
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switching single chain magnet behavior via photoinduced bidirectional metal-to-metal charge transfer†
Wenjing
Jiang
,
Chengqi
Jiao
,
Yinshan
Meng
,
Liang
Zhao
,
Qiang
Liu
and
Tao
Liu
 *
*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, 2 Linggong Rd., 116024, Dalian, China. E-mail: liutao@dlut.edu.cn
First published on 30th October 2017
Abstract
The preparation of single-chain magnets (SCMs) with photo-switchable bistable states is essential for the development of high-density photo-recording devices. However, the reversible switching of the SCM behavior upon light irradiation is a formidable challenge. Here we report a well-isolated double zigzag chain {[Fe(bpy)(CN)4]2[Co(phpy)2]}·2H2O (bpy = 2,2′-bipyridine, phpy = 4-phenylpyridine), which exhibits reversible redox reactions with interconversion between FeIIILS(μ-CN)CoIIHS(μ-NC)FeIIILS (LS = low-spin, HS = high-spin) and FeIIILS(μ-CN)CoIIILS(μ-NC)FeIILS linkages under alternating irradiation with 808 and 532 nm lasers. The bidirectional photo-induced metal-to-metal charge transfer results in significant changes of anisotropy and intrachain magnetic interactions, reversibly switching the SCM behavior. The on-switching SCM behavior driven by light irradiation at 808 nm could be reversibly switched off by irradiation at 532 nm. The results provide an additional and independent way to control the bistable states of SCMs by switching in the 0 → 1 → 0 sequence, with potential applications in high density storage and molecular switches.
Introduction
Single chain magnets (SCMs) possess bistable states (relative to binary 0 and 1) and slow magnetic relaxation,1–3 rendering them promising candidates for high-density information storage, quantum computing and spintronic devices.4–7 Thus, SCMs have attracted considerable attention and been widely studied in chemistry, physics and materials science. Various SCM systems have been developed,8–21 wherein particular interest is paid to designing switchable SCMs that are responsive to external stimuli, such as guest molecules, pressure, light and heat, etc.15–21 The unique magnetic properties of SCMs stem from large single-ion anisotropy, strong intrachain interactions and weak interchain interactions. When externally stimulated changes in single-ion anisotropy and strong intrachain interactions are embedded, it becomes possible to reversibly generate or cancel the bistable states of SCMs. Such switchable SCMs provide an additional and independent way to control the magnetic states by switching in the 0 → 1 → 0 sequence, which is supposed to increase the performance of currently used standard molecular model device-systems that exploit electronic read-out and transverse field techniques.6,7 Thus, the swift and reversible production and elimination of the SCM behavior is of great importance in the face of future technological demand for data storage and processing.Light-induced switchable nanomagnets provide the opportunity to achieve this goal.17–26 This is because the light-induced process usually occurs on the picosecond scale,27 enabling a rapid response of functional signals upon light irradiation. Moreover, not only is light convenient to be turned on/off and switched between different wavelengths, but it also offers matched energy for the interconversion of different electronic states. Benefiting from these, the light-driven SCMs were achieved in cyanide-bridged Fe–Co assemblies,17–19 where the SCM properties in the sequence of off → on were induced by the light-induced metal-to-metal charge transfers (MMCTs)28–38 or excited spin-state trapping (LIESST) processes.20 However, examples of light-driven SCMs are still limited due to the difficulty in combining SCM behavior with the photo-responsive property. Moreover, the manipulation of light-driven SCMs is all unidirectional for the reported examples, wherein the SCM behavior can be activated via the light irradiation process but deactivated only via a time consuming thermal relaxation process upon heating.17–20 No reversible photo-switchable SCMs have been reported, and the development of such systems remains a big challenge to fulfil the reversible control of magnetic states by lights analogous to the switch in the off → on → off sequences.
In this study, we seek to design a system that displays the on/off-switching SCM properties tuned through light irradiation. There are two challenges for the realization of this goal. One crucial problem is the introduction of photo-responsive units in which the electronic states of the constituent metal ions can be reversibly photo-switched. The other is to suitably assemble the photo-sensitive units into a magnetically well-isolated 1D chain associated with large uniaxial magnetic anisotropy and strong intrachain magnetic interactions. An applicable example is the Fe/Co Prussian blue analogues,28,39–41 as the bidirectional light-induced MMCTs were discovered in several Fe2Co2 square compounds.40,41 This discovery inspired us to introduce such a reversibly photo-switchable Fe2Co2 square into a double zigzag chain, wherein SCM behavior42–45 and photo-driven SCMs have been reported.17,18 Herein, we selected Li[FeIII(bpy)(CN)4] (bpy = 2,2′-bipyridine) as the building block to react with CoII ions and 4-phenylpyridine (phpy), forming a well-isolated 1D complex {[Fe(bpy)(CN)4]2Co(phpy)2}·2H2O (1). The cyanide bridges enabled the linkage of the FeIII and CoII ions in a 1D alignment and provided super-exchange pathways for strong magnetic interactions. Phpy was adopted as the auxiliary ligand to adjust the coordination sphere of the CoII sites, providing an appropriate ligand field and redox potential for the interconversion between the paramagnetic FeIIILS(μ-CN)CoIIHS (HS = high-spin, LS = low-spin) and diamagnetic FeIILS(μ-CN)CoIIILS linkages. The HS CoII ion (S = 3/2) is paramagnetic and is supposed to have strong anisotropy,42–46 whereas the LS CoIII ion is diamagnetic (S = 0) and has no anisotropy, offering the potential for photo-induced anisotropy.17–19,26,47–49 The interconversion between the FeIIILS(μ-CN)CoIIHS and FeIILS(μ-CN)CoIIILS linkages will switch the anisotropy and intrachain magnetic interactions that are essential for the SCM behavior, providing the possibility of switchable SCM behavior via a photo-induced bidirectional MMCT upon light irradiation. As a result, 1 represents the first example showing SCM behavior which could be switched on/off via bidirectional MMCT under alternating irradiation with 808 nm and 532 nm lasers.
Results and discussion
X-ray crystal structure
The crystal of 1 suitable for X-ray diffraction was obtained by the diffusion method from the methanol solution of Li[Fe(bipy)(CN)4]·H2O and phpy to the water solution of Co(ClO4)2·6H2O. A single-crystal X-ray diffraction experiment revealed that 1 crystalized in the monoclinic space group C2/c. The repeating unit comprised {[Fe(bpy)(CN)4]2Co(phpy)2} and the units were connected with each other via cyanide bridges, forming an infinite double zigzag chain along the c axis (Fig. 1a). Uncoordinated water molecules were located between the chains (Fig. 1b). Within the repeating unit, the cobalt ion was coordinated to four cyanide nitrogen atoms from two adjacent [Fe(bpy)(CN)4]− entities and two nitrogen atoms from the monodentate phpy ligands, forming an elongated octahedral environment. At 220 K, the Co–Ncyanide and Co–Nphpy distances were 1.885(3)–1.892(3) and 1.975(3) Å, respectively, which are characteristic of the LS CoIII ions. The Fe–C and Fe–N bond distances were 1.882(3)–1.955(4) and 1.981(3)–1.985(3) Å, respectively. These structural characteristics and charge compensations suggested the existence of FeIIILS(μ-CN)CoIIILS(μ-NC)FeIILS linkages in 1. However, due to the inversion symmetry requirement, the FeIIILS and FeIILS can not be distinguished and may be randomly located in the Fe2Co entity.17,18 Between the repeating units, the planes of the triangular Fe2Co units were arranged in an interlaced pattern with the dihedral angle of 34.2°. The shortest interchain Co⋯Co distance was 15.099 Å. | ||
| Fig. 1 (a) Side view of the 1D chain. (b) Packing diagram of the 1D chains along the c axis. The hydrogen atoms are omitted for clarity. Fe, green; Co, pink; C, gray; N, blue. | ||
Such a large distance would diminish the interchain magnetic interactions, meeting the requirements of isolation for SCM behavior well.
Mössbauer spectra analysis
57Fe Mössbauer spectroscopy is very powerful for determining the electronic states of the Fe centers. As shown in Fig. S1 in the ESI,† two quadrupole doublets were observed at 78 K. One is characteristic of the LS FeIII species with Mössbauer parameters of δ (isomer shift) = −0.03 and ΔEQ (quadrupole splitting) = 1.69 mm s−1, and another can be attributed to the LS FeII species with δ = 0.07 and ΔEQ = 0.77 mm s−1. The FeIII/FeII peak area ratio was 0.51/0.49, confirming the LS state of 1 with the FeIIILS(μ-CN)CoIIILS(μ-NC)FeIILS linkages.Magnetic characterization
Magnetic measurements further confirmed the LS state of 1. The temperature dependence of the magnetic susceptibilities was measured over the 2–300 K temperature range in the heating mode (Fig. S2a, ESI†). As the temperature increased, the χT values for each Fe2Co remained close to 0.51 cm3 K mol−1, corresponding to one LS FeIII ion. The results suggested that the electronic state of the intrachain is described as FeIIILSCoIIILSFeIILS. The field-dependent magnetization at 1.8 K increased slowly to 1.01 Nβ at 50 kOe, consistent with the value expected for one LS FeIII ion (Fig. S2b, ESI†). The alternating current (ac) magnetic susceptibilities were studied as a function of both temperature and frequency. No temperature and frequency dependence were observed for both in-phase (χ′) and out-of-phase (χ′′) components, corresponding to the pure paramagnetic behavior of the FeIIILSCoIIILSFeIILS unit (Fig. S2c, ESI†).On-switching SCM (808 nm)
The UV-vis spectra of 1 showed a broad band at λ = 750 nm at 298 K (Fig. S3, ESI†), which can be assigned to the FeII → CoIII intervalence charge transfer (IVCT).35,40,41 Therefore, an 808 nm laser was used to irradiate 1 at 10 K to explore the possibility of photoinduced transformation from FeIIILS(μ-CN)CoIIILS(μ-NC)FeIILS linkages to FeIIILS(μ-CN)CoIILS(μ-NC)FeIIILS linkages. The time-dependent χT values increased significantly from 0.47 cm3 mol−1 K before irradiation, reaching a saturation value of 5.62 cm3 mol−1 K after 9000 s under irradiation at 808 nm (Fig. S4, ESI†). This result indicated the generation of the metastable paramagnetic FeIIILS(μ-CN)CoIIHS(μ-NC)FeIIILS units (HS* state) transforming from the diamagnetic FeIILS(μ-CN)CoIIILS(μ-NC)FeIIILS units (LS state). The temperature-dependent χT product was then collected for the photo-induced HS* state from 1.8 to 120 K (Fig. 2a). The χT values increased steeply to a sharp maximum of 10.5 cm3 mol−1 K at 5.0 K before the HS* phase relaxed back to the LS phase on thermal treatment up to 80 K. The maximum χT value after irradiation indicated the occurrence of intrachain ferromagnetic interactions between FeIIILS (S = 1/2) and photo-induced CoIIHS (S = 3/2).17–19,26 Simultaneously, the field-dependent magnetization recorded after irradiation at 1.8 K showed a steep increase below 5 kOe, followed by an almost linear increase to 2.03 Nβ at 50 kOe (Fig. S5, ESI†). This evolution of the field-dependent magnetization replicated the production of ferromagnetic interactions in the photo-induced HS* chain. Considering the negative anisotropy of the CoII ion and the intrachain ferromagnetic interactions transmitted via the cyanide bridges, the SCM behavior was expected.17–19,42–45 Alternating current (ac) magnetic susceptibility measurements were then carried out as a function of both temperature and frequency to investigate the magnetization dynamics of the photo-generated HS* phase. Both the in-phase (χ′) and the out-of-phase (χ′′) signals showed strong frequency dependence (Fig. 3 and S6a, ESI†). The shift of the peak temperature (Tp) was characterized by the parameter Φ = (ΔTp/Tp)/Δ(log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) f) = 0.13, within the expected range for SCMs, thus excluding the possibility of spin-glass behavior and magnetic ordering.
f) = 0.13, within the expected range for SCMs, thus excluding the possibility of spin-glass behavior and magnetic ordering.
 | ||
| Fig. 2 (a) Plots of χT vs. temperature for 1 before irradiation (LS) and after irradiation at 808 or 532 nm (LS → HS*), and the 808 nm-induced metastable state irradiated at 532 nm (HS* → LS*). (b) Plots of χT vs. time under cycles of successive irradiation at 808 nm and 532 nm at 10 K for 1. | ||
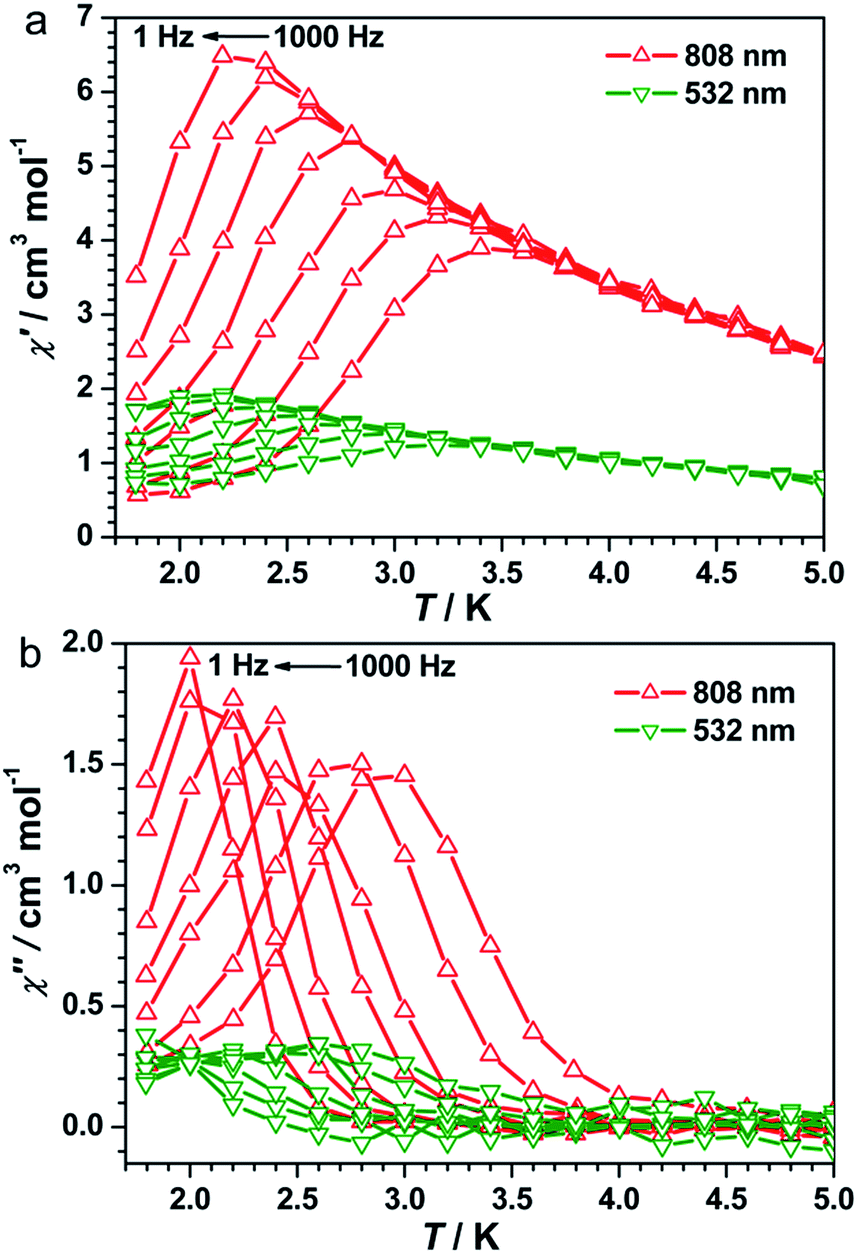 | ||
| Fig. 3 Temperature dependence of the in-phase (a) and out-of-phase (b) components of ac magnetic susceptibility after irradiation at 808 nm and 532 nm in a zero dc field at various ac frequencies and with a 3.5 Oe ac field. | ||
Detailed parameters were estimated and fitted by the Arrhenius law on the basis of the peak values of χ′′ (Fig. S7, ESI†), giving a pre-exponential factor of τ0 = 2.48 × 10−10 s and the relaxation energy barrier of Δ/kB = 40.21 K. The value of τ0 provides a quantitative estimation of the attempt time of relaxation from the chain bath and the obtained value is in good agreement with those reported for other photo-induced SCMs.17–20 A semicircular Cole–Cole diagram was constructed from the variable-frequency data collected at 2.7 K and was fitted with a generalized Debye model (Fig. S8, ESI†), yielding an α value of 0.33 that is similar to those obtained for other photo-induced SCMs.17–20 The one-dimensional behavior after irradiation at 808 nm was further checked with ac data at 1 Hz in a zero dc field following the correlation length that is proportional to the χ′T product in any 1D classical system. The ln(χ′T) vs. T−1 plot exhibited a clear linear region between 7 and 25 K (Fig. S9a, ESI†), supporting an anisotropic Ising-like 1D behavior.50–52 The linear region was fitted according to the equation χT = Ceffexp(Δξ/kBT), where Ceff is the effective Curie constant and Δξ is the energy of creating a domain wall along the chain, giving Δξ/kB = 12.08 K and Ceff = 1.71 cm3 mol−1 K. Below 4.0 K, ln(χ′T) reached saturation owing to the finite-size effects with [χ′T]max = 15.35 cm3 mol−1 K which was equal to nCeff, where n was the unit number in the chain. Hence, n could be estimated to be approximately 9.0, which is comparable to the values for reported SCMs.53 Therefore, all analyses together confirmed that compound 1 became a photo-driven SCM after irradiation at 808 nm.
Off-switching SCM (532 nm)
To exploit the photo-reversibility of the photo-driven SCM behavior, the photo-induced metastable HS* state was irradiated at 405, 532, 473, and 671 nm followed by measuring the magnetic susceptibilities.40,41 The most significant effect was observed at 532 nm, where χT values decreased obviously and dropped to the minimum value after 10000 s, from 5.62 to 2.85 cm3 mol−1 K (Fig. S4, ESI†). This phenomenon could be ascribed to the partial elimination of the HS* FeIIILS–CoIIHS metastable state under irradiation at 532 nm, causing partial recovery of the diamagnetic FeIILS–CoIIILS pairs. After deactivating the 532 nm laser, the temperature-dependent χT values were measured from 1.8 to 120 K. The maximum value of χT decreased to 4.00 cm3 mol−1 K at 5 K. This value was much smaller than that obtained after 808 nm irradiation (Fig. 2a). The field-dependent magnetization decreased to 1.58 Nβ at 50 kOe (Fig. S5, ESI†). Different to the dynamic behavior of the HS* state, the LS* state showed a poor frequency dependence behavior and weak intensity in χ′ and χ′′ signals (Fig. 3 and S6b, ESI†). It was probably because most of the effectively coupled chains have been destroyed, therefore the SCM behavior is lost. As shown in Fig. S9b,† the typical linear region for the 1D chains disappeared, definitely confirming that the SCM behavior driven by irradiation at 808 nm was broken up by the 532 nm laser. Due to the incomplete conversion from the HS* state to LS* state, the remnant frequency dependence may possibly result from the segments of coupled spins.
As found in other [FeCo] cluster complexes, the 532 nm light irradiation could also induce the transformation of both HS* → LS and LS → HS* owing to the partial overlap of the CoII → FeIII IVCT band and the broad FeII → CoIII IVCT band. Herein, the LS state of 1 was irradiated directly at 532 nm40,41 and the χT values reached a saturation value of 2.39 cm3 mol−1 K after 360 min (Fig. S10a, ESI†). The temperature-dependent χT values measured from 1.8 to 120 K show a maximum of 3.32 cm3 mol−1 K at 5 K (Fig. 2a). The ac magnetic susceptibilities displayed poor frequency dependence behavior and weak χ′ and χ′′ signals as well (Fig. S10b, ESI†). Therefore, the 532 nm laser irradiation had effects on both transformable directions.
The repeatability of the photo-switchable SCM behavior was demonstrated by tracing the time-dependent magnetic susceptibilities under successive and alternating irradiation at 808 and 532 nm (Fig. 2b). Furthermore, the temperature-dependent ac magnetic susceptibilities were measured in the third cycle. The ac signals are well repeatable (Fig. S11, ESI†), confirming the photo-reversible SCM behavior. We also performed the magnetization decay of the photo-induced HS* state at a series of temperatures (Fig. S12, ESI†) to see the stability. At high-temperatures (30–50 K), the relaxation time was strongly dependent on temperature, and in the temperature region of 10–20 K, it exhibited a weak temperature-dependence, suggesting that the relaxation from the metastable FeIIILSCoIIHSFeIIILS to the stable FeIILSCoIIILSFeIIILS was dominated by a quantum tunnelling mechanism in the low temperature region. Then, the effect of 532 nm irradiation on the metastable HS* state was compared with the natural relaxation cases at several temperatures. As shown in Fig. S13,† the χT values under irradiation at 532 nm decreased much faster than the nonirradiated ones at 10 K, 15 K and 20 K, suggesting that the 532 nm laser irradiation has a pronounced effect on the transformation from the metastable FeIIILSCoIIHSFeIIILS to the stable FeIILSCoIIILSFeIIILS. Above 30 K, this difference became smaller and almost disappeared at 50 K, indicating that the thermally induced relaxation is more predominant than the 532 nm irradiation. Thus, we have verified that complex 1 possessed the unique nature of the on/off-switching SCMs controlled by bidirectional photoinduced charge transfer at the given temperature.
IR spectra analysis
The infrared (IR) spectra of 1 at 200 K displayed vCN absorptions of 2073 and 2085 cm−1, characteristic of terminal groups of [FeIILS(bpy)(CN)4]2−. The absorption at 2122 cm−1 belonged to the terminal cyano groups of [FeIIILS(bpy)(CN)4]−. The νCN absorptions at 2192 and 2204 cm−1 were attributed to the bridging cyano groups of the FeIILS(μ-CN)CoIIILS and FeIIILS(μ-CN)CoIIILS linkages, respectively. The νCN stretches supported the assignment of the LS state in 1 (Fig. S14, ESI†).17,35In addition, the IR spectra were recorded after irradiation by the 808 and 532 nm lasers at 10 K to provide further evidence for the photo-reversibility in 1 (Fig. S15, ESI†). After irradiating at 808 nm, the intensities of the bridging νCN absorptions (2192 and 2204 cm−1) decreased, while a new band appeared at 2173 cm−1, indicating that part of the FeIILS(μ-CN)CoIIILS linkages were photochemically transformed into FeIIILS(μ-CN)CoIIHS species.17,35 The new νCN absorption at 2105 cm−1 was typical of the intermediate state of the bridging FeIILS(μ-CN)CoIIHS.54 Then, a 532 nm laser was used to irradiate the metastable state, and the intensity of the νCN absorption for the FeIILS(μ-CN)CoIIILS linkage increased whereas that for the FeIIILS(μ-CN)CoIIHS decreased, suggesting that transformation from FeIIILS(μ-CN)CoIIHS to FeIILS(μ-CN)CoIIILS occurred via 532 nm laser irradiation. The intensity of the additional νCN absorption for the FeIILS(μ-CN)CoIIHS state decreased relatively. The optical study visually displayed that the bidirectional photo-response occurred in 1 at different wavelengths. In agreement with the cyanide-bridged [FeCo] analogues,40,41 the LS FeIILS–CoIIILS pairs and the photo-induced HS* FeIIILS–CoIIHS pairs in the 1D chain were sensitive to red and green light, respectively, in the region where the FeII → CoIII and CoII → FeIII IVCT bands are usually observed. Importantly, the bidirectionally photo-induced charge transfer processes were coupled with the concomitant change of anisotropy at the Co sites and intrachain magnetic coupling interactions between cyanide-bridged Fe and Co centers that are the crucial factors for the magnetic dipole alignment. Therefore, in contrast to the reported systems in which the light-driven SCM behavior could merely relax back on heating, the excitation and deexcitation of the SCM behavior studied here could be switched bidirectionally by two light sources at a given temperature based on the MMCT mechanism. The magnetic bistable states switched under light irradiations in either the present system or the light-actuated SMM55 provide the possibility of using molecular nanomagnetic materials for the read-in and erasure of high density computer memory or storage devices, which is directly analogous to current binary storage systems.
Conclusions
In summary, the bidirectional photo-switchable SCM was successfully realized in the well isolated Fe(μ-CN)Co 1D chain for the first time, in which the on-switching SCM behavior driven by laser irradiation at 808 nm can be reversibly switched off by irradiation at 532 nm. The electronic structure evolution upon heating and light irradiations was well demonstrated and confirmed the reversible photo-switchable SCM behavior. This result has shown that the spin topology of SCMs can also be reversibly tuned via the photo-induced MMCT process, enabling access to turn on/off the SCM behavior with the potential application as high-density information storage materials for writing and erasing information upon light irradiations.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was partly supported by the NSFC (Grants 21421005, 91422302, and 21322103), and the Fundamental Research Funds for the Central Universities, China.Notes and references
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford Univ. Press, New York, 2006 Search PubMed.
- C. Coulon, V. Pianet, M. Urdampilleta and R. Clérac, Structure and Bonding, Springer-Verlag, Berlin Heidelberg, 2014, vol. 164, pp. 143–184 Search PubMed.
- R. Glauber, J. Math. Phys., 1963, 4, 294–307 CrossRef.
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, P. Sainctavit, M. A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417–421 CrossRef CAS PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194–197 CrossRef CAS PubMed.
- A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregoril, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760–1763 CrossRef CAS PubMed.
- R. Clérac, H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837–12844 CrossRef.
- H. Miyasaka, M. Julve, M. Yamashita and R. Clérac, Inorg. Chem., 2009, 48, 3420–3427 CrossRef CAS PubMed.
- A. V. Palii, O. S. Reu, S. M. Ostrovsky, S. I. Klokishner, B. S. Tsukerblat, Z.-M. Sun, J.-G. Mao, A. V. Prosvirin, H.-H. Zhao and K. R. Dunbar, J. Am. Chem. Soc., 2008, 130, 14729–14738 CrossRef CAS PubMed.
- T. D. Harris, M. V. Bennett, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2010, 132, 3980–3988 CrossRef CAS PubMed.
- L. Bogani, A. Vindigni, R. Sessoli and D. Gatteschi, J. Mater. Chem., 2008, 18, 4750–4758 RSC.
- W.-X. Zhang, R. Ishikawa, B. Breedlove and M. Yamashita, RSC Adv., 2013, 3, 3772–3798 RSC.
- W. Ouellette, A. V. Prosvirin, K. Whitenack, K. R. Dunbar and J. Zubieta, Angew. Chem., Int. Ed., 2009, 121, 2174–2177 CrossRef.
- M. Mito, H. Deguchi, T. Tajiri, S. Takagi, M. Yamashita and H. Miyasaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 144421 CrossRef.
- T. Liu, Y.-J. Zhang, S. Kanegawa and O. Sato, J. Am. Chem. Soc., 2010, 132, 8250–8251 CrossRef CAS PubMed.
- D.-P. Dong, T. Liu, S. Kanegawa, S. Kang, O. Sato, C. He and C.-Y. Duan, Angew. Chem., Int. Ed., 2012, 51, 5119–5123 CrossRef CAS PubMed.
- N. Hoshino, F. Iijima, G. N. Newton, N. Yoshida, T. Shiga, H. Nojiri, A. Nakao, R. Kumai, Y. Murakami and H. Oshio, Nat. Chem., 2012, 4, 921–926 CrossRef CAS PubMed.
- T. Liu, H. Zheng, S. Kang, Y. Shiota, S. Hayami, M. Mito, O. Sato, K. Yoshizawa, S. Kanegawa and C.-Y. Duan, Nat. Commun., 2013, 4, 2826 Search PubMed.
- E. Heintze, F. E. Hallak, A. Rettori, M. G. Pini, F. Totti, M. Dressel and L. Bogani, Nat. Mater., 2013, 12, 202–206 CrossRef CAS PubMed.
- C. Mathonière, H.-J. Lin, D. Siretanu, R. Clérac and J. M. Smith, J. Am. Chem. Soc., 2013, 135, 19083–19086 CrossRef PubMed.
- R. Ababei, C. Pichon, O. Roubeau, Y.-G. Li, N. Bréfuel, L. Buisson, P. Guionneau, C. Mathonière and R. Clérac, J. Am. Chem. Soc., 2013, 135, 14840–14853 CrossRef CAS PubMed.
- T. Shiga, H. Miyasaka, M. Yamashita, M. Morimotob and M. Irie, Dalton Trans., 2011, 40, 2275–2282 RSC.
- M. Morimoto, H. Miyasaka, M. Yamashita and M. Irie, J. Am. Chem. Soc., 2009, 131, 9823–9835 CrossRef CAS PubMed.
- M. Nihei, Y. Okamoto, Y. Sekine, N. Hoshino, T. Shiga, I. P. C. Liu and H. Oshio, Angew. Chem., Int. Ed., 2012, 51, 6361–6364 CrossRef CAS PubMed.
- R. Bertoni, M. Cammarata, M. Lorenc, S. F. Matar, J. F. Létard, H. T. Lemke and E. Collet, Acc. Chem. Res., 2015, 48, 774–781 CrossRef CAS PubMed.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704–705 CAS.
- T. Liu, D.-P. Dong, S. Kanegawa, S. Kang, O. Sato, Y. Shiota, K. Yoshizawa, S. Hayami, S. Wu, C. He and C.-Y. Duan, Angew. Chem., Int. Ed., 2012, 51, 4367–4370 CrossRef CAS PubMed.
- D.-F. Li, R. Clérac, O. Roubeau, E. Harté, C. Mathonière, R. L. Bris and S. M. Holmes, J. Am. Chem. Soc., 2008, 130, 252–258 CrossRef CAS PubMed.
- Y.-Z. Zhang, D.-F. Li, R. Clérac, M. Kalisz, C. Mathonière and S. M. Holmes, Angew. Chem., Int. Ed., 2010, 49, 3752–3756 CrossRef CAS PubMed.
- K. E. Funck, A. V. Prosvirin, C. Mathonière, R. Clérac and K. R. Dunbar, Inorg. Chem., 2011, 50, 2782–2789 CrossRef CAS PubMed.
- I.-R. Jeon, S. Calancea, A. Panja, D. M. Piñero Cruz, E. S. Koumousi, P. Dechambenoit, C. Coulon, A. Wattiaux, P. Rosa, C. Mathonière and R. Clérac, Chem. Sci., 2013, 4, 2463–2470 RSC.
- E. S. Koumousi, I. R. Jeon, Q. Gao, P. Dechambenoit, D. N. Woodruff, P. Merzeau, L. Buisson, X.-L. Jia, D.-F. Li, F. Volatron, C. Mathonière and R. Clérac, J. Am. Chem. Soc., 2014, 136, 15461–15464 CrossRef CAS PubMed.
- Y.-Z. Zhang, P. Ferko, D. Siretanu, R. Ababei, N. P. Rath, M. J. Shaw, R. Clérac, C. Mathonière and S. M. Holmes, J. Am. Chem. Soc., 2014, 136, 16854–16864 CrossRef CAS PubMed.
- D. Aguilà, Y. Prado, E. S. Koumousi, C. Mathonière and R. Clérac, Chem.Soc. Rev., 2016, 45, 203–224 RSC.
- M. Nihei, Y. Sekine, N. Suganami, K. Nakazawa, A. Nakao, H. Nakao, Y. Murakami and H. Oshio, J. Am. Chem. Soc., 2011, 133, 3592–3600 CrossRef CAS PubMed.
- N. Mirco, C. Sebastiano and S. Franco, Chem. Soc. Rev., 2014, 43, 4005–4018 RSC.
- O. Sato, Y. Einaga, T. Yidoa, A. Fujishima and K. Hashimoto, J. Electrochem. Soc., 1997, 144, L11 CrossRef CAS.
- A. Mondal, Y.-L. Li, M. Seuleiman, M. Julve, L. Toupet, M. B. L. Cointe and R. Lescouëzec, J. Am. Chem. Soc., 2013, 135, 1653–1656 CrossRef CAS PubMed.
- Y. Sekine, M. Nihei, R. Kumai, H. Nakao, Y. Murakami and H. Oshio, Inorg. Chem. Front., 2014, 1, 540–543 RSC.
- R. Lescouëzec, J. Vaissermann, C. Ruiz-Pérez, F. Lloret, R. Carrasco, M. Julve, M. Verdaguer, Y. Dromzee, D. Gatteschi and W. Wernsdorfer, Angew. Chem., Int. Ed., 2003, 42, 1483–1486 CrossRef PubMed.
- L. M. Toma, R. Lescouëzec, J. Pasán, C. Ruiz-Pérez, J. Vaissermann, J. Cano, R. Carrasco, W. Wernsdorfer, F. Lloret and M. Julve, J. Am. Chem. Soc., 2006, 128, 4842–4853 CrossRef CAS PubMed.
- L. M. Toma, C. Ruiz-Pérez, F. Lloret and M. Julve, Inorg. Chem., 2012, 51, 1216–1218 CrossRef CAS PubMed.
- L. M. Toma, C. Ruiz-Pérez, J. Pasán, W. Wernsdorfer, F. Lloret and M. Julve, J. Am. Chem. Soc., 2012, 134, 15265–15268 CrossRef CAS PubMed.
- F. Lloret, M. Julve, J. Cano, R. Ruiz-García and E. Pardo, Inorg. Chim. Acta, 2008, 361, 3432–3445 CrossRef CAS.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- Y.-Z. Zhang, S. Gómez-Coca, A. J. Brown, M. R. Saber, X. Zhang and K. R. Dunbar, Chem. Sci., 2016, 7, 6519–6527 RSC.
- T. J. Woods, M. F. Ballesteros-Rivas, S. Gómez-Coca, E. Ruiz and K. R. Dunbar, J. Am. Chem. Soc., 2016, 138, 16407–16416 CrossRef CAS PubMed.
- J. M. Loveluck, S. W. Lovesey and S. Aubry, J. Phys. C: Solid State Phys., 1975, 8, 3841–3856 CrossRef.
- K. Nakamura and T. Sasada, J. Phys. C: Solid State Phys., 1978, 11, 331–333 CrossRef CAS.
- C. Coulon, R. Clérac, L. Lecren, W. Wernsdorfer and H. Miyasaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 132408 CrossRef.
- H. Miyasaka, T. Madanbashi, A. Saitoh, N. Motokawa, R. Ishikawa, M. Yamashita, S. Bahr, W. Wernsdorfer and R. Clérac, Chem.–Eur. J., 2012, 18, 3942–3954 CrossRef CAS PubMed.
- V. Escax, A. Bleuzen, C. C. d. Moulin, F. Villain, A. Goujon, F. Varret and M. Verdagure, J. Am. Chem. Soc., 2001, 123, 12536–12543 CrossRef CAS PubMed.
- X.-W. Feng, C. Mathonière, I. R. Jeon, M. Rouzières, A. Ozarowski, M. L. Aubrey, M. I. Gonzalez, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2013, 135, 15880–15884 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and physical measurement details. Crystal data in CIF format and additional figures (Fig. S1–S15). CCDC 1528877. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc03401f |
| This journal is © The Royal Society of Chemistry 2018 |