
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBrominated graphene as a versatile precursor for multifunctional grafting†
Heather
Au
 ,
Noelia
Rubio
and
Milo S. P.
Shaffer
*
,
Noelia
Rubio
and
Milo S. P.
Shaffer
*
Departments of Chemistry & Materials, Imperial College London, London, SW7 2AZ, UK. E-mail: m.shaffer@imperial.ac.uk
First published on 29th September 2017
Abstract
A non-destructive and versatile chemical reduction method was used to dissolve and subsequently brominate few-layer graphene sheets (FLGs); the direct covalent attachment of bromine to the graphene framework was demonstrated by X-ray photoelectron spectroscopy (XPS). The brominated few-layer graphenes (FLG-Br) provide a convenient, stable, liquid-phase precursor, suitable for the synthesis of a variety of directly functionalised graphenes. As an example, the FLG-Br species was used to initiate atom transfer radical polymerisation (ATRP), to obtain poly(methyl methacrylate) (PMMA)-grafted graphene (FLG-PMMA), which was six times more dispersible in acetone than controls. In addition, the FLG-Br is active for nucleophilic substitution reactions, as illustrated by the preparation of methoxypolyethylene glycol (mPEG)- and OH-substituted derivatives. The products were characterised by thermogravimetric analysis coupled with mass spectrometry (TGA-MS), XPS and Raman spectroscopy. Grafting ratios (GR) for these polymer-grafted materials varied between 6 and 25%; even at these GRs, all graphene derivatives showed increased solubility in organic solvents.
Introduction
Graphene's exceptional intrinsic properties have been widely exploited in applications ranging from nanocomposites1,2 and conductive inks3 to drug delivery4 and sensors.5 However, pristine graphene itself has a low solubility in common solvents,6 lacking in surface functional groups which enable dispersion in many bulk applications. Increased dispersibility in organic solvents or polymer matrices may be achieved by functionalisation with compatibilising groups, and can enable the processing of, for example, coatings, membranes, and electrodes.7 In application, modified surface chemistry can enhance wettability by the desired phases8 or introduce specific functions, such as fluorescence, selective binding sites, or stable doping species.9,10 Covalent modification has previously been achieved by a wide range of chemistries,9,11–13 producing various modified graphenes including alkylated,14 arylated,15–17 amine-,18 hydroxyl- and epoxide-,19 polymer-20,21 and halogen-grafted22 derivatives.Halogenation of carbon nanomaterials (CNMs)22–24 is a useful route to obtain highly reactive species which may be used in further reactions. Covalently-bound bromine is particularly attractive as a synthetic handle, although intercalation or adsorption of molecular bromine is common.25 Bromine has been introduced onto various CNMs by several methods, including exposure to bromine vapour,26 long sonication27 or microwave irradiation22 in bromine liquid, reaction with dibromocarbene,28 and photocatalytic addition using N-bromosuccinimide as a bromine precursor.29 However, direct covalent attachment of bromine to graphene is rare and usually achieved by harsh methods such as plasma30 or microwave22 treatment, or refluxing in bromine,31 and/or on poorly exfoliated materials. Nevertheless, bromine-activated carbon species can be used in subsequent nucleophilic substitution reactions, as reported for brominated highly-oriented pyrolytic graphite (HOPG) and multi-walled carbon nanotubes (MWCNTs); for example, following plasma-chemical bromination, the C–Br bonds underwent hydrolysis, or further grafting with 1,6-diaminohexane and (3-aminopropyl)triethoxysilane.30 Bromination of single-walled carbon nanotubes (SWCNTs) by reductive functionalisation also showed that nucleophilic substitution of sidewall bromine addends was possible, introducing 2,2,2-trifluoroethanolate and hydroxyl groups at the brominated sites.32
In general, reductive functionalisation is a versatile route to obtaining CNMs grafted with a wide variety of moieties.33–35 For graphene, in particular, charging the carbon framework can be achieved by vapour transport,36,37 direct metal (alloy) contact,38 or through the use of charge transfer agents39 to form alkali-metal graphite intercalation compound (GIC) precursors. Under appropriate conditions, in dry aprotic solvents, individual anionic ‘graphenide’ sheets can be dissolved and processed. Reaction with a wide variety of different electrophiles yields individualised, covalently-grafted graphene derivatives. The connectivity of the carbon lattice is retained, thereby reducing the detrimental effects that covalent grafting and exfoliation can have on graphene's intrinsic mechanical and electrical properties. This method has resulted in successful grafting of alkyl,14,39,40 aryl,41 and polyethylene glycol (PEG)42 addends, as well as hydrogenation;43 however, bromination of graphene is not yet reported by this route.
Polymer grafting can greatly improve the solubility of CNMs, additionally limiting their restacking and agglomeration on drying.44 Using polymers with defined molecular weight and an activated end group, in a ‘grafting-to’ approach, allows control of polymer properties;45 however, the large steric bulk of the chains tends to limit grafting density. Conversely, surface-initiated anionic polymerisation can achieve densely-grown polymers,46 but requires rigorously dry reaction conditions to allow full control over molecular weight and dispersity. Furthermore, the choice of monomers is limited to only those which are stable under reducing conditions. Alternative grafting-from approaches such as atom transfer radical polymerisation (ATRP) afford more control over polymer growth,47 whilst retaining a high graft density. Previous examples of ATRP from nanomaterial surfaces include the polymerisation of styrene, methyl methacrylate (MMA) and glycidyl methacrylate from graphene oxide (GO), CNTs and boron nitride nanotubes (BNNTs).8,48–52 In these procedures, bromine-containing initiator molecules are grafted first (usually via multi-step reactions), rather than directly attaching an active bromine atom to the surface.
The aim of this work was to prepare directly brominated graphene by reductive chemistry as an air-stable precursor for a variety of further transformations. As an illustration, brominated few-layer graphene (FLG-Br) was used to obtain poly(methyl methacrylate) (PMMA)-grafted graphene (FLG-PMMA), via ATRP; in this approach, the grafting reaction is initiated directly from the graphene framework, without an intervening molecular linker. In addition, the potential for nucleophilic substitution was explored using OH-substituted reagents poly(ethylene glycol)methyl ether (mPEG) and water. In comparison with direct reactions with graphenide, both methods offer greater versatility, reflected in their accessibility to a wide range of potential functionalities, including those unstable to reducing conditions. This approach provides an easy means to access modified few-layer graphene (FLG) with improved and tunable dispersibility, critical in bulk applications requiring compatibility with common organic solvents and polymer matrices.
Experimental
Materials
FLG was obtained from Cambridge Nanosystems and used without any further purification. Tetrahydrofuran (THF), dried in-house in a solvent-drying tower packed with alumina, was degassed via a freeze–pump–thaw method then further dried over 20 vol% 4 Å activated molecular sieves. Naphthalene (99%, Sigma-Aldrich) was dried under vacuum in the presence of phosphorus pentoxide before use. Sodium (99.95%, ingot) and bromine (reagent grade) were purchased from Sigma-Aldrich and used as received. MMA (99%, Sigma-Aldrich) was passed through an alumina column to remove stabilisers, before degassing via freeze–pump–thaw and drying over 20 vol% 4 Å activated molecular sieves. Acetone (≥99.9%, Sigma-Aldrich) was distilled over CaH2 and stored under nitrogen. Immediately before use, both monomer and solvent were purged with nitrogen for 30 min. Copper(I) bromide (CuBr, 98%, Sigma-Aldrich) was purified by washing with glacial acetic acid, followed by 2-propanol, and then dried under vacuum.47N,N,N′,N′′,N′′-Pentamethyldiethylenetriamine (PMDETA, 99%) and copper(II) bromide (CuBr2, 99%) were obtained from Sigma-Aldrich and used as received. mPEG (MW 2000 Da, Sigma Aldrich) was dried under vacuum with phosphorus pentoxide prior to use. Dichloromethane (VWR) was used as received.Procedures
Equipment and characterisation
Thermogravimetric analysis coupled with mass spectrometry (TGA-MS) was performed using a Mettler Toledo TGA/DSC 1 instrument integrated with a Hiden HPR-20 QIC EGA mass spectrometer under nitrogen atmosphere. Samples were held at 100 °C for 30 min, then heated from 100 °C to 850 °C at 10 °C min−1 (N2 flow rate = 60 mL min−1). X-ray photoelectron spectroscopy (XPS) data were recorded using a K-alpha+ XPS spectrometer equipped with an MXR3 Al Kα monochromated X-ray source (hν = 1486.6 eV). X-ray gun power was set to 72 W (6 mA and 12 kV). Charge compensation was achieved with the FG03 flood gun using a combination of low energy electrons and the ion flood source. Survey scans were acquired using 200 eV pass energy, 1 eV step size and 100 ms (50 ms × 2 scans) dwell times. All high resolution spectra were acquired using 20 eV pass energy, 0.1 eV step size and 1 s (50 ms × 20 scans) dwell times. Samples were prepared by pressing the sample onto carbon-based double-sided tape. Pressure during measurement acquisition was ≤1 × 10−8 mbar. Atomic compositions were calculated from averaged spectra obtained from at least 3 areas per sample. Raman spectra were collected on a Renishaw inVia micro-Raman (1000–3000 cm−1), using a 50 mW 532 nm laser at 10% laser power. Statistical Raman data were obtained from measurements carried out in Streamline mode of at least 500 areas per sample. Samples were prepared by drop casting dispersions on a glass slide or silicon wafer. UV-Vis-NIR absorption spectra were measured with a Perkin Elmer Lambda 950 UV/Vis spectrophotometer, using a quartz cuvette with 1 cm pathlength. Ultrasonication was performed using an ultrasonic cleaner (USC300T, 80 W). Transmission electron microscopy (TEM) was carried out using a JEOL 2100Plus TEM at 200 kV operating voltage. Samples were prepared on 300 copper mesh holey carbon grids (Agar Scientific) by drop-casting dilute graphene dispersions onto a grid supported by filter paper and drying under vacuum.Results and discussion
Synthesis of brominated graphene
Following a previously established functionalisation sequence,39 reduced FLG was prepared by treatment with sodium naphthalide in THF, with a charging ratio (number of framework carbon atoms per sodium atom, C/Na) of 12, and an absolute sodium concentration of 0.01 M. This reductive treatment generates a black, graphenide-containing dispersion, via electrostatically-driven exfoliation. The resulting charged graphene sheets were reacted with bromine liquid to yield brominated graphene (Scheme 1). Stable bromine-GICs are well-known in the literature, where bromine molecules adsorb within the interlayer galleries;25,53–55 reductive activation of FLG to form a reactive intermediate is therefore required for covalent functionalisation to take place.32 The FLG starting material is already reasonably exfoliated (<5 layers), with a crumpled morphology, and small lateral flake size of 0.25–1 μm (Fig. S1†). The flake surfaces and edges are accessible for functionalisation, whilst non-covalently adsorbed bromine is less likely to remain trapped between graphene layers after processing; compared to larger, flatter sheets, quantification of grafting is more straightforward.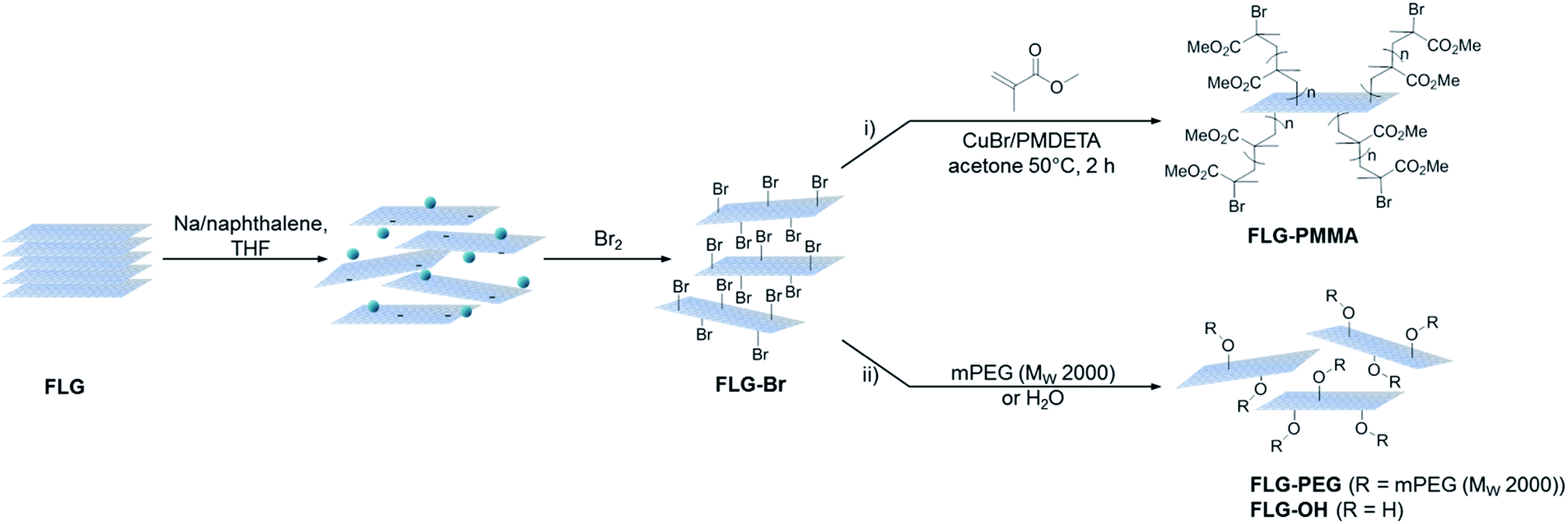 | ||
| Scheme 1 Bromine functionalisation of FLG to FLG-Br via reduction with sodium naphthalide in THF, followed by (i) synthesis of FLG-PMMA via ATRP; (ii) nucleophilic substitution of FLG-Br with mPEG or water, yielding FLG-PEG and FLG-OH, respectively. | ||
TGA-MS under nitrogen indicates successful covalent functionalisation of the FLG starting material (Fig. 1a). The weight loss in the region between 200 and 450 °C can be assigned to the detachment of bromine addends29,32 from mass fragments m/z 79, 81, which show a characteristic isotopic distribution of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Bromine is known to intercalate graphite to form stable Br2-GICs, but desorption occurs at relatively low temperatures (80–180 °C).56 A reference experiment mixing bromine with uncharged FLG (FLG + Br2) showed that bromine was fully removed by the same washing procedure as used for the functionalised product, suggesting that bromine species detected are covalently-bound (Fig. S2a†). In FLG-Br, MS fragments attributed to residual THF were detected, due to some solvent trapped within the sample (Fig. S2b†); it is therefore difficult to quantify the degree of grafting definitively from TGA alone. However, from the mass loss during pyrolysis (8.4 wt%), an upper bound for the grafting ratio is 9.3 wt%, corresponding to a C/Br of 71 (Table 1).
:1. Bromine is known to intercalate graphite to form stable Br2-GICs, but desorption occurs at relatively low temperatures (80–180 °C).56 A reference experiment mixing bromine with uncharged FLG (FLG + Br2) showed that bromine was fully removed by the same washing procedure as used for the functionalised product, suggesting that bromine species detected are covalently-bound (Fig. S2a†). In FLG-Br, MS fragments attributed to residual THF were detected, due to some solvent trapped within the sample (Fig. S2b†); it is therefore difficult to quantify the degree of grafting definitively from TGA alone. However, from the mass loss during pyrolysis (8.4 wt%), an upper bound for the grafting ratio is 9.3 wt%, corresponding to a C/Br of 71 (Table 1).
 | ||
| Fig. 1 (a) TGA-MS profile of FLG (black) and FLG-Br (orange), m/z 79, 81 –Br+; (b) wide survey XPS spectrum of FLG-Br; (c) core level Br 3d spectrum for FLG; (d) core level C 1s spectrum for FLG; (e) core level Br 3d spectrum for FLG-Br, dashed lines denote 3d3/2, solid 3d5/2; (f) core level C 1s spectrum for FLG-Br. | ||
Further evidence for covalent functionalisation was obtained by XPS. In the wide survey XPS spectrum of FLG-Br (Fig. 1b), the major peak at 284.6 eV arises from C 1s of sp2 hybridised carbon. Additional peaks corresponding to Br 3s, 3p1/2/3p3/2 and 3d appear at 257.1, 190.1/184.1 and 70.2 eV, respectively. In the high resolution bromine spectrum (Fig. 1e), the deconvoluted Br 3d peaks at 70.1 and 71.1 eV are characteristic of covalently-bound Br,26 although a small fraction (∼12%) of adsorbed bromine or residual sodium bromide was detected, around 68.5 eV,22,57–59 consistent with these species being intercalated between graphene layers. A corresponding C–Br peak at 285.3 eV can be identified in the deconvoluted C 1s core-level spectrum of FLG-Br,28 but is absent in FLG, confirming that Br atoms have been covalently attached to the graphene sheets (Fig. 1d and f). Additional peaks at 286.1 and 286.9 eV are assigned to C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively, also present in the starting material. No Br 3d peak was detected in the as-received sample (Fig. 1c) or control (Fig. S3†). The XPS data therefore present strong evidence for covalent Br attachment, consistent with reports in the literature concerning bromine functionalisation of graphite by microwave irradiation15 and of various CNMs by plasma-chemical treatment.22,28,30 Analysis of the higher resolution data indicates a composition of 0.9 at% covalent Br to 95.6 at% C (and 3.4 at% O) after functionalisation, giving a C/Br of 108 and grafting ratio of 6.2 wt% (Table 1). The relative amount of oxygen and carbon does not vary substantially compared to FLG starting material (96.5 at% C, 3.5 at% O) suggesting that the THF content is insignificant. The GRs by TGA and XPS are in good agreement, given the differences in surface sensitivity, and are comparable to the degree of grafting achieved for alkylated and arylated graphenes obtained following similar reductive treatment.14,39,41 The number density (number of grafted sites per unit area) of bromine calculated from XPS data (3.5 × 1013 cm−2), therefore, also agrees well with that obtained from TGA (5.4 × 1013 cm−2) (see ESI for calculations†). This degree of functionalisation remains below the critical threshold for preserving the band structure of graphene,60 in contrast to harsher methods of bromination, which achieve stoichiometries of up to 40% Br/C but degrade the connectivity of the graphitic network, reflected in the significant broadening and weakening of characteristic Raman signals, and greatly decreased thermal stability.31
O, respectively, also present in the starting material. No Br 3d peak was detected in the as-received sample (Fig. 1c) or control (Fig. S3†). The XPS data therefore present strong evidence for covalent Br attachment, consistent with reports in the literature concerning bromine functionalisation of graphite by microwave irradiation15 and of various CNMs by plasma-chemical treatment.22,28,30 Analysis of the higher resolution data indicates a composition of 0.9 at% covalent Br to 95.6 at% C (and 3.4 at% O) after functionalisation, giving a C/Br of 108 and grafting ratio of 6.2 wt% (Table 1). The relative amount of oxygen and carbon does not vary substantially compared to FLG starting material (96.5 at% C, 3.5 at% O) suggesting that the THF content is insignificant. The GRs by TGA and XPS are in good agreement, given the differences in surface sensitivity, and are comparable to the degree of grafting achieved for alkylated and arylated graphenes obtained following similar reductive treatment.14,39,41 The number density (number of grafted sites per unit area) of bromine calculated from XPS data (3.5 × 1013 cm−2), therefore, also agrees well with that obtained from TGA (5.4 × 1013 cm−2) (see ESI for calculations†). This degree of functionalisation remains below the critical threshold for preserving the band structure of graphene,60 in contrast to harsher methods of bromination, which achieve stoichiometries of up to 40% Br/C but degrade the connectivity of the graphitic network, reflected in the significant broadening and weakening of characteristic Raman signals, and greatly decreased thermal stability.31
Raman spectroscopy is commonly used to provide semi-quantitative analysis of the degree of grafting in carbon nanomaterials. In heterogeneous bulk samples, Raman mapping is necessary to obtain statistically relevant conclusions regarding the degree and distribution of functionalisation. Graphitic materials typically exhibit three characteristic bands: D band at ∼1350 cm−1, which is defect activated; G band at ∼1580 cm−1, related to the in-plane graphitic bonding; and 2D band at ∼2700 cm−1, from which information about stacking and layers can be determined.61 In general, the relative intensities of the defect and graphitic bands (ID/IG) are used to estimate the number of grafted sites. Statistical Raman analysis of the bulk sample shows an ID/IG ratio of 0.40 ± 0.03 in the FLG starting material (Fig. 2b); the large D peak arises from edge scattering,62 because many flakes are smaller than the laser spot size. For charged, unfunctionalised FLG directly quenched with dry oxygen/nitrogen, and for FLG + Br2 control, the ID/IG ratios (0.43 ± 0.05 and 0.45 ± 0.09, respectively) remain essentially unchanged, confirming that no sp3 defects have been introduced (Fig. S4†). In contrast, ID/IG increases to 0.58 ± 0.02 in FLG-Br (Fig. 2b orange) suggesting that bulk covalent functionalisation has indeed occurred.39,63 However, at the high grafting densities implied by the TGA and XPS data,64,65 the ID/IG trend might be expected to reverse, and the peaks broaden, since the defect spacing should be below the Tuinstra–Koenig limit,66,67 if uniformly distributed. Since, in fact, the spectra for FLG-Br show very well-defined peaks with narrow full width at half maximum (FWHM, Γ, Fig. 2a and Table S1†), it is likely that grafting occurs in clusters, possibly nucleating from existing defects or edges, as has been proposed previously for Birch-type reductions of SWCNTs.68 Additional analysis of the D′ band at ∼1615 cm−1 shows an increase in ID/ID′ after functionalisation (Fig. S5†) which is consistent with an increase in the ratio of sp3 defects to edge-type defects,69 and therefore indicates that covalent grafting occurs, with retention of lattice connectivity. The width of the ID/IG ratio distribution remains essentially unchanged (Fig. 2b), suggesting that all the flakes in the sample react equally. The 2D peaks indicate the extent of exfoliation and are discussed below.
 | ||
| Fig. 2 (a) Averaged Raman spectra, and (b) Raman histograms of ID/IG ratio, of FLG (black), FLG-Br (orange), FLG-PMMA (red) and FLG-PEG (blue); λexc = 532 nm, spectra normalised to the G peak intensity and offset for clarity. | ||
FLG-Br as a precursor to polymer-grafted graphene
Polymer grafting can improve the dispersibility of graphene in common organic solvents or matrices,70 and potentially enhance stress-transfer in nanocomposites.71 In particular, PMMA has been used as a compatibiliser for various fillers in epoxy resins, such as glass fibre72 and nanoclays,73 in addition, PMMA is a well-known ATRP target. FLG-Br was therefore used as a precursor to obtain PMMA-grafted graphene as both a model system and for practical purposes. Using FLG-Br as the initiator molecule and following standard ATRP procedures in the literature, FLG-PMMA was obtained from the polymerisation of MMA74 (Scheme 1). Briefly, under inert conditions, distilled MMA was stirred with CuBr, CuBr2, PMDETA, and FLG-Br in acetone, at 50 °C for 2 h. FLG-PMMA was obtained as a black powder after washing with THF and drying under vacuum.TGA-MS analysis of the product shows an increased mass loss (16.2 wt%), and correspondingly larger grafting ratio (19.5 wt%), as compared to FLG-Br, in the range 200 to 550 °C (Fig. 3a); the accompanying mass fragments correspond to PMMA polymer, including m/z 41 (–CH2C(CH3)–+), 59 (–COOCH3+), 69 (–CH2C(CH3)(CO)–+), and 100 (–CH2C(CH3)(COOCH3)–+). This weight loss temperature is also consistent with the expected thermal degradation of PMMA.8 XPS data for FLG-PMMA show an increase in oxygen content (Fig. S6†) with a composition of 92.6 at% C, 6.9 at% O and 0.5 at% Br (Table 1); assuming, therefore, that approximately half the bromine addends served as initiators in the ATRP reaction, the polymer chains are estimated to have an average molecular weight in the range 370 to 590 Da, based on the C/Br limits obtained from TGA and XPS (see ESI for calculations†). The carbon and oxygen contents from XPS are in good agreement with calculated compositional values based on C/PMMA = 250, Mn ∼ 590; the estimated grafting ratio from XPS is 21.4 wt%, close to the value obtained by TGA. TGA-MS shows only a trace amount of THF in FLG-PMMA; the compositional values from XPS also suggest that no THF remains. Introduction of polymer on the graphene surface likely preserves an open pathway between layers, allowing evaporation of solvent.
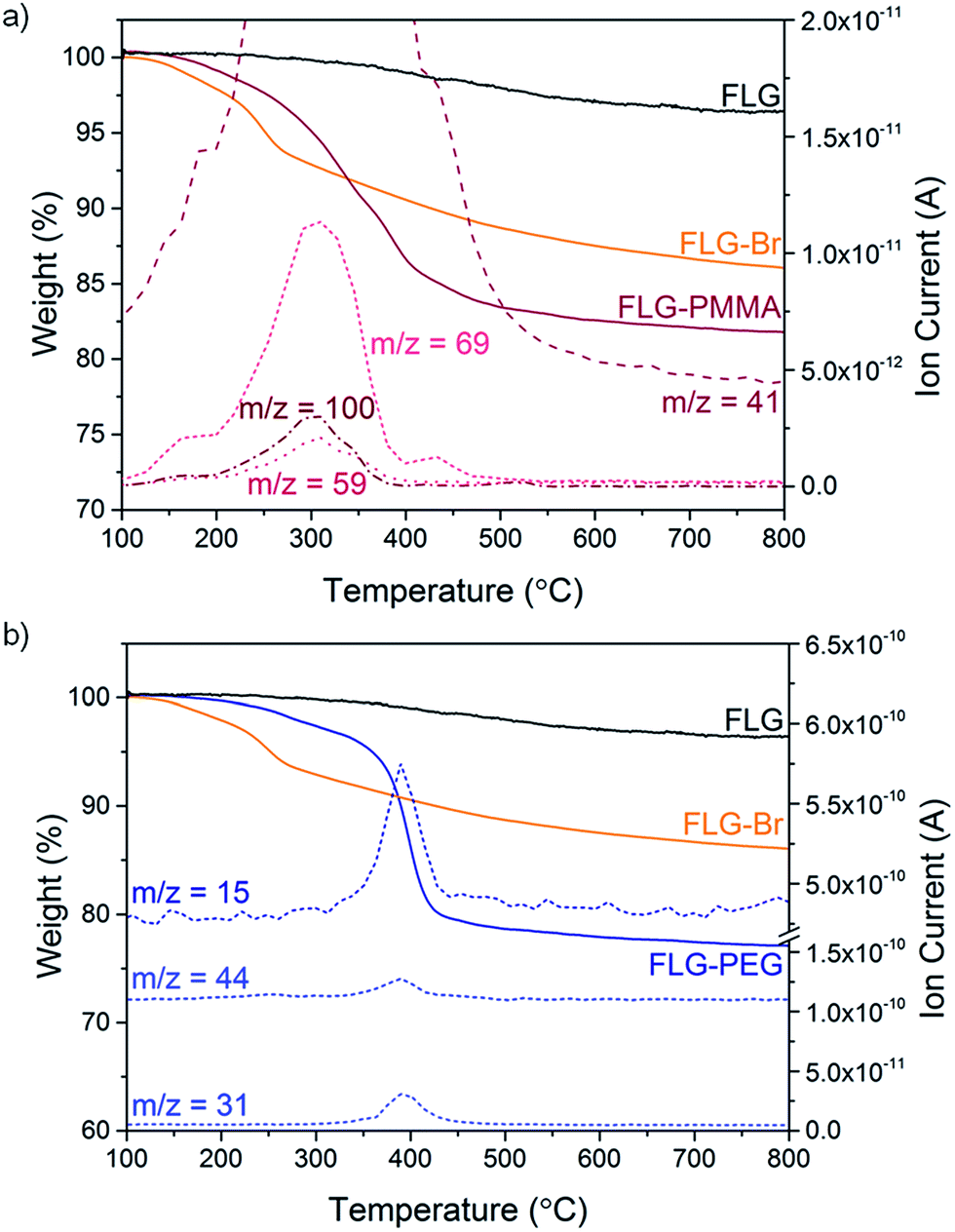 | ||
| Fig. 3 (a) TGA-MS profile of FLG-PMMA, m/z 41 (–CH2C(CH3)–+), 59 (–COOCH3+), 69 (–CH2C(CH3)(CO)–+), 100 (–CH2C(CH3)(COOCH3)–+); (b) TGA-MS profile of FLG-PEG, m/z 15 (–CH3+), 31 (–OCH3+), 44 (–CH2CH2O–+). | ||
Raman analysis of FLG-PMMA product shows that the initial density of defects in FLG-Br is hardly changed (ID/IG 0.58 ± 0.02 to 0.53 ± 0.03) after PMMA growth from the surface (Fig. 2 orange and red); therefore, no additional sp3 centres were introduced during this part of the reaction sequence. A reference experiment (FLG + MMA control) using FLG as the ATRP initiator, rather than FLG-Br, was carried out under the same conditions; the TGA-MS and Raman data (Fig. S7†) show that the reaction does not take place on the original FLG surface, demonstrating that brominated sites are necessary for polymerisation initiation.
An alternative route to polymer-grafted graphene, and further proof of the versatility of bromine functionalisation, is provided by nucleophilic substitution of the bromine addends with polyethylene glycol (Scheme 1). To obtain the PEG-substituted product, FLG-Br was dispersed in dry THF and dichloromethane by brief and mild bath sonication. After addition of mPEG (MW 2000 Da), the suspension was stirred at 40 °C for 2.5 h, and FLG-PEG was obtained after filtering and washing with THF, water and ethanol, and drying under vacuum. The weight loss during TGA pyrolysis of FLG-PEG in the range 300–500 °C is accompanied by mass fragments which can be attributed to mPEG (m/z 15 (–CH3+), 31 (–OCH3+), 44 (–CH2CH2O–+)), confirming successful introduction of PEG on the graphene surface (Fig. 3b). The mass loss of 18.7 wt% implies an incomplete reaction of ∼1 in 6 bromine addends (based on C/Br from XPS); this partial substitution by mPEG may be explained by the steric bulk of the polymer chain which may prevent substitution of less accessible bromine-grafted sites. A large increase in oxygen content can be seen by XPS (Fig. S6†); C and O atomic percentages obtained by XPS (Table 1) are consistent with the proposed substitution of 1 in 6 addends, although the Br content is lower than expected, possibly due to the layer of long PEG chains on the surface. The grafting ratio from TGA of 23.8 wt% is in good agreement with that implied by the XPS data (26.2 wt%). To explore whether substitution efficiency was affected by nucleophile size, FLG-Br was reacted with water under the same conditions for PEG substitution, to obtain FLG-OH. XPS data show that a greater degree of substitution occurs in this case, with over half (0.6) of the bromine addends replaced with hydroxyl groups (Table 1). The reduced TGA weight loss (6.1 wt%, Fig. S8†) is also consistent with the substitution of this proportion of addends (see ESI for calculations†). Hydroxyl groups and water (m/z 17, 18) are detected by TGA-MS (Fig. S8†), as well as some residual solvent. However, bromine fragments are not seen, possibly because the low concentration cannot be detected by the MS; even in FLG-Br, the bromine m/z fragments give only a very weak signal.
As is the case for FLG-PMMA, in the Raman spectra for the PEG- and OH-substituted products, the ID/IG ratios of 0.56 ± 0.03 and 0.57 ± 0.09 (Fig. 2 blue, and Fig. S9†), respectively, indicate that no further defects are introduced during subsequent reaction, and that substitution occurs exclusively at existing grafted sites. Overall, the TGA, Raman and XPS data indicate that successful growth of PMMA by ATRP, or nucleophilic substitution, illustrated by mPEG and water, can be achieved from brominated FLG, without the creation of further defects.
Exfoliation and dispersion
Analysis of the 2D band in Raman spectra provides valuable information about the exfoliated nature of the different functionalised materials. The shape, intensity (I2D) and full width at half maximum (Γ2D) of the 2D peak can be used to evaluate the number of graphene layers present.61 FLG shows a symmetric 2D band at 2694 cm−1 with a Γ2D of 58 cm−1, characteristic of the loosely-stacked few-layer nature of the starting material. After bromine grafting, the I2D/IG ratio increases only slightly (from 0.97 ± 0.04 to 1.02 ± 0.05), since bromine addends are not large and do not significantly aid further exfoliation (Fig. 4a). Following PMMA functionalisation, this value increases again to 1.06 ± 0.05 suggesting that as ATRP progresses, graphene layers are pushed apart and prevented from restacking and aggregation by grafted polymer chains. An accompanying decrease in Γ2D (Fig. 4b and Table S1†) also implies that the grafted samples are more exfoliated than the starting material.75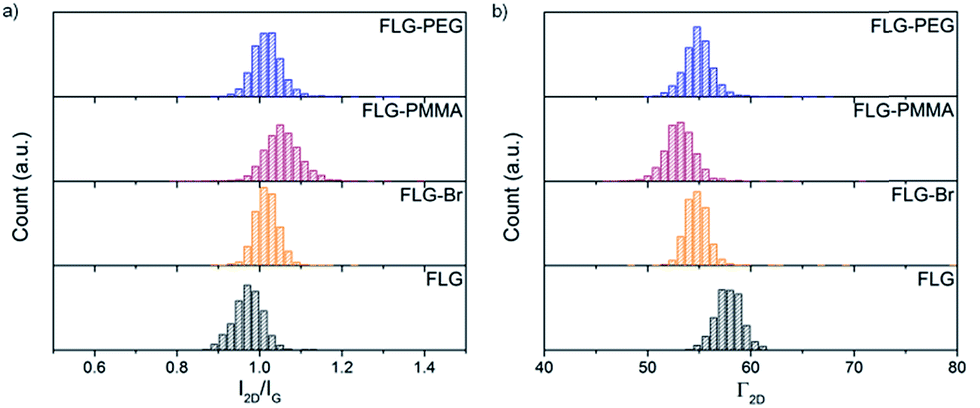 | ||
| Fig. 4 Raman histograms of (a) I2D/IG ratio, and (b) Γ2D, of FLG (black), FLG-Br (orange), FLG-PMMA (red) and FLG-PEG (blue); λexc = 532 nm. | ||
In FLG-PEG, there is no significant change in either I2D/IG or Γ2D (Fig. 4) relative to the brominated FLG; it is likely that in this ‘graft-to’ reaction the large polymer chains cannot diffuse within the interlayer galleries so easily and therefore only substitute bromines grafted on the edges and surfaces of a few-layer stack. As a result, no further exfoliation occurs, and the I2D/IG ratio remains unchanged. There is little change in I2D/IG after –OH substitution; hydroxyl addends are small and do not significantly prevent layer restacking after drying.
The dispersibility of the polymer-grafted graphenes was measured by UV-Vis absorbance (Fig. S10†). The samples were dispersed in different solvents by bath ultrasonication for 15 minutes and allowed to sediment over two days. The extinction coefficient6 for dispersed graphene in solution (α660 = 2460 L g−1 m−1) was used to estimate the concentrations of the different solutions. Covalent grafting significantly improved the dispersibility of the material, with FLG-PMMA reaching a concentration of 200 μg mL−1 in acetone, and FLG-PEG 255 μg mL−1 in ethanol. In the case of FLG-PMMA, even the addition of low molecular weight polymer chains was enough to increase dispersibility six-fold in acetone. FLG-PEG was three and a half times more soluble than FLG in ethanol; despite a low grafting density, the longer mPEG chains could effectively stabilise the graphene sheets. Hydroxyl groups increased the solubility of FLG in water, from 8.51 μg mL−1 to 96.4 μg mL−1 (Table S2†).
Conclusions
Brominated graphene was synthesised by reacting exfoliated Na-based GICs with bromine, as confirmed by TGA-MS, XPS and Raman spectroscopy. This reductive method results only in a low concentration of sp3 defects and the underlying connectivity of the carbon network, and therefore graphene's intrinsic properties,60 are retained. The brominated FLG serves as a convenient and versatile precursor to modified graphenes. The FLG-Br can act as an initiator in the polymerisation of MMA by ATRP, resulting in PMMA-grafted graphene. In contrast to previous examples of ATRP on GO, CNTs and BNNTs,8,48–52 polymerisation is activated directly from the brominated graphene surface, rather than mediated by a specific grafted molecular group, providing an interesting new approach for ATRP reactions on nanocarbon surfaces. FLG-PMMA showed improved dispersibility in acetone, with the addition of only low molecular weight polymers; optimising polymerisation conditions could further increase dispersibility in various solvents. In contrast to direct anionic polymerisation from reduced graphite/graphenides, this methodology offers greater versatility with a wide choice of monomers including those that are unstable to graphenide, for example glycidyl methacrylate or aminoethyl methacrylate. In addition, the bromine addends may easily undergo nucleophilic substitutions, as illustrated by reactions with mPEG and water, without introducing any additional sp3 centres on the carbon framework. In this generalised reaction, brominated graphenes represent versatile, air-stable starting materials for the generation of grafted graphenes with a wide variety of functional moieties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Ignacio Villar-Garcia for his help interpreting and processing XPS data, to Catharina Paukner (FGV Cambridge Nanosystems Limited) for providing the FLG starting material, and thank the EPSRC (EP/G007314/1) for financial support.References
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- W. K. Chee, H. N. Lim, N. M. Huang and I. Harrison, RSC Adv., 2015, 5, 68014–68051 RSC.
- K. Arapov, E. Rubingh, R. Abbel, J. Laven, G. de With and H. Friedrich, Adv. Funct. Mater., 2016, 26, 586–593 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- R. S. Edwards and K. S. Coleman, Nanoscale, 2013, 5, 38–51 RSC.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- X. Cui, C. Zhang, R. Hao and Y. Hou, Nanoscale, 2011, 3, 2118–2126 RSC.
- G. Goncalves, P. A. A. P. Marques, A. Barros-Timmons, I. Bdkin, M. K. Singh, N. Emami and J. Gracio, J. Mater. Chem., 2010, 20, 9927–9934 RSC.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- J. Liu, J. Tang and J. J. Gooding, J. Mater. Chem., 2012, 22, 12435–12452 RSC.
- L. Rodriguez-Perez, M. a. A. Herranz and N. Martin, Chem. Commun., 2013, 49, 3721–3735 RSC.
- S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720–7738 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2013, 42, 3222–3233 RSC.
- J. M. Englert, K. C. Knirsch, C. Dotzer, B. Butz, F. Hauke, E. Spiecker and A. Hirsch, Chem. Commun., 2012, 48, 5025–5027 RSC.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS PubMed.
- E. Bekyarova, M. E. Itkis, P. Ramesh, C. Berger, M. Sprinkle, W. A. de Heer and R. C. Haddon, J. Am. Chem. Soc., 2009, 131, 1336–1337 CrossRef CAS PubMed.
- R. Sharma, J. H. Baik, C. J. Perera and M. S. Strano, Nano Lett., 2010, 10, 398–405 CrossRef CAS PubMed.
- M. Quintana, K. Spyrou, M. Grzelczak, W. R. Browne, P. Rudolf and M. Prato, ACS Nano, 2010, 4, 3527–3533 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- J. Wang, Z. Shi, Y. Ge, Y. Wang, J. Fan and J. Yin, Mater. Chem. Phys., 2012, 136, 43–50 CrossRef CAS.
- M. Castelain, G. Martinez, G. Ellis and H. J. Salavagione, Chem. Commun., 2013, 49, 8967–8969 RSC.
- J. Zheng, H.-T. Liu, B. Wu, C.-A. Di, Y.-L. Guo, T. Wu, G. Yu, Y.-Q. Liu and D.-B. Zhu, Sci. Rep., 2012, 2, 662 CrossRef PubMed.
- R. Stine, W.-K. Lee, K. E. Whitener, J. T. Robinson and P. E. Sheehan, Nano Lett., 2013, 13, 4311–4316 CrossRef CAS PubMed.
- K. S. Coleman, A. K. Chakraborty, S. R. Bailey, J. Sloan and M. Alexander, Chem. Mater., 2007, 19, 1076–1081 CrossRef CAS.
- P. C. Eklund, N. Kambe, G. Dresselhaus and M. S. Dresselhaus, Phys. Rev. B: Solid State, 1978, 18, 7069–7079 CrossRef CAS.
- L. G. Bulusheva, A. V. Okotrub, E. Flahaut, I. P. Asanov, P. N. Gevko, V. O. Koroteev, Y. V. Fedoseeva, A. Yaya and C. P. Ewels, Chem. Mater., 2012, 24, 2708–2715 CrossRef CAS.
- J. Gao, F. Bao, Q. Zhu, Z. Tan, T. Chen, H. Cai, C. Zhao, Q. Cheng, Y. Yang and R. Ma, Polym. Chem., 2013, 4, 1672–1679 RSC.
- T. Sainsbury, M. Passarelli, M. Naftaly, S. Gnaniah, S. J. Spencer and A. J. Pollard, ACS Appl. Mater. Interfaces, 2016, 8, 4870–4877 CAS.
- D. Hines, M. H. Rummeli, D. Adebimpe and D. L. Akins, Chem. Commun., 2014, 50, 11568–11571 RSC.
- J. F. Friedrich, S. Wettmarshausen, S. Hanelt, R. Mach, R. Mix, E. B. Zeynalov and A. Meyer-Plath, Carbon, 2010, 48, 3884–3894 CrossRef CAS.
- O. Jankovsky, P. Simek, K. Klimova, D. Sedmidubsky, S. Matejkova, M. Pumera and Z. Sofer, Nanoscale, 2014, 6, 6065–6074 RSC.
- F. Hof, F. Hauke and A. Hirsch, Chem. Commun., 2014, 50, 6582–6584 RSC.
- D. Voiry, O. Roubeau and A. Penicaud, J. Mater. Chem., 2010, 20, 4385–4391 RSC.
- A. Molina-Ontoria, M. N. Chaur, M. E. Plonska-Brzezinska and L. Echegoyen, Chem. Commun., 2013, 49, 2406–2408 RSC.
- S. Chakraborty, J. Chattopadhyay, W. Guo and W. E. Billups, Angew. Chem., Int. Ed., 2007, 46, 4486–4488 CrossRef CAS PubMed.
- D. E. Nixon and G. S. Parry, J. Phys. D: Appl. Phys., 1968, 1, 291 CAS.
- Y. Wang, P. Puech, I. Gerber and A. Pénicaud, J. Raman Spectrosc., 2014, 45, 219–223 CrossRef CAS.
- M. Zanini, S. Basu and J. E. Fischer, Carbon, 1978, 16, 211–212 CrossRef CAS.
- T. Morishita, A. J. Clancy and M. S. P. Shaffer, J. Mater. Chem. A, 2014, 2, 15022–15028 CAS.
- J. Chattopadhyay, A. Mukherjee, S. Chakraborty, J. Kang, P. J. Loos, K. F. Kelly, H. K. Schmidt and W. E. Billups, Carbon, 2009, 47, 2945–2949 CrossRef CAS.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H.-P. Steinrück, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279–286 CrossRef CAS PubMed.
- H. S. Leese, L. Govada, E. Saridakis, S. Khurshid, R. Menzel, T. Morishita, A. J. Clancy, E. R. White, N. E. Chayen and M. S. P. Shaffer, Chem. Sci., 2016, 7, 2916–2923 RSC.
- R. A. Schäfer, J. M. Englert, P. Wehrfritz, W. Bauer, F. Hauke, T. Seyller and A. Hirsch, Angew. Chem., Int. Ed., 2013, 52, 754–757 CrossRef PubMed.
- N. Rubio, H. Au, H. S. Leese, S. Hu, A. J. Clancy and M. S. P. Shaffer, Macromolecules, 2017, 50, 7070–7079 CrossRef CAS.
- Y.-S. Ye, Y.-N. Chen, J.-S. Wang, J. Rick, Y.-J. Huang, F.-C. Chang and B.-J. Hwang, Chem. Mater., 2012, 24, 2987–2997 CrossRef CAS.
- F. Liang, J. M. Beach, K. Kobashi, A. K. Sadana, Y. I. Vega-Cantu, J. M. Tour and W. E. Billups, Chem. Mater., 2006, 18, 4764–4767 CrossRef CAS.
- T. Sarbu, K.-Y. Lin, J. Ell, D. J. Siegwart, J. Spanswick and K. Matyjaszewski, Macromolecules, 2004, 37, 3120–3127 CrossRef CAS.
- H. Roghani-Mamaqani, RSC Adv., 2015, 5, 53357–53368 RSC.
- M. Ejaz, S. C. Rai, K. Wang, K. Zhang, W. Zhou and S. M. Grayson, J. Mater. Chem. C, 2014, 2, 4073–4079 RSC.
- S. H. Lee, D. R. Dreyer, J. An, A. Velamakanni, R. D. Piner, S. Park, Y. Zhu, S. O. Kim, C. W. Bielawski and R. S. Ruoff, Macromol. Rapid Commun., 2010, 31, 281–288 CrossRef CAS PubMed.
- H. Kong, C. Gao and D. Yan, J. Am. Chem. Soc., 2004, 126, 412–413 CrossRef CAS PubMed.
- D. Baskaran, J. W. Mays and M. S. Bratcher, Angew. Chem., Int. Ed., 2004, 43, 2138–2142 CrossRef CAS PubMed.
- E. Widenkvist, D. W. Boukhvalov, S. Rubino, S. Akhtar, J. Lu, R. A. Quinlan, M. I. Katsnelson, K. Leifer, H. Grennberg and U. Jansson, J. Phys. D: Appl. Phys., 2009, 42, 112003 CrossRef.
- D. A. Young, Carbon, 1977, 15, 373–377 CrossRef CAS.
- G. A. Saunders, A. R. Ubbelohde and D. A. Young, Proc. R. Soc. London, Ser. A, 1963, 271, 499–511 CrossRef CAS.
- J. S. Culik and D. D. L. Chung, Mater. Sci. Eng., 1980, 44, 129–137 CrossRef CAS.
- E. Papirer, R. Lacroix, J.-B. Donnet, G. Nanse and P. Fioux, Carbon, 1994, 32, 1341–1358 CrossRef CAS.
- J. Li, L. Vaisman, G. Marom and J.-K. Kim, Carbon, 2007, 45, 744–750 CrossRef CAS.
- A. E. Mansour, S. Dey, A. Amassian and M. H. Tanielian, ACS Appl. Mater. Interfaces, 2015, 7, 17692–17699 CAS.
- A. J. Marsden, P. Brommer, J. J. Mudd, M. A. Dyson, R. Cook, M. Asensio, J. Avila, A. Levy, J. Sloan, D. Quigley, G. R. Bell and N. R. Wilson, Nano Res., 2015, 8, 2620–2635 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- K. C. Knirsch, J. M. Englert, C. Dotzer, F. Hauke and A. Hirsch, Chem. Commun., 2013, 49, 10811–10813 RSC.
- S. Eigler, F. Hof, M. Enzelberger-Heim, S. Grimm, P. Müller and A. Hirsch, J. Phys. Chem. C, 2014, 118, 7698–7704 CAS.
- J. M. Englert, P. Vecera, K. C. Knirsch, R. A. Schäfer, F. Hauke and A. Hirsch, ACS Nano, 2013, 7, 5472–5482 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- L. G. Cançado, A. Jorio, E. H. M. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
- S. Deng, Y. Zhang, A. H. Brozena, M. L. Mayes, P. Banerjee, W.-A. Chiou, G. W. Rubloff, G. C. Schatz and Y. Wang, Nat. Commun., 2011, 2, 382 CrossRef PubMed.
- A. Eckmann, A. Felten, A. Mishchenko, L. Britnell, R. Krupke, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 3925–3930 CrossRef CAS PubMed.
- T. Gatti, N. Vicentini, M. Mba and E. Menna, Eur. J. Org. Chem., 2016, 2016, 1071–1090 CrossRef CAS.
- R. J. Young, I. A. Kinloch, L. Gong and K. S. Novoselov, Compos. Sci. Technol., 2012, 72, 1459–1476 CrossRef CAS.
- D. Olmos, K. Bagdi, J. Mózcó, B. Pukánszky and J. González-Benito, J. Colloid Interface Sci., 2011, 360, 289–299 CrossRef CAS PubMed.
- M. Hernandez, B. Sixou, J. Duchet and H. Sautereau, Polymer, 2007, 48, 4075–4086 CrossRef CAS.
- T. Sarbu, K.-Y. Lin, J. Spanswick, R. R. Gil, D. J. Siegwart and K. Matyjaszewski, Macromolecules, 2004, 37, 9694–9700 CrossRef CAS.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM, TGA-MS, Raman and XPS characterisation, grafting calculations and solubility data. Further supporting data may be requested from the corresponding author, but may be subject to confidentiality obligations. See DOI: 10.1039/c7sc03455e |
| This journal is © The Royal Society of Chemistry 2018 |