields four different arenes, with o-propyltoluene as the major product.80
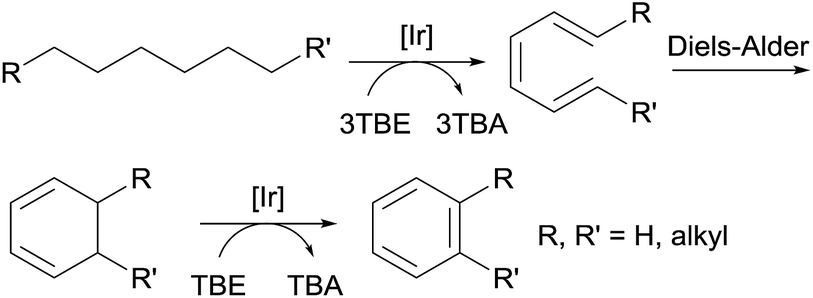 | ||
| Fig. 7 Dehydroaromatization of alkanes. | ||
Another strategy for the synthesis of n-alkylarenes was reported by Schrock and Goldman.91 The cross-metathesis between an n-alkane and ethylbenzene using a combination of the dehydrogenation Ir catalyst 1a and a thermally stable W-based metathesis catalyst enables the selective formation of n-alkylarenes over alkanes.91
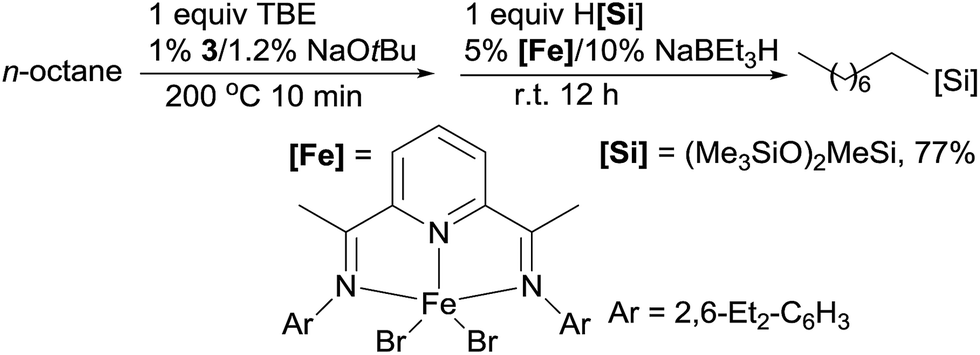
Linear alkylsilanes are valuable synthetical intermediates in the silicone industry. In 2016, Huang reported the net terminal silylation of alkanes to form linear alkylsilanes using a dual Ir/Fe catalyst system.92 The pincer Ir 3 catalysed dehydrogenation gives internal olefins as the major products, which, in the presence of iron catalysts, are isomerized to α-olefins for anti-Markovnikov hydrosilylation. The ineffectiveness of the bis(imino)pyridine Fe complex (9) for the hydrosilylation of internal olefins precludes the formation of branched alkylsilanes. For example, the TD of n-octane with TBE at 200 °C for 10 min, followed by the isomerization–hydrosilylation with (Me3SiO)2MeSiH, generates the linear alkylsilane in a good yield (eqn (20)).
5. Conclusion and outlook
The period of the last four decades has witnessed significant progress in homogeneously catalyzed alkane transformations through C–H bond functionalisation using methods including radical pathways or carbene/nitrene insertions. In practice, these approaches using highly reactive free radicals or carbene/nitrene intermediates may be primarily applicable to the small-scale production of specialized chemicals, given the use of relatively expensive and/or forcing reagents and inherent regioselectivity. The preference of such methods for the functionalisation of secondary or tertiary C–H over primary C–H bonds renders them more suitable for cyclic substrates, rather than linear alkanes. Moreover, one specific C–H bond of an alkyl chain in a more functionalised molecule may react with a radical or carbene/nitrene with useful selectivity. Thus, one may expect that some transformations, such as C–H fluorination and enantioselective carbene insertion, will find applications in the late-stage labeling of biologically active molecules or in the synthesis of fine chemicals.From a practical point of view, the functionalisation of the primary sites in simple alkanes is especially attractive for the large-scale synthesis of specialty or commodity chemicals. In this respect, the transition metal-catalysed selective activation of primary C–H bonds holds great potential. The challenge ahead now lies in the development of practical processes that utilize low-cost and environmentally benign reagents. Although highly regioselective, the Rh- or Ru-catalysed alkane borylations using boron reagents are not economically viable. While the formal terminal silylation of alkanes with relatively low-cost silanes using a two-step dehydrogenation–hydrosilylation process has been developed by the author’s group,92 the direct silylation of alkanes higher than methane93 to form valuable linear alkylsilanes remains to be exploited.
Most important bulk oxygenates contain the functional group at the end of an alkyl chain, therefore, the selective oxidation of alkanes to linear alkyl alcohols, aldehydes, or carboxylic acids is a goal of great interest. Unfortunately, the known methods for alkane oxidation via electrophilic C–H activation feature low regioselectivity. An ultimate challenge in this important field is to use molecular oxygen or hydrogen peroxide for selective oxidations, generating water as the only by-product.
Catalytic alkane dehydrogenations typically need a sacrificial hydrogen acceptor to gain high turnover numbers, while the existing systems for acceptorless dehydrogenations suffer from very low efficiency. Another challenge is the poor selectivity for the formation of α-olefins due to the facile isomerization of the kinetic terminal products to internal olefin mixtures. Encouragingly, as noted in the body of this perspective, Goldman and co-workers have found that using practical hydrogen acceptors, ethylene or propylene, can effectively eliminate olefin isomerization, leading to significantly enhanced yields of α-olefins. Thus, further development of long-lived highly efficient catalysts in conjunction with an appropriate hydrogen acceptor and reaction vessel may provide a practical catalytic system for regioselective alkane dehydrogenation. Moreover, efficient dehydrogenation catalysts based on earth-abundant and low-cost base-metals will be a welcome addition.
Regardless of the nature of the different classes of method for alkane functionalisations, catalyst development will continue to be the central theme of research. In-depth mechanistic understanding of the factors controlling reactivity and selectivity will lead to the future development of more efficient catalysts and the discovery of new catalytic reactions. Yet, one must not overlook the power of cooperative catalysis that involves two or more catalysts for new alkane transformations. Moreover, the design of enantioselective catalytic systems will provide a framework for the synthesis of high-value fine chemicals using a simple hydrocarbon as the starting material. One may also expect that developing appropriate catalysts and conditions for photocatalysis and electrocatalysis will be an emerging topic in the area of alkane chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Key R&D Program of China (2016YFA0202900, 2015CB856600), National Natural Science Foundation of China (21422209, 21432011, 21421091), Strategic Priority Research Program of CAS (XDB20000000), and Science and Technology Commission of Shanghai Municipality (17JC1401200) support this work.Notes and references
- (a) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed; (b) A. S. Goldman and K. I. Goldberg, Activation and Functionalization of C–H Bonds, American Chemical Society, 2004, vol. 885, ch. 1, pp. 1–43 Search PubMed; (c) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed; (d) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2 CrossRef CAS PubMed; (e) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281 CrossRef CAS PubMed; (f) K. I. Goldberg and A. S. Goldman, Acc. Chem. Res., 2017, 50, 620 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255 CrossRef CAS PubMed.
- (a) A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567 RSC; (b) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC.
- (a) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947 CrossRef CAS PubMed; (b) J. T. Groves, in Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, Springer US, Boston, MA, 2005, pp. 1–43 Search PubMed; (c) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932 CrossRef CAS PubMed; (d) M. Bordeaux, A. Galarneau and J. Drone, Angew. Chem., Int. Ed., 2012, 51, 10712 CrossRef CAS PubMed.
- (a) A. Company, J. Lloret, L. Gómez and M. Costas, Alkane C–H Activation by Single-Site Metal Catalysis, ed. P. J. Pérez, Springer Netherlands, Dordrecht, 2012, pp. 143–228 Search PubMed; (b) M. Grau and G. J. P. Britovsek, in Iron Catalysis II, ed. E. Bauer, Springer International Publishing, Cham, 2015, pp. 145–171 Search PubMed; (c) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS PubMed.
- C. Walling and B. B. Jacknow, J. Am. Chem. Soc., 1960, 82, 6108 CrossRef CAS.
- B. L. Tran, B. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 2555 CrossRef CAS PubMed.
- B. R. Cook, T. J. Reinert and K. S. Suslick, J. Am. Chem. Soc., 1986, 108, 7281 CrossRef CAS.
- J. T. Groves, T. E. Nemo and R. S. Myers, J. Am. Chem. Soc., 1979, 101, 1032 CrossRef CAS.
- (a) J. T. Groves, J. Inorg. Biochem., 2006, 100, 434 CrossRef CAS PubMed; (b) J. T. Groves, R. Quinn, T. J. McMurry, G. Lang and B. Boso, J. Chem. Soc., Chem. Commun., 1984, 1455 RSC; (c) J. E. Penner-Hahn, K. Smith Eble, T. J. McMurry, M. Renner, A. L. Balch, J. T. Groves, J. H. Dawson and K. O. Hodgson, J. Am. Chem. Soc., 1986, 108, 7819 CrossRef CAS PubMed; (d) K. Jayaraj, A. Gold, R. N. Austin, D. Mandon, R. Weiss, J. Terner, E. Bill, M. Muether and A. X. Trautwein, J. Am. Chem. Soc., 1995, 117, 9079 CrossRef CAS.
- W. Liu and J. T. Groves, J. Am. Chem. Soc., 2010, 132, 12847 CrossRef CAS PubMed.
- W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322 CrossRef CAS PubMed.
- X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300 CrossRef CAS PubMed.
- C.-W. Tse, T. W.-S. Chow, Z. Guo, H. K. Lee, J.-S. Huang and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 798 CrossRef CAS PubMed.
- (a) Y. Li, F. Zhu, Z. Wang and X.-F. Wu, ACS Catal., 2016, 6, 5561 CrossRef CAS; (b) L. Lu, R. Shi, L. Liu, J. Yan, F. Lu and A. Lei, Chem.–Eur. J., 2016, 22, 14484 CrossRef CAS PubMed; (c) S. Tang, P. Wang, H. Li and A. Lei, Nat. Commun., 2016, 7, 11676 CrossRef PubMed; (d) D. Liu, Y. Li, X. Qi, C. Liu, Y. Lan and A. Lei, Org. Lett., 2015, 17, 998 CrossRef CAS PubMed; (e) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef CAS PubMed.
- (a) B. L. Tran, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 17292 CrossRef CAS PubMed; (b) T. K. Salvador, C. H. Arnett, S. Kundu, N. G. Sapiezynski, J. A. Bertke, M. Raghibi Boroujeni and T. H. Warren, J. Am. Chem. Soc., 2016, 138, 16580 CrossRef CAS PubMed; (c) Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547 CrossRef CAS PubMed; (d) P. K. Chikkade, Y. Kuninobu and M. Kanai, Chem. Sci., 2015, 6, 3195 RSC; (e) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600 CrossRef CAS PubMed; (f) J. G. West, T. A. Bedell and E. J. Sorensen, Angew. Chem., Int. Ed., 2016, 55, 8923 CrossRef CAS PubMed.
- S. Dupuy, K.-F. Zhang, A.-S. Goutierre and O. Baudoin, Angew. Chem., Int. Ed., 2016, 55, 14793 CrossRef CAS PubMed.
- F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed.
- (a) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379 CrossRef PubMed; (b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (c) A. Caballero, M. M. Diaz-Requejo, M. R. Fructos, A. Olmos, J. Urbano and P. J. Pérez, Dalton Trans., 2015, 44, 20295 RSC.
- L. T. Scott and G. J. DeCicco, J. Am. Chem. Soc., 1974, 96, 322 CrossRef CAS.
- (a) A. Demonceau, A. F. Noels, A. J. Hubert and P. Teyssie, J. Chem. Soc., Chem. Commun., 1981, 688 RSC; (b) A. Demonceau, A. F. Noels, A. J. Hubert and P. Teyssié, Bull. Soc. Chim. Belg., 1984, 93, 945 CrossRef CAS; (c) A. Demonceau, A. F. Noels, P. Teyssie and A. J. Hubert, J. Mol. Catal., 1988, 49, 13 CrossRef; (d) A. Demonceau, A. F. Noels and A. J. Hubert, J. Mol. Catal., 1989, 57, 149 CrossRef CAS.
- M. M. Díaz-Requejo, T. R. Belderraín, M. C. Nicasio, S. Trofimenko and P. J. Pérez, J. Am. Chem. Soc., 2002, 124, 896 CrossRef.
- A. Caballero, M. M. Díaz-Requejo, T. R. Belderraín, M. C. Nicasio, S. Trofimenko and P. J. Pérez, J. Am. Chem. Soc., 2003, 125, 1446 CrossRef CAS PubMed.
- (a) M. R. Fructos, P. de Frémont, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2006, 25, 2237 CrossRef CAS; (b) E. Despagnet-Ayoub, K. Jacob, L. Vendier, M. Etienne, E. Álvarez, A. Caballero, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2008, 27, 4779 CrossRef CAS.
- (a) H. V. R. Dias, R. G. Browning, S. A. Richey and C. J. Lovely, Organometallics, 2005, 24, 5784 CrossRef CAS; (b) M. Á. Fuentes, B. K. Muñoz, K. Jacob, L. Vendier, A. Caballero, M. Etienne and P. J. Pérez, Chem.–Eur. J., 2013, 19, 1327 CrossRef CAS PubMed.
- H.-Y. Thu, G. S.-M. Tong, J.-S. Huang, S. L.-F. Chan, Q.-H. Deng and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9747 CrossRef CAS PubMed.
- (a) H. M. L. Davies and T. Hansen, J. Am. Chem. Soc., 1997, 119, 9075 CrossRef CAS; (b) H. M. L. Davies, T. Hansen and M. R. Churchill, J. Am. Chem. Soc., 2000, 122, 3063 CrossRef CAS.
- K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230 CrossRef CAS PubMed.
- D. S. Breslow and M. F. Sloan, Tetrahedron Lett., 1968, 9, 5349 CrossRef.
- D. H. R. Barton, R. S. Hay-Motherwell and W. B. Motherwell, J. Chem. Soc., Perkin Trans. 1, 1983, 445 RSC.
- (a) I. Nägeli, C. Baud, G. Bernardinelli, Y. Jacquier, M. Moraon and P. Müllet, Helv. Chim. Acta, 1997, 80, 1087 CrossRef; (b) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS PubMed; (c) C. Liang, F. Collet, F. Robert-Peillard, P. Müller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343 CrossRef CAS PubMed; (d) C. Liang, F. Robert-Peillard, C. Fruit, P. Müller, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2006, 45, 4641 CrossRef CAS PubMed.
- (a) Z. Li, D. A. Capretto, R. Rahaman and C. He, Angew. Chem., Int. Ed., 2007, 46, 5184 CrossRef CAS PubMed; (b) Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961 CrossRef CAS PubMed; (c) B. P. Gómez-Emeterio, J. Urbano, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2008, 27, 4126 CrossRef.
- (a) M. M. Díaz-Requejo, T. R. Belderraín, M. C. Nicasio, S. Trofimenko and P. J. Pérez, J. Am. Chem. Soc., 2003, 125, 12078 CrossRef PubMed; (b) R. A. Olsen, J. Struppe, D. W. Elliott, R. J. Thomas and L. J. Mueller, J. Am. Chem. Soc., 2003, 125, 11784 CrossRef CAS PubMed.
- B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154 CrossRef CAS.
- (a) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS PubMed; (b) N. M. West and J. L. Templeton, Can. J. Chem., 2009, 87, 288 CrossRef CAS; (c) J. A. Labinger, Chem. Rev., 2017, 117, 8483 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (e) J. A. Labinger and J. E. Bercaw, in Higher Oxidation State Organopalladium and Platinum Chemistry, ed. A. J. Canty, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 29–59 Search PubMed.
- (a) P. L. Watson, J. Am. Chem. Soc., 1983, 105, 6491 CrossRef CAS; (b) M. E. Thompson, S. M. Baxter, A. R. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero, W. P. Schaefer and J. E. Bercaw, J. Am. Chem. Soc., 1987, 109, 203 CrossRef CAS.
- (a) R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578 CrossRef CAS PubMed; (b) K. Hoshi, A. Tahara, Y. Sunada, H. Tsutsumi, R. Inoue, H. Tanaka, Y. Shiota, K. Yoshizawa and H. Nagashima, Bull. Chem. Soc. Jpn., 2017, 90, 613 CrossRef CAS.
- (a) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (c) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed; (d) D. G. Musaev, T. M. Figg and A. L. Kaledin, Chem. Soc. Rev., 2014, 43, 5009 RSC.
- A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1983, 105, 3929 CrossRef CAS.
- W. D. Jones and F. J. Feher, J. Am. Chem. Soc., 1984, 106, 1650 CrossRef CAS.
- (a) E. S. Aleksandr and B. S. p. Georgiy, Russ. Chem. Rev., 1987, 56, 442 CrossRef; (b) A. E. Shilov and G. B. Shul’pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed.
- G. A. Luinstra, L. Wang, S. S. Stahl, J. A. Labinger and J. E. Bercaw, J. Organomet. Chem., 1995, 504, 75 CrossRef CAS.
- (a) N. F. Goldshleger, V. V. Lavrushko, A. P. Khrushch and A. A. Shteinman, Izv. Akad. Nauk SSSR, Ser. Khim., 1976, 2174 CAS; (b) I. Bar-Nahum, A. M. Khenkin and R. Neumann, J. Am. Chem. Soc., 2004, 126, 10236 CrossRef CAS PubMed; (c) D. R. Weinberg, J. A. Labinger and J. E. Bercaw, Organometallics, 2007, 26, 167 CrossRef CAS; (d) J. E. Kreutz, A. Shukhaev, W. Du, S. Druskin, O. Daugulis and R. F. Ismagilov, J. Am. Chem. Soc., 2010, 132, 3128 CrossRef CAS PubMed; (e) J. A. Labinger, Alkane C–H Activation by Single-Site Metal Catalysis, ed. P. J. Pérez, Springer Netherlands, Dordrecht, 2012, pp. 17–71 Search PubMed.
- (a) R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560 CrossRef CAS PubMed; (b) R. A. Periana, D. J. Taube, E. R. Evitt, D. G. Löffler, P. R. Wentrcek, G. Voss and T. Masuda, Science, 1993, 259, 340 CAS; (c) R. A. Periana, O. Mironov, D. Taube, G. Bhalla and C. Jones, Science, 2003, 301, 814 CrossRef CAS PubMed; (d) B. G. Hashiguchi, M. M. Konnick, S. M. Bischof, S. J. Gustafson, D. Devarajan, N. Gunsalus, D. H. Ess and R. A. Periana, Science, 2014, 343, 1232 CrossRef CAS PubMed.
- (a) N. F. Goldshleger, M. B. Tyabin, A. E. Shilov and A. A. Shteinman, Russ. J. Phys. Chem. USSR., 1969, 43, 1222 Search PubMed; (b) N. F. Goldshleger, A. A. Shteinman, A. E. Shilov and V. V. Eskova, Russ. J. Phys. Chem. USSR., 1972, 46, 785 Search PubMed; (c) N. F. Goldshleger, V. V. Eskova, A. E. Shilov and A. A. Shteinman, Zh. Fiz. Khim., 1972, 46, 1353 CAS.
- M. Lin, C. Shen, E. A. Garcia-Zayas and A. Sen, J. Am. Chem. Soc., 2001, 123, 1000 CrossRef CAS PubMed.
- M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 12796 CrossRef CAS PubMed.
- Y. V. Geletii and A. E. Shilov, Kinet. Catal., 1983, 24, 413 Search PubMed.
- (a) J. F. Hartwig, Acc. Chem. Res., 2011, 45, 864 CrossRef PubMed; (b) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- K. M. Waltz and J. F. Hartwig, Science, 1997, 277, 211 CrossRef CAS.
- H. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391 CrossRef CAS PubMed.
- H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS PubMed.
- C. Bae, J. F. Hartwig, N. K. Boaen Harris, R. O. Long, K. S. Anderson and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 767 CrossRef CAS PubMed.
- J. M. Murphy, J. D. Lawrence, K. Kawamura, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 13684 CrossRef CAS PubMed.
- J. F. Hartwig, K. S. Cook, M. Hapke, C. D. Incarvito, Y. Fan, C. E. Webster and M. B. Hall, J. Am. Chem. Soc., 2005, 127, 2538 CrossRef CAS PubMed.
- (a) T. Sakakura and M. Tanaka, J. Chem. Soc., Chem. Commun., 1987, 758 RSC; (b) T. Sakakura, T. Sodeyama, K. Sasaki, K. Wada and M. Tanaka, J. Am. Chem. Soc., 1990, 112, 7221 CrossRef CAS.
- (a) W. T. Boese and A. S. Goldman, J. Am. Chem. Soc., 1992, 114, 350 CrossRef CAS; (b) G. P. Rosini, K. Zhu and A. S. Goldman, J. Organomet. Chem., 1995, 504, 115 CrossRef CAS.
- B. S. Jaynes and C. L. Hill, J. Am. Chem. Soc., 1995, 117, 4704 CrossRef CAS.
- (a) B. M. Weckhuysen and R. A. Schoonheydt, Catal. Today, 1999, 51, 223 CrossRef CAS; (b) R. P. O’Connor, E. J. Klein, D. Henning and L. D. Schmidt, Appl. Catal., A, 2003, 238, 29 CrossRef; (c) U. Olsbye, A. Virnovskaia, Ø. Prytz, S. J. Tinnemans and B. M. Weckhuysen, Catal. Lett., 2005, 103, 143 CrossRef CAS.
- (a) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS PubMed; (b) Y. Zhang, W. Yao, H. Fang, A. Hu and Z. Huang, Sci. Bull., 2015, 60, 1316 CrossRef CAS.
- R. H. Crabtree, J. M. Mihelcic and J. M. Quirk, J. Am. Chem. Soc., 1979, 101, 7738 CrossRef CAS.
- (a) M. J. Burk, R. H. Crabtree, C. P. Parnell and R. J. Uriarte, Organometallics, 1984, 3, 816 CrossRef CAS; (b) R. H. C., M. J. Burk and D. V. McGrath, J. Chem. Soc., Chem. Commun., 1985, 1829 Search PubMed.
- (a) D. Baudry, M. Ephritikhine, H. Felkin and R. Holmes-Smith, J. Chem. Soc., Chem. Commun., 1983, 788 RSC; (b) H. Felkin, T. Fillebeen-Khan, Y. Gault, R. Holmes-Smith and J. Zakrzewski, Tetrahedron Lett., 1984, 25, 1279 CrossRef CAS.
- M. Gupta, C. Hagen, R. J. Flesher, W. C. Kaska and C. M. Jensen, Chem. Commun., 1996, 2083 RSC.
- W.-w. Xu, G. P. Rosini, K. Krogh-Jespersen, A. S. Goldman, M. Gupta, C. M. Jensen and W. C. Kaska, Chem. Commun., 1997, 2273 RSC.
- F. Liu and A. S. Goldman, Chem. Commun., 1999, 655 RSC.
- F. Liu, E. B. Pak, B. Singh, C. M. Jensen and A. S. Goldman, J. Am. Chem. Soc., 1999, 121, 4086 CrossRef CAS.
- (a) S. Kundu, Y. Choliy, G. Zhuo, R. Ahuja, T. J. Emge, R. Warmuth, M. Brookhart, K. Krogh-Jespersen and A. S. Goldman, Organometallics, 2009, 28, 5432 CrossRef CAS; (b) B. Punji, T. J. Emge and A. S. Goldman, Organometallics, 2010, 29, 2702 CrossRef CAS; (c) K. Krogh-Jespersen, M. Czerw, K. Zhu, B. Singh, M. Kanzelberger, N. Darji, P. D. Achord, K. B. Renkema and A. S. Goldman, J. Am. Chem. Soc., 2002, 124, 10797 CrossRef CAS PubMed; (d) K. Zhu, P. D. Achord, X. Zhang, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2004, 126, 13044 CrossRef CAS PubMed; (e) J. J. Adams, N. Arulsamy and D. M. Roddick, Organometallics, 2012, 31, 1439 CrossRef CAS.
- (a) I. Göttker-Schnetmann and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 9330 CrossRef PubMed; (b) I. Göttker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804 CrossRef PubMed; (c) Z. Huang, M. Broohart, A. S. Goldman, S. Kundu, A. Ray, S. L. Scott and B. C. Vicente, Adv. Synth. Catal., 2009, 351, 188 CrossRef CAS.
- W. Yao, Y. Zhang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 1390 CrossRef CAS PubMed.
- (a) M. W. Haenel, S. Oevers, K. Angermund, W. C. Kaska, H.-J. Fan and M. B. Hall, Angew. Chem., Int. Ed., 2001, 40, 3596 CrossRef CAS; (b) S. A. Kuklin, A. M. Sheloumov, F. M. Dolgushin, M. G. Ezernitskaya, A. S. Peregudov, P. V. Petrovskii and A. A. Koridze, Organometallics, 2006, 25, 5466 CrossRef CAS; (c) Y. Shi, T. Suguri, C. Dohi, H. Yamada, S. Kojima and Y. Yamamoto, Chem.–Eur. J., 2013, 19, 10672 CrossRef CAS PubMed; (d) D. Bézier and M. Brookhart, ACS Catal., 2014, 4, 3411 CrossRef.
- (a) M. Raynal, R. Pattacini, C. S. J. Cazin, C. Vallée, H. Olivier-Bourbigou and P. Braunstein, Organometallics, 2009, 28, 4028 CrossRef CAS; (b) A. R. Chianese, M. J. Drance, K. H. Jensen, S. P. McCollom, N. Yusufova, S. E. Shaner, D. Y. Shopov and J. A. Tendler, Organometallics, 2014, 33, 457 CrossRef CAS; (c) K. Tanoue and M. Yamashita, Organometallics, 2015, 34, 4011 CrossRef CAS; (d) X. Jia, L. Zhang, C. Qin, X. Leng and Z. Huang, Chem. Commun., 2014, 50, 11056 RSC; (e) D. F. Brayton, P. R. Beaumont, E. Y. Fukushima, H. T. Sartain, D. Morales-Morales and C. M. Jensen, Organometallics, 2014, 33, 5198 CrossRef CAS.
- D. Bezier, C. Guan, K. Krogh-Jespersen, A. S. Goldman and M. Brookhart, Chem. Sci., 2016, 7, 2579 RSC.
- C. Six, B. Gabor, H. Görls, R. Mynott, P. Philipps and W. Leitner, Organometallics, 1999, 18, 3316 CrossRef CAS.
- (a) B. C. Gruver, J. J. Adams, S. J. Warner, N. Arulsamy and D. M. Roddick, Organometallics, 2011, 30, 5133 CrossRef CAS; (b) J. J. Adams, B. C. Gruver, R. Donohoue, N. Arulsamy and D. M. Roddick, Dalton Trans., 2012, 41, 12601 RSC.
- B. C. Gruver, J. J. Adams, N. Arulsamy and D. M. Roddick, Organometallics, 2013, 32, 6468 CrossRef CAS.
- Y. Zhang, H. Fang, W. Yao, X. Leng and Z. Huang, Organometallics, 2016, 35, 181 CrossRef CAS.
- E. Khaskin, D. L. Lew, S. Pal and A. N. Vedernikov, Chem. Commun., 2009, 6270 RSC.
- D. P. Solowey, M. V. Mane, T. Kurogi, P. J. Carroll, B. C. Manor, M.-H. Baik and D. J. Mindiola, Nat. Chem., 2017, 9, 1126 CrossRef PubMed.
- R. Ahuja, B. Punji, M. Findlater, C. Supplee, W. Schinski, M. Brookhart and A. S. Goldman, Nat. Chem., 2011, 3, 167 CrossRef CAS PubMed.
- S. Kundu, T. W. Lyons and M. Brookhart, ACS Catal., 2013, 3, 1768 CrossRef CAS.
- A. Kumar, T. Zhou, T. J. Emge, O. Mironov, R. J. Saxton, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2015, 137, 9894 CrossRef CAS PubMed.
- M. J. Burk, R. H. Crabtree and D. V. McGrath, J. Chem. Soc., Chem. Commun., 1985, 1829 RSC.
- K. Nomura and Y. Saito, J. Chem. Soc., Chem. Commun., 1988, 161 RSC.
- J. A. Maguire, W. T. Boese and A. S. Goldman, J. Am. Chem. Soc., 1989, 111, 7088 CrossRef CAS.
- A. D. Chowdhury, N. Weding, J. Julis, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 6477 CrossRef PubMed.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257 CrossRef CAS PubMed.
- Z. Huang, E. Rolfe, E. C. Carson, M. Brookhart, A. S. Goldman, S. H. El-Khalafy and A. H. R. MacArthur, Adv. Synth. Catal., 2010, 352, 125 CrossRef CAS.
- (a) D. C. Leitch, Y. C. Lam, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2013, 135, 10302 CrossRef CAS PubMed; (b) D. C. Leitch, J. A. Labinger and J. E. Bercaw, Organometallics, 2014, 33, 3353 CrossRef CAS.
- T. W. Lyons, D. Guironnet, M. Findlater and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 15708 CrossRef CAS PubMed.
- G. E. Dobereiner, J. Yuan, R. R. Schrock, A. S. Goldman and J. D. Hackenberg, J. Am. Chem. Soc., 2013, 135, 12572 CrossRef CAS PubMed.
- X. Jia and Z. Huang, Nat. Chem., 2016, 8, 157 CrossRef CAS PubMed.
- A. D. Sadow and T. D. Tilley, Angew. Chem., Int. Ed., 2003, 42, 803 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |