
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide nucleic acid-templated selenocystine–selenoester ligation enables rapid miRNA detection†
Jessica
Sayers
 ab,
Richard J.
Payne
*a and
Nicolas
Winssinger
*b
ab,
Richard J.
Payne
*a and
Nicolas
Winssinger
*b
aSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: richard.payne@sydney.edu.au
bDepartment of Organic Chemistry, Faculty of Science, NCCR Chemical Biology, University of Geneva, Quai Ernest Ansermet 30, 1211 Geneva, Switzerland. E-mail: nicolas.winssinger@unige.ch
First published on 22nd November 2017
Abstract
The development of a rapid and chemoselective selenocystine–selenoester peptide ligation that operates at nanomolar reactant concentrations has been developed by utilising PNA templation. Kinetic analysis of the templated peptide ligation revealed that the selenocystine–selenoester reaction was 10 times faster than traditional native chemical ligation at cysteine and to our knowledge is the fastest templated ligation reaction reported to date. The efficiency and operational simplicity of this technology is highlighted through the formation of hairpin molecular architectures and in a novel paper-based lateral flow assay for the rapid and sequence specific detection of oligonucleotides, including miRNA in cell lysates.
Inspired by nature, oligonucleotide-templated reactions are designed to promote a specific chemical transformation upon adjacent hybridisation of reactants to a single template.1,2 A key feature of templated chemistry is that bimolecular reactions, that would otherwise be unfavourable at high dilution, are promoted by enhanced effective molarity in a templated reaction manifold. As such, templated chemistry has become an enormously useful tool in the fundamental sciences, e.g. in programmed organic synthesis,3–6 and in biomedicine and device development, e.g. in nucleic acid sensing technologies.7–10 Importantly, the use of an oligonucleotide template also significantly improves the biocompatibility of the transformation by kinetically favouring the designed reaction over parasitic background reactions with functionalities present in native biomolecules. Considering the fast hybridisation kinetics of short oligonucleotide probes (k ≈ 106 M−1 s−1),11–13 the reaction that occurs once the reagents are aligned on the template is rate limiting and proceeds with pseudo-first order kinetics. Therefore, it is critical that this reaction proceeds with fast kinetics to take full advantage of the templation.
Peptide ligation chemistry, in particular native chemical ligation between peptide thioesters and N-terminal cysteine-derived peptides, has revolutionised the synthesis of large polypeptides and proteins. The rate limiting step of native chemical ligation (and most other ligation techniques) is the bimolecular transthioesterification reaction between the side-chain thiol functionality of the Cys residue and the thioester fragment, and therefore proceeds at prohibitively slow reaction rates at high dilution. This factor, coupled with competing thioester hydrolysis, leads to poor overall reaction yields when ligations are performed at low reactant concentrations.14,15 To overcome this limitation, Seitz and co-workers pioneered nucleic acid-templated native chemical ligation reactions16,17 that have found utility in nucleic acid sensing and bioactive compound synthesis.18–21 In a templated format, native chemical ligation has been shown to proceed significantly faster than the corresponding non-templated transformation (e.g. 70% completion in 2 h templated vs. >48 h non-templated for a specific example).16 In this work by Seitz and co-workers, peptide nucleic acid (PNA) tags were employed to serve as the oligonucleotide template. Two probes were designed and synthesised, one bearing a thioester at the C-terminus of a short PNA tag and the second complementary PNA sequence functionalised with an N-terminal Cys residue. PNAs are a particularly attractive oligonucleotide surrogate for use in templated chemistry. This is due, in major part, to their chemical and enzymatic stability, but also because the solid-phase chemistry that is employed for their synthesis is highly reliable.6,22 PNAs maintain the sequence specific hybridisation qualities of DNA but offer enhanced binding affinity due to the replacement of the anionic ribose-phosphate backbone of DNA with a charge-free polyamide scaffold, thereby reducing electrostatic repulsion upon hybridisation. PNA therefore exhibits remarkable stability when forming duplexes with complementary DNA/RNA strands and enables the use of comparatively short PNA segments as high-affinity tags.
We have an interest in the development of tools for the high-sensitivity, sequence-specific detection of DNA/RNA targets in biological samples and, as such, we sought to develop a novel PNA-templated reaction that could be applied in this context (Scheme 1). To achieve this, we sought chemistry that proceeds with faster reaction kinetics than native chemical ligation to enable accelerated read-out of a detection assay, whilst being operationally simple to perform. Towards this end, we turned to the recently reported diselenide-selenoester ligation at selenocystine23 which we rationalised could be used for rapid PNA-templated reactions at low concentrations but with faster kinetics than native chemical ligation. Indeed, this transformation has been shown to be chemoselective in the presence of unprotected peptide fragments and proceeds remarkably quickly in aqueous phosphate buffer (1–10 min) without the addition of any exogenous additives. Based on the increased rate of the bimolecular reaction, it was hypothesised that a PNA-templated variant of the selenium-mediated reaction, with enhanced effective molarity of the selenocystine and selenoester peptide fragments, would lead to faster reaction kinetics than currently available templated ligation technologies.
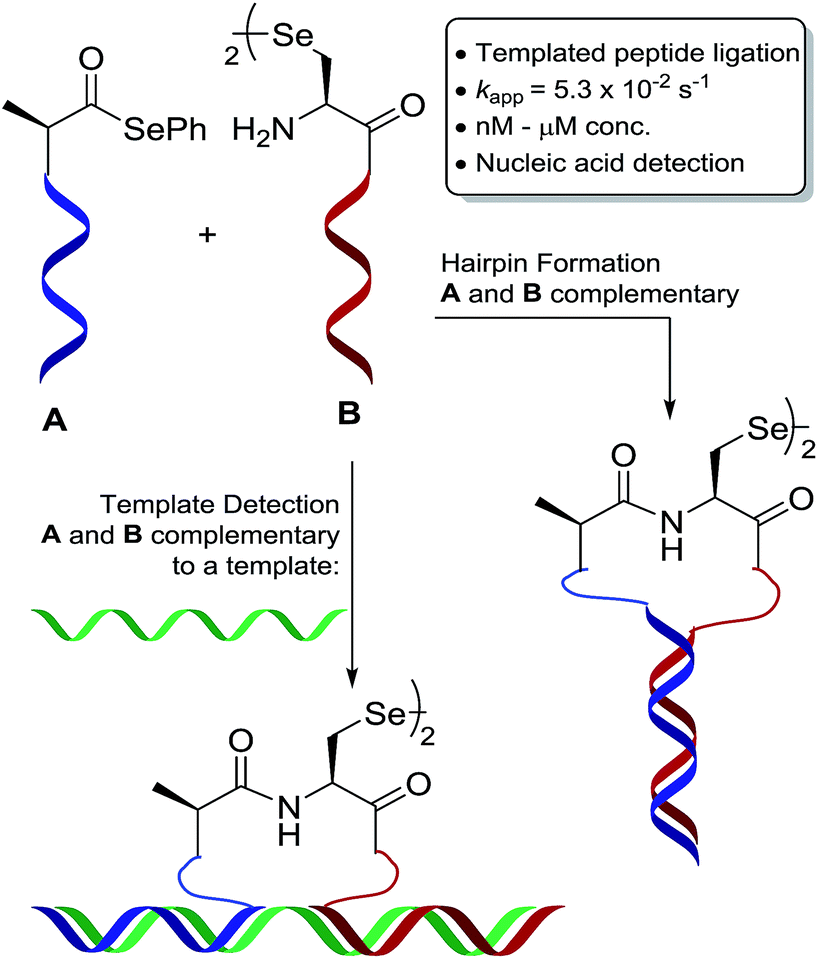 | ||
| Scheme 1 Schematic representation of oligonucleotide-templated selenocystine–selenoester ligation reaction. | ||
To test our hypothesis, we initially designed a model reaction system comprising of a tetra nucleobase PNA bearing a C-terminal phenyl selenoester moiety 1 and a complementary tetra nucleobase PNA sequence attached to a tetrapeptide with an N-terminal selenocystine [(Sec)2] residue 2 (Scheme 2). Upon hybridisation of the PNA tags, the resulting “hairpin” molecular architecture would position the reactive functionalities in close proximity to facilitate the ligation reaction, which we proposed would occur rapidly irrespective of initial fragment concentration. To synthesise selenoester fragment 1, the PNA chain was elongated on 2-chlorotrityl chloride resin using standard iterative solid-phase synthesis procedures24 to afford resin-bound PNA 3 (see ESI† for details). Mild acidolytic cleavage from the solid support was then effected via treatment with a 30 vol% HFIP solution in dichloromethane to ensure that the side-chain protecting groups remained intact. Selenoesterification of the C-terminus was then performed in solution via treatment with diphenyldiselenide (DPDS) and tributylphosphine. Following global acidic cleavage and purification by reverse-phase HPLC, PNA selenoester 1 was obtained. The complementary PNA sequence was prepared on Rink amide resin using the same PNA oligomerisation methodology and was extended with a short L-Ala-L-Ser-L-Ala tripeptide to afford resin-bound PNA 4. The selenocystine [(Sec)2] residue was finally incorporated at the N-terminus, under standard peptide coupling conditions. Following cleavage from the solid support and purification by reverse-phase HPLC, PNA fragment 2 was afforded.
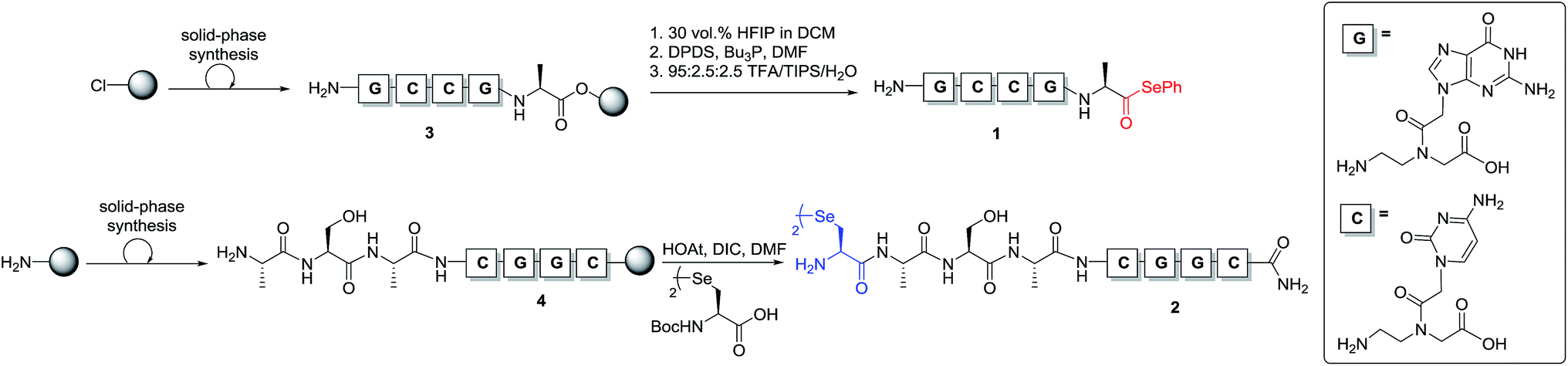 | ||
| Scheme 2 Synthesis of PNA-selenoester 1 and PNA-diselenide dimer 2. | ||
With complementary PNA reactants 1 and 2 in hand, we could now investigate the templated ligation reaction. Stock solutions (10 mM and 1 mM) of the functionalised PNAs were prepared in anhydrous DMF (to avoid selenoester hydrolysis) and stored at −80 °C.‡ Ligation experiments were performed by simply adding equal volumes of the selenocystine and complementary selenoester fragment stock solutions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry with respect to the monomeric form of diselenide 2) to aqueous phosphate buffer at pH 7.0 to achieve the desired concentration for the reaction. Initially, reactions were carried out at 0.5 mM concentration of both fragments 1 and 2 (Scheme 3A). Gratifyingly, immediate LCMS analysis of the reaction mixture showed complete conversion to the desired “hairpin” ligation product. It should be noted that due to the redox potential of selenocysteine it exists predominantly as the diselenide dimer, i.e. selenocystine, in solution. As such, the major product of the ligation reaction is also the diselenide dimer. We were also able to show that selenocystine can be chemoselectively converted to a native alanine residue via an in situ deselenization protocol following the templated ligation.25 This was achieved via the addition of TCEP (50 equiv.) and DTT (5 equiv.) to the crude reaction mixture once the ligation reaction had reached completion to provide product 5 (Scheme 3A).
:1 stoichiometry with respect to the monomeric form of diselenide 2) to aqueous phosphate buffer at pH 7.0 to achieve the desired concentration for the reaction. Initially, reactions were carried out at 0.5 mM concentration of both fragments 1 and 2 (Scheme 3A). Gratifyingly, immediate LCMS analysis of the reaction mixture showed complete conversion to the desired “hairpin” ligation product. It should be noted that due to the redox potential of selenocysteine it exists predominantly as the diselenide dimer, i.e. selenocystine, in solution. As such, the major product of the ligation reaction is also the diselenide dimer. We were also able to show that selenocystine can be chemoselectively converted to a native alanine residue via an in situ deselenization protocol following the templated ligation.25 This was achieved via the addition of TCEP (50 equiv.) and DTT (5 equiv.) to the crude reaction mixture once the ligation reaction had reached completion to provide product 5 (Scheme 3A).
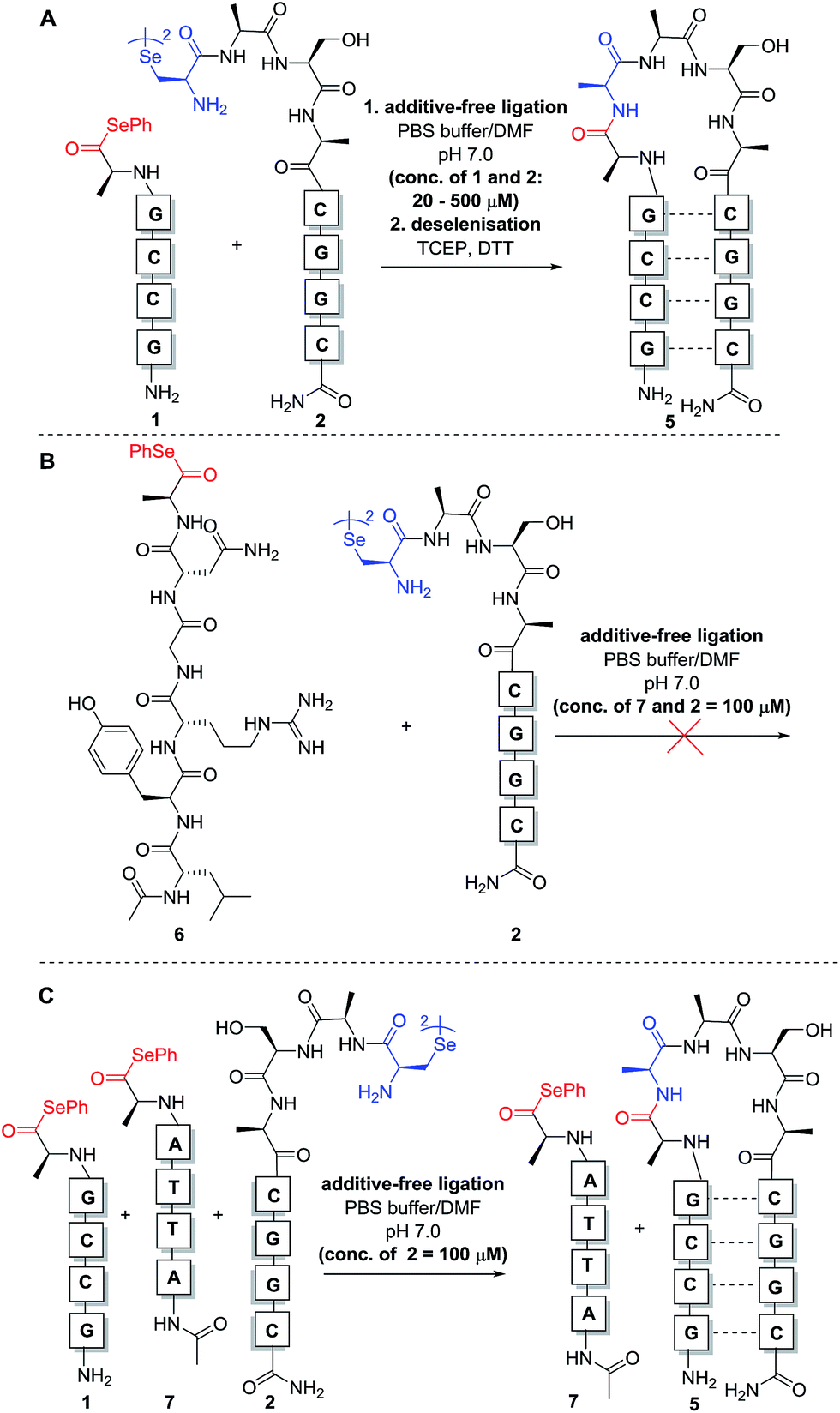 | ||
| Scheme 3 Model PNA-templated selenocystine–selenoester ligation reactions. (A) Ligation of complementary fragments 1 and 2. (B) Unsuccessful reaction between peptide selenoester 6 and PNA diselenide dimer 2 at 100 μM concentration. (C) Competitive ligation between complementary selenoester 1, non-complementary selenoester 7 and diselenide dimer 2. | ||
The model templated ligation reaction to generate the hairpin shown in Scheme 3A was subsequently performed with incremental increases in dilution. At 20 μM, the ligation product could be detected immediately by LC-MS analysis with complete consumption of 1 and 2, although at this concentration the signal was relatively weak in the chromatogram due to the low concentrations of reactants (see ESI†). Nonetheless, these dilution experiments indicate that the templated reaction likely proceeds in a concentration-independent manner as predicted. Given the difficulties for reproducible UV-based detection and reaction monitoring by LC-MS at concentrations at or below 20 μM, all further model experiments were conducted at a standard concentration of 100 μM.
In the original report of the untemplated selenocystine–selenoester ligation methodology, the reaction did not proceed at concentrations of 100 μM or less.23 Our preliminary results show that this reaction proceeds rapidly in a templated format at concentrations considerably lower than 100 μM, suggesting that the adjacent hybridisation of the reactive partners is likely the driving force for the success of the reaction (Scheme 3A). In order to confirm this, we performed further control experiments. Firstly, model peptide selenoester 6 (missing the PNA tag) was synthesised and reacted with PNA diselenide dimer 2 (Scheme 3B). The reaction was monitored over 24 h, however, in the absence of hybridisation, the ligation reaction did not proceed at 100 μM concentration of reactants and only led to selenoester hydrolysis over this time. As a further control experiment, a non-complementary PNA selenoester 7 was synthesised and allowed to react with diselenide dimer 2 in competition with the complementary PNA selenoester 1 (Scheme 3C). Strikingly, the only ligation product observed in this competition experiment was that bearing complementary PNA sequences, confirming that the ligation reaction is facilitated by the hybridisation-driven increase in effective molarity of the reactants. Additionally, a control experiment with non-complementary probes 2 and 7 (in the absence of complementary PNA selenoester 1) under the same conditions did not yield detectable ligation product. This result is consistent with the control ligation reaction between peptide selenoester 6 and PNA diselenide 2 and suggests that the discrimination observed in the competition reaction is not simply the result of kinetic selectivity. Taken together, these results rule out the occurrence of bimolecular background reactions at this concentration.
Next, we sought to determine the apparent rate constant of the pseudo-first order templated selenocystine–selenoester ligation reaction and compare it to the homologous templated NCL variant (Fig. 1). To this end, PNA selenoester 8 was synthesised on-resin via an operationally simple side-chain anchoring strategy recently reported by Hanna et al.26 Briefly, Fmoc-Glu-OAll was first loaded to Rink amide resin via the carboxylate side chain. Following elongation of the desired peptide sequence, the C-terminal allyl protecting group was removed followed by en bloc selenoesterification by treatment with DPDS and Bu3P. Acidolytic side chain deprotection and cleavage from the resin followed by HPLC purification then afforded the target peptide selenoester. Importantly, this solid-phase methodology enabled the synthesis of PNA selenoester 8 and PNA thioester 9 (using a thioesterification step instead of selenoesterification) from the same resin-bound PNA peptide precursor (see ESI† for synthetic details). Cysteine-bearing PNA 10 was also produced in an analogous manner to the chalcogenic diselenide dimer 2. In order to determine the half-life of the selenium-mediated reaction, we initially attempted to quench the ligation. Addition of a large excess of competing cysteine, increasing the pH to hydrolyse the selenoester and flash-freezing methods all proved ineffective at quenching the selenium-mediated ligation. With the knowledge that the untemplated selenocystine–selenoester ligation does not proceed at a reactant concentration of 100 μM, we opted for an alternative approach whereby we dehybridised the PNA tags by rapidly reducing the pH to 1.0 through addition of neat TFA to aliquots taken at precise time points. Gratifyingly, the kinetic data obtained via this method proved to be highly reproducible (Fig. 1). A rate constant of 5.3 × 10−2 s−1 was calculated. The reaction has a t1/2 = 13 s, which offers approximately an order of magnitude increase in reaction rate over the equivalent PNA-templated NCL reaction (t1/2 = 166 s, Fig. 1).
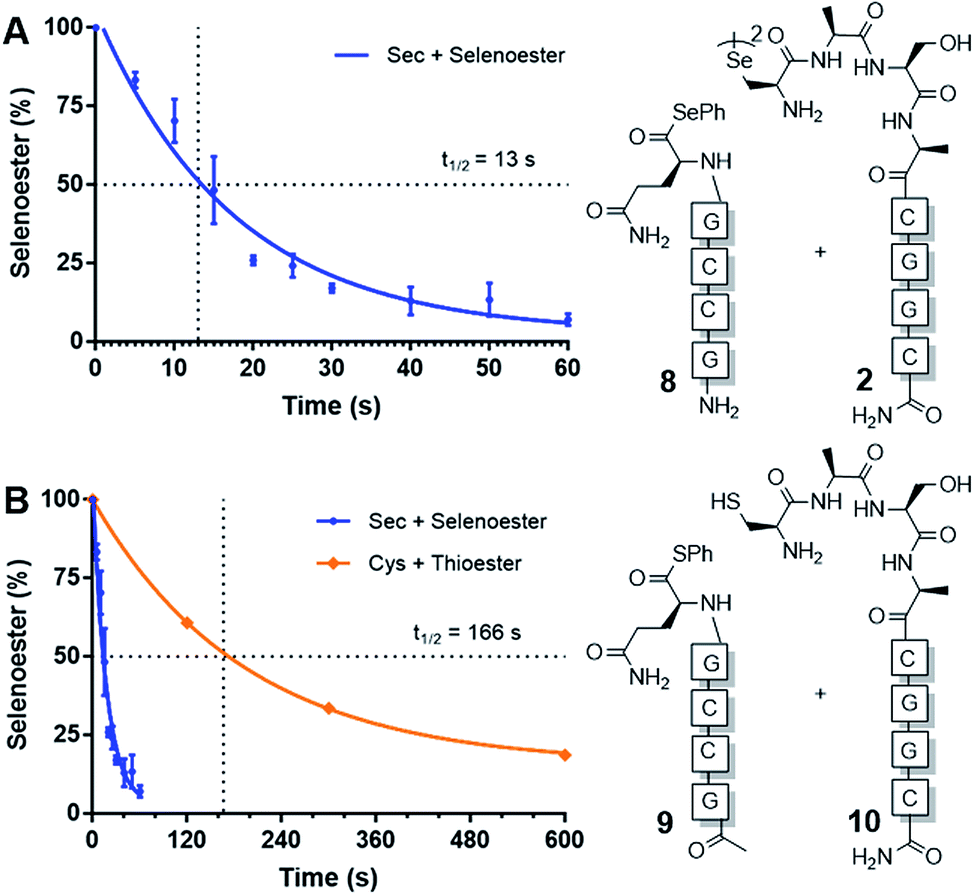 | ||
| Fig. 1 Rate of model PNA-templated ligation reactions. (A) Ligation of diselenide dimer 2 and selenoester 8. (B) Rate comparison of selenocystine–selenoester ligation and NCL (Cys 10 + thioester 9). | ||
Having demonstrated that templated selenium-mediated ligation reactions proceed with fast kinetics at high dilution, we were next interested in exploring the potential application of the reaction in immediate read-out nucleic acid detection assays. Nucleic acids represent an important class of target biomolecules and current detection approaches, such as real-time polymerase chain reaction, northern blotting and microarray analysis, offer highly accurate and sensitive detection. However, most of these conventional methods are hindered by complicated and costly procedures, time consuming target amplification steps and/or diminishing sensitivity of fluorophore-based detection methods. These limitations severely restrict the practical applications of such methods to specialised environments, such as in the field. There is therefore a need for the development of a rapid, widely-applicable and operationally simple nucleic acid detection system. Paper-based diagnostics have proven to be a robust format for economical point-of-care diagnostics and field application devices and was the device modality pursued here.27,28 Due to the mass production of lateral-flow immunochromatographic assays (LFA),29 such as commercially available pregnancy test strips, we opted for this format (Fig. 2). More specifically, we designed a nucleic acid-templated reaction utilising two synthetic PNA-peptide probes, one functionalised with a biotin and a selenocystine residue, and one with fluorescein (FITC) and a selenoester moiety that could react under a selenium-mediated ligation manifold upon hybridisation to the desired analyte (Scheme 4). It was proposed that upon addition of the two probes at a given concentration to a sample solution containing the oligonucleotide sequence of interest, the probes would adjacently hybridise to the target analyte and react to form a covalent peptide linkage. Given that the product of the ligation affords a junction between the probes that is the same length as a single PNA residue, the reactions were designed to leave a single unpaired nucleobase on the template between the hybridisation sites (Scheme 4). Previous studies with NCL have shown that this spacing improves sequence fidelity of templated reactions.30 Insertion of a lateral flow test strip into the sample solution would then provide a rapid indication of the presence/absence of the oligonucleotide target sequence through the capture of biotin by streptavidin in the test zone and by subsequent binding of the anti-FITC antibody coated gold nanoparticles to the FITC moiety that are then visible to the naked eye. When a single species containing both a FITC and a biotin tag is present, the Au-nanoparticles are bound to the test zone and indicate a positive result as a red line. Each strip also contains an internal control band.
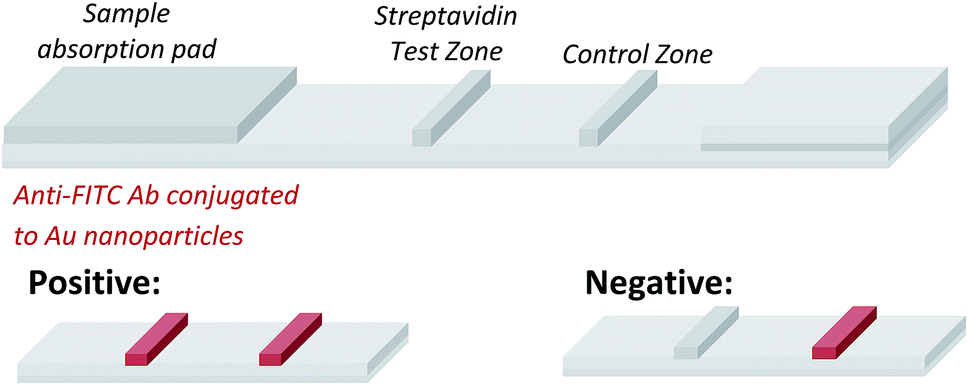 | ||
| Fig. 2 Illustrative representation of the principle of the lateral flow test strips employed. | ||
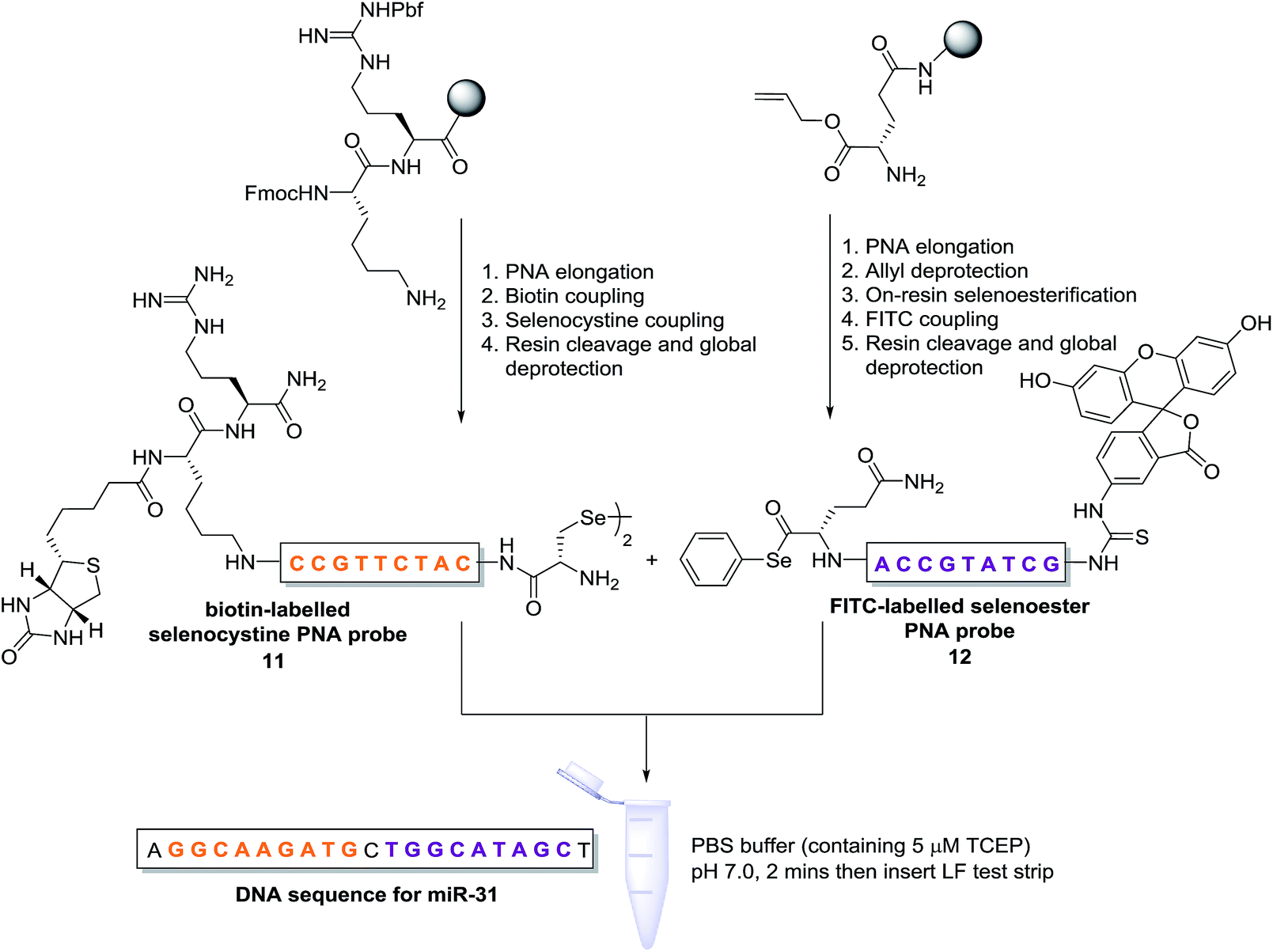 | ||
| Scheme 4 Synthesis of PNA-diselenide dimer probe 11 and PNA-selenoester probe 12 (for detailed synthetic procedures see ESI†) and general miRNA detection assay conditions. | ||
In an initial proof of concept study we chose to target the miRNA-31 (microRNA) sequence which has been characterised as a tumour suppressor, with altered expression levels of the miRNA detected in a large variety of tumour types.31 A pair of labelled PNA probes were therefore designed and synthesised possessing complementary sequences for hybridisation to the analyte of interest. More specifically, two complementary ligation partner probes were synthesised, a biotin-labelled PNA diselenide-dimer probe 11 and a FITC-labelled PNA bearing a selenoester functionality 12 (see Scheme 4 and ESI† for synthetic details). The PNA sequences of 11 and 12 were designed to ensure reaction in the presence of the specific miR-31 analyte. Having successfully prepared 11 and 12, the assay was performed by simultaneous addition of the two probes as stock solutions in DMF (100 nM final concentration) to the sampling buffer solution containing the purified DNA version of the miR-31 target analyte (10 nM). It should be noted that aqueous buffer containing TCEP (5 μM) as a reductant was used to ensure diselenide probe 11 was reduced and existed in the monomeric form. The analyte-templated ligation reaction was allowed to proceed for 2 min at room temperature before the LF test strip was inserted into the solution. Once the solution had migrated through the test strip beyond the control zone, the strip was removed and allowed to dry at room temperature. Visual inspection of the strip revealed a positive result in the test zone indicating that the hybridisation-driven ligation reaction had proceeded and was capable of confirming the presence of the miR-31 target analyte. A control experiment performed under identical conditions with the carboxylic acid of probe 12 rather than the selenoester failed to produce a positive result indicating that a covalent adduct between the probes must be formed, consistent with the fact that dissociation kinetics of the 9-mer PNAs and the analyte is fast relative to the LF strip readout (see ESI†). Next, in order to determine the detection limits of the LF strips, a concentration gradient of the ligation product was set up by dilution of the product solution from 100 nM down to 33 pM. We found that the streptavidin test zone reached saturation of the biotin-labelled ligation product at approximately 10 nM and the positive result was undetectable to the naked eye at a concentration of 33 pM (Fig. 3A). Imaging software, ImageJ, was used to process digital photographs of the test strips in order to gain a semi-quantitative reading of the outcome and to enable presentation of the results in a graphical format. With the standard product concentration gradient established, we next determined the effect of varying the miR-31 template concentration (Fig. 3B). The assay was conducted at a constant concentration of both probes (10 nM, 1:1 stoichiometry with respect to monomeric 11) and a range of template concentrations between 10 and 0.1 nM. After 2 min, the LF strips were inserted and the sample solution migrated by capillary flow through the strip. The resulting test strips were imaged and processed in order to measure the intensity of the test zone band for each strip. The results were then plotted on the standard titration curve to estimate the concentration of the ligation product (Fig. 3B). At analyte concentrations of 1 nM and below, the estimated analyte concentrations were in excellent agreement with the experimental values (see ESI†). These results demonstrate that the assay provides a method of estimating the concentration of the ligation product and hence the concentration of the analyte within the sample solution in a semi-quantitative manner.
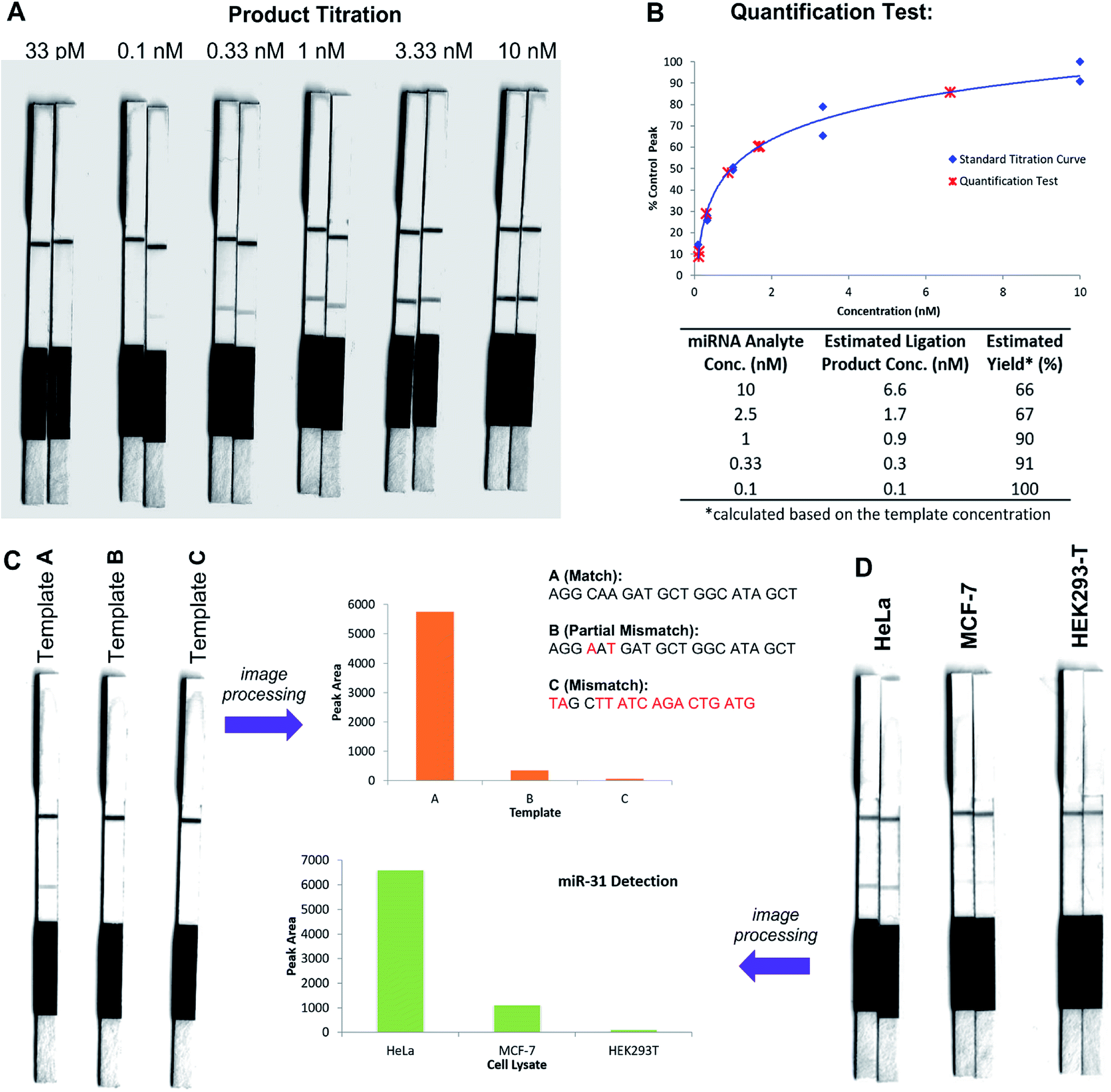 | ||
| Fig. 3 Lateral flow miRNA detection assays using probes 11 and 12. (A) Titration of ligation product showing upper and lower detection limits of test strips. (B) Quantification tests: assays performed at a constant probe concentration (10 nM) and varied miR-31 analyte concentration (10–0.1 nM). Test strip images processed and plotted on standard titration curve. Table shows estimated ligation product concentrations determined from standard titration curve and resulting conversion to ligation reaction yield (%). (C) Image of exemplar test strip results from sequence fidelity test at 40 °C and processed image in graphical format. (D) Image of test strip results (n = 2) from detection of miR-31 in lysates of HeLa, MCF-7 and HEK293-T cell lines and processed image in graphical format of averaged test zone intensities. | ||
In order to investigate the sequence fidelity of our synthetic PNA probes, we used the miR-31 PNA probes to detect three different template sequences (Fig. 3C). Following addition of the probes to the different template-containing buffer solutions at room temperature, 2 min later test strips were inserted. A positive result was obtained for the ligation product in the presence of the matched sequence template (A), a significantly less intense signal was produced for the ligation product in the presence of partially mismatched template (B) and with completely mismatched template (C) a negative result was returned, thus indicating the absence of the selenocystine–selenoester ligation product. In an attempt to refine the sequence specificity of the hybridisation-driven reaction we next investigated the effect of raising the temperature (see ESI†). Pleasingly, at 40 °C the resolution was improved dramatically and clearly indicated the presence of ligation product with template (A) compared to the reaction in the presence of template (B) and (C) which did not produce a signal visible to the naked eye (Fig. 3C). Remarkably, the reaction proceeded equally well at 70 °C (see ESI†). The ability to perform nucleic acid templated reactions at higher temperature may be important in certain settings to disrupt secondary structure or other interactions that would shield the template.
Having demonstrated that the templated ligation-based LF assays could be used to detect miRNA at low concentration with sequence fidelity, we next sought to assess the viability of the assay in a more complex environment. To this end, lysates of three different cell lines (HeLa, MCF-7, HEK293-T) with differing reported levels of miRNA expression were cultured. The miR-31 detection assay was performed in HeLa cell lysate, a cervical cancer cell line which overexpresses miR-31,32 by addition of the appropriately labelled synthetic probes to a final concentration of 10 nM. Upon insertion of the LF test strip, visual inspection of the results indicated a positive reading for the presence of miR-31 in HeLa lysate (Fig. 3D), consistent with the result obtained for the in vitro experiments described in Fig. 3A and B. The same experiment was next performed using MCF-7 and HEK293-T lysates which do not overexpress miR-31. Both experiments yielded negative results upon visual inspection of the test zone (Fig. 3D), suggesting that the LFA could be used to detect threshold concentrations of specific miRNA sequences. Finally, to highlight the versatility of the technology for miRNA detection we synthesised new probes bearing appropriate PNA sequences for miR-21 hybridisation and detection. It has been previously reported that miR-21 is over-expressed in MCF-7 cells.33 The LF assay was carried out on the lysates of the same three cell lines (HeLa, MCF-7 and HEK293T) and importantly showed a higher concentration of the miR-21 analyte in the lysate of the MCF-7 cell line compared to both HeLa and HEK293T lysates (see ESI†). These results are in agreement with prior qPCR quantification of the miRNA targets.34
Conclusions
In summary, we have developed a rapid and chemoselective PNA-templated selenocystine–selenoester peptide ligation reaction. We have shown that under highly dilute conditions, the reaction does not proceed in the absence of the PNA template and is therefore facilitated by the sequence-specific hybridisation of the PNA tags. The pseudo-first order rate constant of the ligation reaction was calculated to be 5.3 × 10−2 s−1 which, to our knowledge, represents the fastest peptide ligation method reported to date.30 We also demonstrate that selenocystine can be converted to a native alanine residue following the templated reactions, which is an important asset for the application of this reaction in templated synthesis of peptide libraries or hairpin loops.35–37 This novel templated ligation technology was showcased through the rapid detection of specific miRNA analytes using an operationally simple paper-based lateral flow assay. Importantly, the assays were also effective for rapid miRNA detection in crude lysates demonstrating that the technology operates within a complex biological and analytical environment. The assay described here is the first enzyme-free method to detect nucleic acids in such a format and, given the considerable interest in fast and inexpensive analytical methods based on paper microfluidics e.g. pregnancy test strips, the technology platform described may find use in simple and rapid detection assays for a range of oligonucleotide analytes.Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Y. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848–4870 CrossRef CAS PubMed.
- K. Gorska and N. Winssinger, Angew. Chem., Int. Ed., 2013, 52, 6820–6843 CrossRef CAS PubMed.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, R. K. O'Reilly and A. J. Turberfield, J. Am. Chem. Soc., 2012, 134, 1446–1449 CrossRef CAS PubMed.
- Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed.
- C. Zambaldo, S. Barluenga and N. Winssinger, Curr. Opin. Chem. Biol., 2015, 26, 8–15 CrossRef CAS PubMed.
- S. Barluenga and N. Winssinger, Acc. Chem. Res., 2015, 48, 1319–1331 CrossRef CAS PubMed.
- A. P. Silverman and E. T. Kool, Chem. Rev., 2006, 106, 3775–3789 CrossRef CAS PubMed.
- A. Shibata, H. Abe and Y. Ito, Molecules, 2012, 17, 2446–2463 CrossRef CAS PubMed.
- C. Percivalle, J. F. Bartolo and S. Ladame, Org. Biomol. Chem., 2013, 11, 16–26 CAS.
- J. Michaelis, A. Roloff and O. Seitz, Org. Biomol. Chem., 2014, 12, 2821–2833 CAS.
- S. Howorka, L. Movileanu, O. Braha and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12996–13001 CrossRef PubMed.
- U. Christensen, N. Jacobsen, V. K. Rajwanshi, J. Wengel and T. Koch, Biochem. J., 2001, 354, 481–484 CAS.
- D. Chang, E. Lindberg and N. Winssinger, J. Am. Chem. Soc., 2017, 139, 1444–1447 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and B. H. K. Stephen, Science, 1994, 266, 776–779 CAS.
- X. Liu, L. R. Malins, M. Roche, J. Sterjovski, R. Duncan, M. L. Garcia, N. C. Barnes, D. A. Anderson, M. J. Stone, P. R. Gorry and R. J. Payne, ACS Chem. Biol., 2014, 9, 2074–2081 CrossRef CAS PubMed.
- S. Ficht, A. Mattes and O. Seitz, J. Am. Chem. Soc., 2004, 124, 9970–9981 CrossRef PubMed.
- O. Vázquez and O. Seitz, J. Pept. Sci., 2014, 20, 78–86 CrossRef PubMed.
- O. Vazquez and O. Seitz, Chem. Sci., 2014, 5, 2850–2854 RSC.
- A. Roloff and O. Seitz, Chem. Sci., 2013, 4, 432–436 RSC.
- A. Erben, T. N. Grossmann and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 2828–2832 CrossRef CAS PubMed.
- T. N. Grossmann, L. Roglin and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 7119–7122 CrossRef CAS PubMed.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566–568 CrossRef CAS PubMed.
- N. J. Mitchell, L. R. Malins, X. Liu, R. E. Thompson, B. Chan, L. Radom and R. J. Payne, J. Am. Chem. Soc., 2015, 137, 14011–14014 CrossRef CAS PubMed.
- S. Pothukanuri, Z. Pianowski and N. Winssinger, Eur. J. Org. Chem., 2008, 2008, 3141–3148 CrossRef.
- N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049–7053 CrossRef CAS PubMed.
- C. C. Hanna, S. S. Kulkarni, E. E. Watson, B. Premdjee and R. J. Payne, Chem. Commun., 2017, 53, 5424–5427 RSC.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3–10 CrossRef CAS PubMed.
- V. Gubala, L. F. Harris, A. J. Ricco, M. X. Tan and D. E. Williams, Anal. Chem., 2012, 84, 487–515 CrossRef CAS PubMed.
- A. Warsinke, Anal. Bioanal. Chem., 2009, 393, 1393–1405 CrossRef CAS PubMed.
- S. Ficht, C. Dose and O. Seitz, ChemBioChem, 2005, 6, 2098–2103 CrossRef CAS PubMed.
- S. Valastyan and R. A. Weinberg, Cell Cycle, 2010, 9, 2124–2129 CrossRef CAS PubMed.
- E. J. Lee, M. Baek, Y. Gusev, D. J. Brackett, G. J. Nuovo and T. D. Schmittgen, RNA, 2007, 14, 35–42 CrossRef PubMed.
- L. N. Fix, M. Shah, T. Efferth, M. A. Farwell and B. Zhang, Cancer Genomics Proteomics, 2010, 7, 261–277 CAS.
- K. Gorska, I. Keklikoglou, U. Tschulena and N. Winssinger, Chem. Sci., 2011, 2, 1969–1975 RSC.
- S. Rapireddy, L. Nhon, R. E. Meehan, J. Franks, D. B. Stolz, D. Tran, M. E. Selsted and D. H. Ly, J. Am. Chem. Soc., 2012, 134, 4041–4044 CrossRef CAS PubMed.
- S. Thurley, L. Roglin and O. Seitz, J. Am. Chem. Soc., 2007, 129, 12693–12695 CrossRef CAS PubMed.
- T. Machida, S. Dutt and N. Winssinger, Angew. Chem., Int. Ed., 2016, 55, 8595–8598 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, compound characterisation, data for ligation reactions and miRNA detection. See DOI: 10.1039/c7sc02736b |
| ‡ During the course of this work, we noted that HPLC purified compounds (including selenoesters) were stable for up to two years in DMF solutions at −20 °C or as lyophilised solids. |
| This journal is © The Royal Society of Chemistry 2018 |