
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsperphenamate biosynthesis reveals a novel two-module NRPS system to synthesize amino acid esters in fungi†
Wei
Li‡
abc,
Aili
Fan‡
a,
Long
Wang
a,
Peng
Zhang
a,
Zhiguo
Liu
a,
Zhiqiang
An
d and
Wen-Bing
Yin
 *abc
*abc
aState Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, 100101 Beijing, China. E-mail: yinwb@im.ac.cn
bSavaid Medical School, University of Chinese Academy of Sciences, Beijing, 100049, China
cState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100050, China
dTexas Therapeutics Institute, The Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center at Houston, Houston, Texas 77030, USA
First published on 24th January 2018
Abstract
Amino acid esters are a group of structurally diverse natural products with distinct activities. Some are synthesized through an inter-molecular esterification step catalysed by nonribosomal peptide synthetase (NRPS). In bacteria, the formation of the intra-molecular ester bond is usually catalysed by a thioesterase domain of NRPS. However, the mechanism by which fungal NRPSs perform this process remains unclear. Herein, by targeted gene disruption in Penicillium brevicompactum and heterologous expression in Aspergillus nidulans, we show that two NRPSs, ApmA and ApmB, are sufficient for the synthesis of an amino acid ester, asperphenamate. Using the heterologous expression system, we identified that ApmA, with a reductase domain, rarely generates dipeptidyl alcohol. In contrast, ApmB was determined to not only catalyse inter-molecular ester bond formation but also accept the linear dipeptidyl precursor into the NRPS chain. The mechanism described here provides an approach for the synthesis of new small molecules with NRPS as the catalyst. Our study reveals for the first time a two-module NRPS system for the formation of amino acid esters in nature.
Introduction
Amino acid esters and their derivatives represent a huge group of pharmaceutically important natural products. These products include the cytotoxic asperphenamate (1),1 anticancer taxol,2 antibiotic daptomycin,3 and the recently identified antibiotic teixobactin, which kills bacteria with no detectable resistance (Fig. 1).41 is a rare linear amino acid ester derived from fungal nonribosomal peptide that exhibits antitumor activity towards a number of cell lines.5,6 It was first isolated from Aspergillus flavipes in 19777 and then identified from a wide range of Aspergillus8 and Penicillium species.9,10 The structure of 1 contains two subunits, N-benzoylphenylalanine (2) and N-benzoylphenylalaninol (3), which are connected by an inter-molecular ester bond. An experiment performed by feeding the labeled precursor [U–14C]-4 into P. brevicompactum proved that the benzoyl groups of 1 are derived from the precursor phenylalanine (4).1 However, no genetic study has been reported for the biosynthesis of 1. The motivation to study the biosynthesis of 1 is not only because of its biological activity but also because of the structural formation of the rare linear amino acid ester bond. The elucidation of the biosynthesis of 1 at the molecular level will provide an opportunity to produce 1 using an engineered microorganism. Moreover, it will provide new insights into the biosynthesis of related natural products. | ||
| Fig. 1 Representations of bioactive amino acid esters from nature: asperphenamate (1), taxol, daptomycin, and teixobactin. The ester bond connecting the subunit of each compound is highlighted in red. | ||
NRPSs usually have modular structures that are responsible for peptide elongation. Typically, an NRPS consists of an adenylation (A) domain, a thiolation (T) domain or PCP (peptidyl carrier protein), and a condensation (C) domain. A previous study demonstrated that bacterial NRPSs utilize thioesterase (TE) or C domains to perform esterification reactions.11 For example, the NRPS subunit DptD with a termination TE domain is involved in the biosynthesis of daptomycin in Streptomyces roseosporus. In this process, the TE domain is required for the formation of the daptomycin lariat structure and releasing of the peptide from the synthetase.3 SgcC5 is a free-standing C-domain. Interestingly, in Streptomyces globisporus this domain catalyses both C–O ester bond and C–N amide bond formations in the biosynthesis of antitumor antibiotic C-1027.12 In contrast, little is known about the mechanism by which NRPS catalyses ester bond formation in fungi. One example is FUM14p, an NRPS containing only PCP and C domains from Fusarium verticillioides. This NRPS catalyses the esterification of fumonisins instead of the typical amide bond formation. Therefore, characterisation of the esterification mechanism will provide useful knowledge for mycotoxin reduction and new insight into understanding the reactions catalysed by NRPS.13
Results and discussion
To investigate the biosynthetic pathway of 1, we worked on the 1 producing strain P. brevicompactum.1 The genome of this strain was sequenced in 2013. First, compound 1 was isolated from a 3 day culture of P. brevicompactum on YES medium by repeated column purification and semi-preparative HPLC (see ESI† for further details). 1 was elucidated by NMR identification and the data corresponded well to the previously published data.14 Analysing the structure of 1 showed that it consisted of two subunits, 2 and 3. Therefore, we postulated that an NRPS containing at least two A-domains was required for catalysing its formation. By searching the genome of P. brevicompactum, we found 73 biosynthetic gene clusters (BGCs), including 14 NRPSs. The NRPS domain prediction was performed using the service NRPSpredictor2.15 The prediction indicated that 6 NRPSs containing two A-domains matched our previous predictions. Interestingly, cluster 1 contained two NRPSs harbouring four A-domains. It was also possible that 1 needed four A-domains for the synthesis because its structure was not symmetric. To narrow down the NRPS target, we assessed their expressions in transcriptional levels by reverse transcriptase PCR (RT-PCR). Five NRPSs, NRPS1.1, NRPS1.2, NRPS9, NRPS10, and NRPS49 were normally expressed under the tested conditions. These NRPSs are indicated as possibly involved in the biosynthesis of 1 (Fig. S1†).To verify which NRPSs were involved in the biosynthesis of 1, we implemented a genetic deletion approach in P. brevicompactum. First, we examined the strain's antibiotic resistance to hygromycin (hph) and determined the growth inhibition concentration of 100 μg ml−1. Next, we made the protoplasts with modifications according to the previously reported method to improve the quality of the protoplasts.16,17 After that, the constructed NRPS deletion cassettes were transformed into P. brevicompactum protoplasts. Following the hygromycin selection and PCR verification, corrected transformants were created (Fig. S2†). HPLC analysis showed that 1 was completely abolished in both NRPS1.1 (renamed ApmA) and NRPS1.2 (renamed ApmB) deletion mutants. HPLC analysis also showed the essential roles of ApmA (KX443597) and ApmB (KX443596) in 1 biosynthesis (Fig. 2A). Therefore, these two NRPSs in cluster 1 (named the apm cluster) were shown to be involved in the biosynthesis of 1.
 | ||
| Fig. 2 Identification of asperphenamate (apm) biosynthetic gene cluster. (A) HPLC analysis of crude extracts from wild type (WT) strain (i) and apm deletion mutants (ii, iii): apmA and apmB deletion strains eliminated 1 production; ΔapmB accumulated 3 production (iii); feeding compound 3 into the apmA deletion strain restored 1 production (iv); traces v, vi show the pure compounds 1, 3. (B) Organization of apm biosynthetic genes in P. brevicompactum and their homologues in A. terreus and A. aculeatus. | ||
Next, we predicted the apm biosynthetic genes by bioinformatics analysis across the sequenced fungal genomes (JGI, http://genome.jgi.doe.gov). The apm cluster was conserved in the genomes of three fungi: P. brevicompactum, A. terreus, and A. aculeatus. The cluster consisted of two NRPS genes, apmA and apmB, one putative aldolase gene (apmC) and one putative epimerase/dehydratase gene (apmD) (Fig. 2B). The genes apmA-D from P. brevicompactum shared 68–80% sequence identities at the amino acid level with their orthologues from A. terreus; they shared 70–76% sequence identities with their orthologues from A. aculeatus (Table 1). Scanning the flanking regions of apmA-D, no biosynthetic genes were found on either side. Furthermore, no homologues were found in the flanking regions of A. aculeatus or upstream of A. terreus. Though there were three homologues in the down stream of A. terreus, they were putative kinesin-like or zinc finger proteins (Table 1). In addition, neither 1 nor its derivative was reported from A. terreus or A. aculeatus. These results indicated that four genes in the cluster are possibly sufficient for the synthesis of 1. To address the question of whether apmC and apmD are involved in the biosynthesis of 1, the same strategy was used for gene deletion, and the extracts from the deletion mutants were analysed (Fig. S2†). HPLC analysis showed that 1 still existed in both apmC and apmD mutants, indicating that they are not essential for the production of 1 (Fig. S3†). Taken together, these results demonstrate that apmA and apmB would be sufficient for the biosynthesis of 1.
| P. brevicom pactum | A. terreus | Coverage/identity | A. aculeatus | Coverage/identity | Putative function |
|---|---|---|---|---|---|
| ORF_06 | ATEG_09![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 071 071 |
— | ORF_07 | — | C6 zinc finger protein |
| ORF_07 | ATEG_09070 |
— | ORF_08 | — | RTA-like protein |
| ORF_08 | ATEG_09069 |
— | ORF_09 | — | HHE domain protein |
| apmA | ATEG_09068 |
99/71 | ORF_10 | 99/71 | NRPS |
| apmC | ATEG_09067 |
100/80 | ORF_11 | 100/75 | Phospho-2-dehydro-3-deoxyheptonate aldolase |
| apmD | ATEG_09065 |
39/80 | ORF_13 | 96/76 | NAD dependent epimerase |
| apmB | ATEG_09064 |
96/68 | ORF_14 | 94/70 | NRPS |
| ORF_14 | ATEG_09063 |
— | ORF_15 | 86/65 | Kinesin-like protein klpA |
| ORF_15 | ATEG_09062 |
— | ORF_16 | 99/72 | Coronin -binding protein |
| ORF_16 | ATEG_09061 |
— | ORF_17 | 90/45 | Zinc finger protein |
To probe the roles of ApmA and ApmB in the biosynthesis of 1, we identified the intermediates from the apm gene deletion mutants. By analysing ΔapmB strains, we found that a peak (named compound 3) was accumulated in 15.2 min (Fig. 2A, trace (iii)). We hypothesized that 3 is the product of ApmA. To identify the structure of 3, it was isolated by a scale-up fermentation of ΔapmB mutant. Its structure was elucidated as N-benzoylphenylalaninol by NMR and MS analysis (see ESI† data), which corresponded well with the previously published data.1 Feeding 3 into the ΔapmA mutant restored 1 production, which confirmed that 3 was a product of ApmA (Fig. 2A, trace iv). It is rare for an NRPS to generate an alcohol. Using antiSMASH analysis, we identified ApmA as an unusual NRPS module consisting of A-T-C-A-T-R with a C-terminal reductase (R)-domain.18 Several studies have demonstrated the function of the R-domain in natural product biosynthesis.19–23 For example, Cox and co-workers revealed that the R domain of PKS-NRPS TENS functions as a Dieckmann cyclase in the formation of tenellin.24 Walsh and Liu demonstrated the role of the R-like domain of PKS-NRPS CpaS for releasing the product in the biosynthesis of cyclopiazonic acid.25 Recently, the R domain in PKS-NRPS was identified as an aryl-aldehyde generator from an aryl-acid in the formation of meroterpenoid LL-Z1272β and 2,4-dihydroxy 5,6-dimethyl benzaldehyde.26,27 Differences of R-domains in NRPSs from bacteria and fungi were demonstrated by a phylogenetic tree analysis (Fig. S4†). Thus, the hypothesis is that the R domain acts in the reductive release of the shunt product 3 in the two-modular NRPS ApmA. 4 and benzoic acid (5) are the possible substrates of ApmA.
To confirm this, apmA was expressed in both Saccharomyces cerevisiae and A. nidulans systems.17,28 HPLC analysis of ApmA-expressing A. nidulans showed a new peak at 12.2 min. This peak was isolated and the structure was elucidated as 3. The final titer was 20 mg L−1 by the scale-up fermentation using 4 and 5 as substrates (Fig. 3A and ESI†). LC-MS analysis of crude extracts from apmA expressed strains in yeast and in A. nidulans revealed its molecular weight as 256.1395 [M + H]+, confirming 3 production (Fig. S5†). Two additional peaks were also observed with masses of 270.2131 [M + H]+ and 254.0875 [M + H]+ by LC-MS analysis. These molecular weights suggested the formations of the acid form of 2 (molecular formula C16H15NO3, calculated molecular weight 269.1052) and the aldehyde form of N-benzolphenylalaninal (6, molecular formula C16H15NO2, calculated molecular weight 253.1103) (Fig. S5†). These data indicate that ApmA catalyses the carrier protein-bound thioester-intermediate, releasing it from the acid form of 2 to the primary alcohol through an aldehyde intermediate 6. Since ApmA harbored an R domain in the C-terminal, we assumed that it had a reduction function. To test this hypothesis, we fed the chemically synthesized 2S-N-acetylcysteamine (2-SNAC) into ApmA-expressing A. nidulans. 3 was detected by LC-MS analysis (Fig. S6†). Our data confirmed that ApmA is a N-benzoylphenylalaninol (3) synthetase using 4 and 5 as substrates and revealed for the first time a two-module NRPS system that acts in the reductive release in the biosynthesis of natural product.
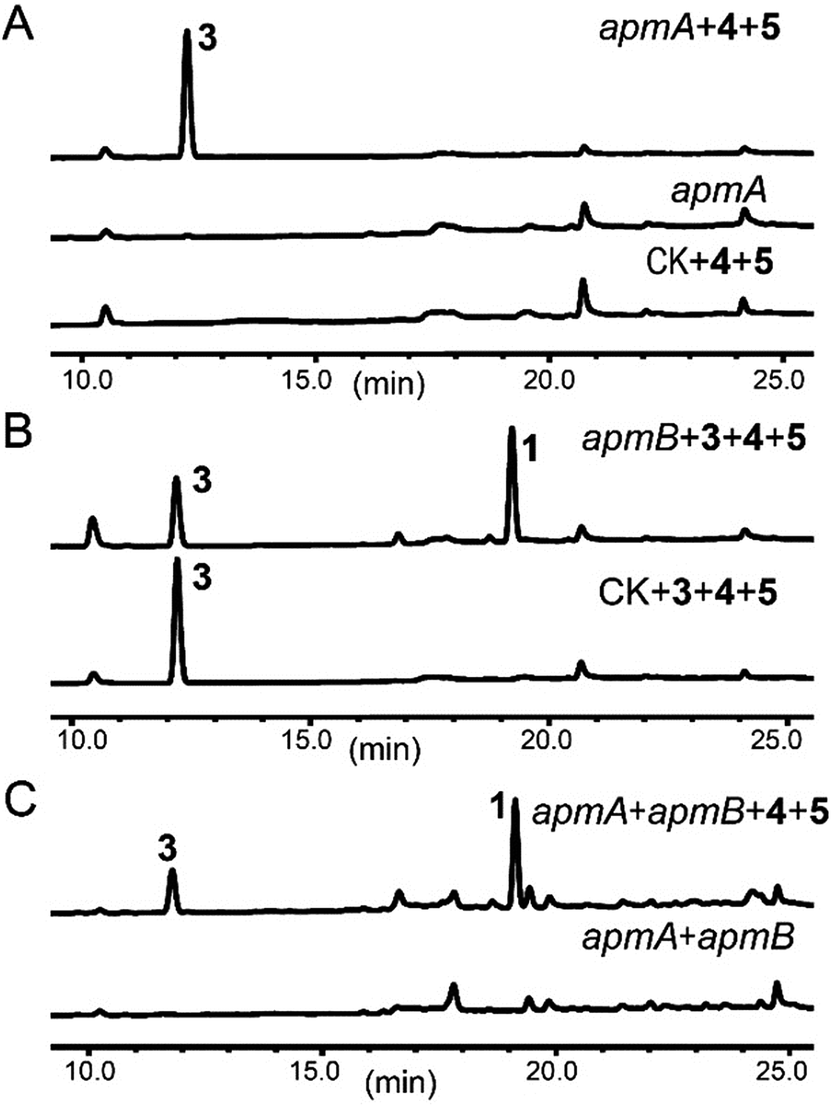 | ||
| Fig. 3 Roles of ApmA and ApmB in the biosynthesis of asperphenamate. ApmA and/or ApmB were expressed in A. nidulans. (A) HPLC analysis of metabolites produced from ApmA-expressing strains with feeding of precursors 4 and 5. (B) HPLC analysis of metabolites produced from ApmB-expressing strains with feeding of precursors 3, 4 and 5. (C) 1 was obtained by co-expressions of ApmA and ApmB. | ||
Next, we assessed the role of ApmB by LC-MS analysis of its expression mutants in both S. cerevisiae and A. nidulans, but no product was detected (data not shown). Bioinformatic analysis showed that ApmB is a regular two modular NRPS with six domains, A-T-C-A-T-C. Analysing the structure of 1, we proposed that ApmB may activate the same substrates as does ApmA, 4 and 5, then we performed a two-step cascade reaction: tethering the linear N-benzolphenylalaninepeptidyl to the ApmB chain and waiting for the attack of 3 before moving to the next step.29 Accordingly, we fed the proposed substrates 3, 4, and 5 into ApmB-expressing strains along with the control strains under the same conditions. Notably, HPLC analysis demonstrated that the peak of 3 decreased, while a new peak with the same retention time and UV spectra as 1 appeared in the ApmB transformant (Fig. 3B). LC-MS analysis of the extracts further confirmed that the compound was 1 (molecular formula C32H30N2O4, measured molecular weight 507.2293 [M + H]+, calculated 506.2206) (Fig. S7†). The feeding of 4 in the culture media greatly increased the yield of 1 (Fig. S8†). Considering all results, we concluded that ApmB acts in the ester bond formation and release of the final product 1. Since substrate 5 is a product of primary metabolism, the biosynthesis of 5 in fungi is rarely reported. One case is the squalestatin biosynthesis in Phoma sp. C2932 using benzoic acid as the substrate. In this example, biosynthesis may have been catalysed by a gene encoding phenylalanine ammonia lyase (PAL) mfM7 as the first step of 4 degradation.30 Searching the P. brevicompactum genome with M7 as a probe, we found the homologue of PAL gene without annotation (identity of 39.0% in amino acid). Interestingly, the PAL encoding gene was not located in the apm cluster. It may be involved in benzoate production.
Because of the unusual function of ApmB, it is logical to ask whether ApmB can use linear dipeptides such as 2 and 3 as substrates. This question was addressed by feeding 2 and 3 into the apmB-expressing A. nidulans strain. 1 was detected by LC-MS analysis (Fig. 4). To exclude the residual 4 in the medium as substrate, 13C,15N-2 (Fig. S10–S11†) was synthesized chemically and used as a substrate for the feeding experiments. LC-MS analysis of the crude extract clearly demonstrated that 13C,15N-1 was the major product with a determined molecular weight of 509.2282 [M + H]+ (Fig. S9†). To characterise the structure of 13C,15N-1, large scale fermentations were conducted by feeding labelled 13C,15N-2 and 3 as substrates, and the structure was elucidated by NMR analysis (Fig. S12–S13†). This result suggested that ApmB activates and accepts the linear dipeptide 2 as a substrate. Finally, we coexpressed apmA and apmB genes in A. nidulans. A clear final product peak 1 was observed by feeding the substrates of 4 and 5 in the transformant (Fig. 3C).
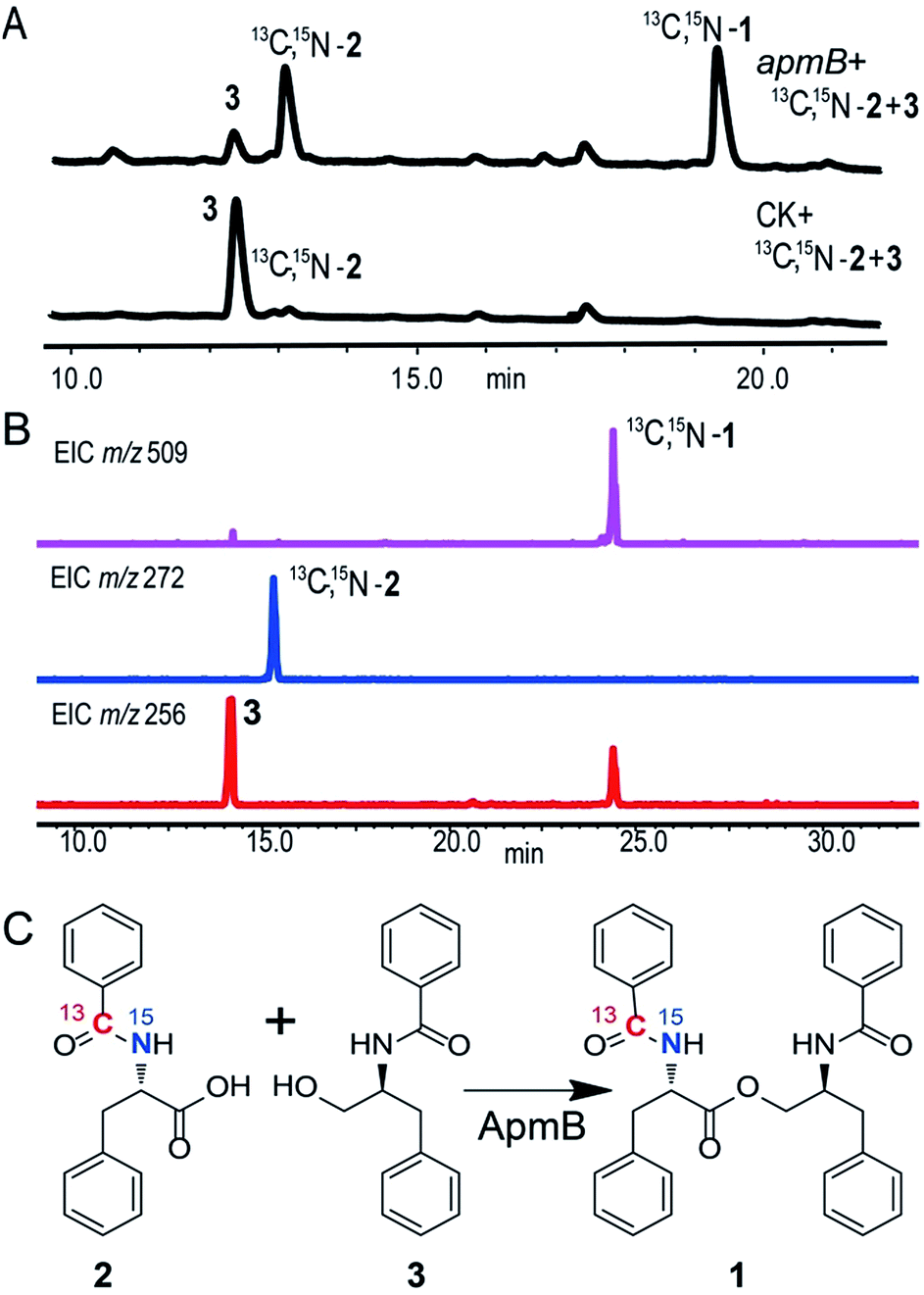 | ||
| Fig. 4 ApmB utilizes linear dipeptidyl precursor to synthesize asperphenamate. (A) HPLC analysis of metabolites produced from ApmB expressing-strain with feeding 13C,15N-2 and 3. 13C,15N-1 was obtained. (B) LC-MS confirmation of labeled 13C,15N-2 integration into final product of 1. (C) ApmB-catalysed reactions by using 13C,15N-2 and 3 as substrates. Labelled 13C,15N-1 was characterised by NMR and MS analysis. | ||
Based on the gene disruption results and feeding studies in heterologous hosts, we proposed a biosynthetic pathway for 1 (Fig. 5). Using 4 and 5 as substrates, ApmA catalysed amide bond formation and tethered the intermediate into the NRPS chain. Then, the terminal R domain of ApmA catalysed the reduction reaction to get the shunt product 3 through the acid form 2 and the aldehyde form 6 (Fig. 5). Subsequently, ApmB activated the same substrates as did ApmA and waited for the attack of 3. After the attack of 3 on the peptidyl-S-N-benzoylphenylalanine, the inter-molecular ester bond was formed and the final product 1 was released by ApmB (Fig. 5). The whole process was completed by the coordination of two NRPSs, ApmA and ApmB. This is the first case of a simple compound formation by the machinery of two NRPSs shown in nature.
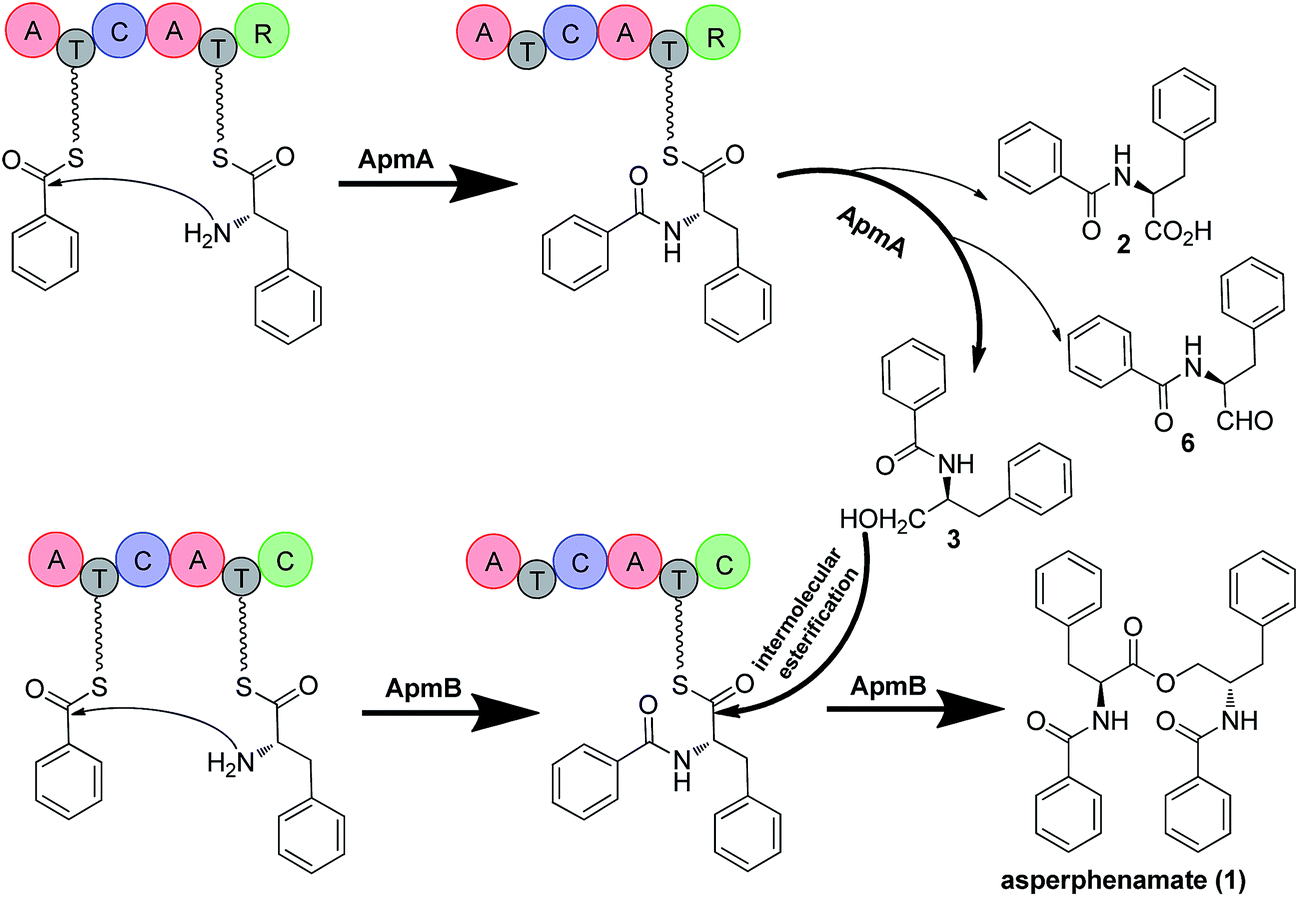 | ||
| Fig. 5 Proposed biosynthetic pathway of 1 and mechanism for reduction and esterification steps in P. brevicompactum. Bold arrow indicates the main steps of ApmA and ApmB coordination for the formation of 1. 2, 6 are marked by light arrows indicating that ApmA catalyses the formation of 3 through reduction steps. | ||
Conclusions
In summary, we identified the biosynthetic gene cluster of asperphenamate (1) and characterised the two NRPSs function using a heterologous expression approach in A. nidulans. We demonstrated that the NRPS enzyme ApmA had an R-domain, which was responsible for the formation of alcohol reduced from the acid through the aldehyde intermediate. Meanwhile, we also elucidated another NRPS enzyme, ApmB, with functions for ester bond formation and the release from the synthetase to form the final product. More interestingly, ApmB was able to accept the linear dipeptide to form the final product 1. Therefore, our findings not only provide new insights into fungal NRPS machinery for the formation of amino acid esters, but also demonstrate a synthetic approach to develop new small molecules catalysed by NRPS.Materials and methods
The strains, plasmids and primers used in this work are described in the ESI Tables S1, S2 and S3.† The HPLC analyses of the feeding experiment in apmB-expressed A. nidulans are listed in the ESI Fig. S7 and S8.† The LC-MS spectra of feeding experiment in apmB-expressed A. nidulans with 13C,15N-2 and 3 as substrates are listed in the ESI Fig. S9.† The NMR spectra for compound 13C, 15N-1, 13C,15N-2, 3 and 2-SNAC are listed in the ESI Fig. S10–S15†. Detailed descriptions of experimental procedures and reagents are provided in detail in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Drs Jinwei Ren, Wenzhao Wang and Haining Lyu (Institute of Microbiology, CAS) for NMR and MS data collection. We thank Dr Xuebing Li for generously providing N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride and 1-hydroxysuccinimide. We thank Dr Georgina Salazar at the University of Texas Health Science Center at Houston for her careful and critical reading of the manuscript. This work was supported by the National Natural Science Foundation of China (31470178 to W. B. Y. and 81502964 to Z. L.) and the State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College (Grant GTZK201506), and the Welch Foundation (Grant AU00024 to Z. A.). W. B. Y. is a scholar of “the 100 Talents Project” of CAS.References
- D. L. Doerfler, B. A. Bird and I. M. Campbell, Phytochemistry, 1981, 20, 2303–2304 CrossRef CAS.
- R. Croteau, R. E. B. Ketchum, R. M. Long, R. Kaspera and M. R. Wildung, Phytochem. Rev., 2006, 5, 75–97 CrossRef CAS PubMed.
- V. Miao, M. F. Coëffet-Legal, P. Brian, R. Brost, J. Penn, A. Whiting, S. Martin, R. Ford, I. Parr, M. Bouchard, C. J. Silva, S. K. Wrigley and R. H. Baltz, Microbiology, 2005, 151, 1507–1523 CrossRef CAS PubMed.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- Y. Li, Q. Luo, L. Yuan, C. Miao, X. Mu, W. Xiao, J. Li, T. Sun and E. Ma, Toxicol. Appl. Pharmacol., 2012, 263, 21–31 CrossRef CAS PubMed.
- L. Yuan, Y. Li, C. Zou, C. Wang, J. Gao, C. Miao, E. Ma and T. Sun, Bioorg. Med. Chem. Lett., 2012, 22, 2216–2220 CrossRef CAS PubMed.
- A. M. Clark, C. D. Hufford and L. W. Robertson, Lloydia, 1977, 40, 146–151 CAS.
- C. J. Zheng, C. L. Shao, L. Y. Wu, M. Chen, K. L. Wang, D. L. Zhao, X. P. Sun, G. Y. Chen and C. Y. Wang, Mar. Drugs, 2013, 11, 2054–2068 CrossRef PubMed.
- J. C. Frisvad, J. Smedsgaard, T. O. Larsen and R. A. Samson, Stud. Mycol., 2004, 49, 201–241 Search PubMed.
- J. C. Frisvad, J. Houbraken, S. Popma and R. A. Samson, FEMS Microbiol. Lett., 2013, 339, 77–92 CrossRef CAS PubMed.
- R. M. Kohli and C. T. Walsh, Chem. Commun., 2003, 297–307 RSC.
- S. Lin, S. G. Van Lanen and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4183–4188 CrossRef CAS PubMed.
- K. Zaleta-Rivera, C. Xu, F. Yu, R. A. E. Butchko, R. H. Proctor, M. E. Hidalgo-Lara, A. Raza, P. H. Dussault and L. Du, Biochemistry, 2006, 45, 2561–2569 CrossRef CAS PubMed.
- J. L. Songue, Kouam, E. Dongo, T. N. Mpondo and R. L. White, Molecules, 2012, 17, 13673–13686 CrossRef CAS PubMed.
- M. Rottig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef PubMed.
- Z. Ren, C. Su, J. Yan, M. Dai, Y. Zhao, H. Wang, M. Dai and J. Zhang, Acta Microbiol. Sin., 2013, 53, 1226–1232 CAS.
- W. B. Yin, Y. H. Chooi, A. R. Smith, R. A. Cacho, Y. Hu, T. C. White and Y. Tang, ACS Synth. Biol., 2013, 2, 629–634 CrossRef CAS PubMed.
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS PubMed.
- B. Silakowski, B. Kunze, G. Nordsiek, H. Bloecker, G. Hoefle and R. Mueller, Eur. J. Biochem., 2000, 267, 6476–6485 CrossRef CAS PubMed.
- J. F. Barajas, R. M. Phelan, A. J. Schaub, J. T. Kliewer, P. J. Kelly, D. R. Jackson, R. Luo, J. D. Keasling and S. C. Tsai, Chem. Biol., 2015, 22, 1018–1029 CrossRef CAS PubMed.
- M. Wang, M. Beissner and H. Zhao, Chem. Biol., 2014, 21, 257–263 CrossRef CAS PubMed.
- D. E. Ehmann, A. M. Gehring and C. T. Walsh, Biochemistry, 1999, 38, 6171–6177 CrossRef CAS PubMed.
- L. Li, W. Deng, J. Song, W. Ding, Q. F. Zhao, C. Peng, W. W. Song, G. L. Tang and W. Liu, J. Bacteriol., 2008, 190, 251–263 CrossRef CAS PubMed.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS PubMed.
- X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 8746–8757 CrossRef CAS PubMed.
- M. Wang, M. Beissner and H. Zhao, Chem. Biol., 2014, 21, 257–263 CrossRef CAS PubMed.
- C. Li, Y. Matsuda, H. Gao, D. Hu, X. S. Yao and I. Abe, ChemBioChem, 2016, 17, 904–907 CrossRef CAS PubMed.
- Y. M. Chiang, M. Ahuja, C. E. Oakley, R. Entwistle, A. Asokan, C. Zutz, C. C. Wang and B. R. Oakley, Angew. Chem., Int. Ed. Engl., 2015, 54, 1–5 CrossRef.
- W. B. Yin, J. A. Baccile, J. W. Bok, Y. Chen, N. P. Keller and F. C. Schroeder, J. Am. Chem. Soc., 2013, 135, 2064–2067 CrossRef CAS PubMed.
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. M. Lazarus, A. M. Bailey, T. J. Simpsonc and R. J. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc02396k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |