
New ultrasonic assisted co-precipitation for high surface area oxide based nanostructured materials†
 *ab
and
Ricardo H. R.
Castro
*a
*ab
and
Ricardo H. R.
Castro
*a
Abstract
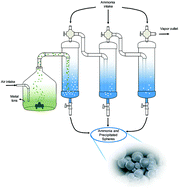
Owing to their enhanced properties as compared to bulk materials, the prospective applications for nanomaterials have experienced unprecedented growth, gaining attention from all levels of industry, from medical to electronics, chemistry, catalysis and mechanics. However, one of the greatest challenges of the nanomaterial industry lies in developing a production system that assures low cost and high production capabilities while maintaining quality standards. Here, we show a new method for the synthesis of metal oxide nanoparticles based on an aqueous precipitation method. The system makes use of ultrasonic probes and continuous precipitation chambers which allow it to operate continuously. Catalyst support materials, such as MgAl2O4 and γ-Al2O3, were synthesized showing high BET surface areas of 338.61 and 366.10 m2 g−1, hollow spherical morphologies and crystallite sizes as small as 3.2 and 2.1 nm, respectively.
 Please wait while we load your content...
Please wait while we load your content...

