
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDelivery of interleukin-22 binding protein (IL-22BP) gene by cationic micelle for colon cancer gene therapy
Ke Men†
 *a,
Rong Huang†a,
Xueyan Zhanga,
Rui Zhanga,
Yuanfa Zhanga,
Yao Penga,
Rongsheng Tongb,
Li Yanga,
Yuquan Weia and
Xingmei Duan*ab
*a,
Rong Huang†a,
Xueyan Zhanga,
Rui Zhanga,
Yuanfa Zhanga,
Yao Penga,
Rongsheng Tongb,
Li Yanga,
Yuquan Weia and
Xingmei Duan*ab
aState Key Laboratory of Biotherapy and Cancer Center/Collaborative Innovation Center for Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, People's Republic of China. E-mail: mendingbob@hotmail.com; duanxingmei2003@163.com
bIndividualized Medication Key Laboratory of Sichuan Province, Department of Pharmacy, Sichuan Provincial People's Hospital, Chengdu, 610072, People's Republic of China
First published on 4th May 2018
Abstract
Gene therapy has provided an alternative strategy for cancer therapy. As an important cytokine, interleukin-22 (IL-22) is not only critical in reinforcing innate immune defenses and tissue regeneration, but also involved in the initial establishment of tumors. A soluble-secreted receptor of the cytokine IL-22, IL-22 binding protein (IL-22BP), binds IL-22 and prevents its binding to the functional transmembrane receptor IL-22R1 complex, inhibiting IL-22-based intracellular cancer proliferation signal. In this work, a novel IL-22BP-based cancer gene therapy strategy was reported for the first time. It was established by delivering IL-22BP gene with a newly developed non-viral gene vector DMP. The DMP cationic micelles were prepared by modifying monomethoxy poly(ethylene glycol)-poly(ε-caprolactone) with DOTAP lipid through self-assembling. The anti-cancer efficacy of the DMP/IL-22BP complex was studied on a colon cancer model by intraperitoneal administration. Our results demonstrated that the secretory expressed IL-22BP cytokine effectively inhibited cancer growth both in vitro and in vivo. Multiple anti-cancer mechanisms including IL-22 blocking, apoptosis inducing, lymphocyte infiltration and angiogenesis inhibition were indicated to be involved while no pathology changes were observed in healthy tissues. These results suggest the DMP/IL-22BP complex to be a potential candidate for cancer gene therapy.
Introduction
Cancer is one of the leading causes of death in both economically developed and developing countries.1 Colon carcinoma is the second most common cause of death among cancers.1,2 Except for conventional treatment methods including surgery, radiotherapy and chemotherapy, gene therapy has provided an alternative method for colon cancer treatment.3,4 Facilitated by various gene delivery systems, cancer gene therapy has been successfully applied through repairing, interfering or replacing target gene or gene product.5,6 Some products have successfully reached clinical stage with high efficacy and safety.4,5,7,8 However, novel therapeutic targets as well as gene delivery vectors with high efficacy and safety are still in high demand.Interleukin-22 binding protein (IL-22BP) is a soluble-secreted receptor of the cytokine IL-22, which binds IL-22 and prevents its binding to the functional cell surface receptor IL-22R1 complex.9–14 Produced by T cells and innate lymphoid cells, IL-22 is critical in reinforcing innate immune defenses and tissue regeneration through pleiotropic effects including pro-survival signaling, cell migration, dysplasia and angiogenesis.9,10,12,15 However, numerous studies have also demonstrated that uncontrolled IL-22 can be a potent inducer of pathological inflammation.10 Indeed, IL-22 can promote tissue inflammation and self-destruction16–18 and is involved in the pathophysiology of several immune-mediated inflammatory diseases, such as psoriasis, intestinal pathogenesis and rheumatoid arthritis.10,17 Thus, IL-22BP exerts this control by specifically neutralize IL-22 activity, suggesting that IL-22BP acts as a natural regulator and preventing exaggerated effects of IL-22 (ref. 9). This biological inhibiting effect of IL-22BP has been testified both in vitro9,19–21 and in vivo,21 and ideal therapeutic effects have been evaluated in the treatment of inflammatory diseases such as chronic inflammatory bowel disease11 and chronic liver diseases.22
On the other hand, IL-22 is also involved in the initial establishment of tumors. By binding with its transmembrane receptor IL-22R1, IL-22 signal activates several pathways including signal transducers and activators of transcription (STATs) 1/3/5, nuclear factor kappa B (NF-κB), mitogen-activated protein kinase (MAPK) and phosphatidylinositide 3-kinase, Akt mammalian target of rapamycin (PI3K-Akt-mTOR).15,16,23,24 Particularly, activation of STAT3 induces expression of a medley of genes principally downstream, resulting in pro-inflammatory, anti-inflammatory, mitogenic, pro-survival, anti-apoptotic or anti-fibrotic effects depending on specific context.15,25 These properties can possibly be hijacked by aggressive cancers to enhance tumor growth and metastasis.15 Given that the critical roles of these pathways such as STAT3 and NF-κB in the development of many tumors,25–28 the IL-22/IL-22R1 axis has attracted great attention as a driver of cancer progression. To this end, IL-22BP is supposed to play a key role in the regulation process. Nevertheless, it has been reported that IL-22BP production is prominent in the steady state except inflammatory conditions, lacking enough intensity for tumor inhibition.10 Therefore, replenishing exogenous IL-22BP and neutralizing IL-22 in tumor microenvironment seems to be a novel and practical strategy for cancer gene therapy.
Appropriate delivery system is also critical for gene therapy. Ideal vectors would contribute to therapeutic efficiency and safety.29 In our previous work, a novel non-viral gene delivery system was developed by one-step modifying monomethoxy poly(ethylene glycol)-poly(ε-caprolactone) (mPEG-PCL, MP) micelles with amphiphilic cationic lipid DOTAP, producing a hybrid cationic micelle (DMP).30 The prepared DMP micelle demonstrated high gene delivery ability with safety and has been successfully applied in anti-cancer studies.30,31 However, our previous works focused on delivering apoptosis inducing gene into cancer cells to trigger “suicide” signals, while secretly expression of extracellular proteins by DMP delivery has never been evaluated. Thus, the application of DMP micelle system was largely restricted. In this work, we established an IL-22BP-based therapy strategy by delivering IL-22BP gene with DMP micelles. We attempt to evaluate the anti-cancer effect of IL-22BP gene on colon cancer model through inhibition of IL-22 signal. We assumed that intentionally expression and secretion of IL-22BP into tumor environment could effectively block the signaling of IL-22/IL-22R1, resulting in tumor growth inhibition. To our best knowledge, this is the first report of IL-22BP-based cancer gene therapy study. The in vivo delivery capacity and safety of DMP/IL-22BP complex through local administration way was characterized. Moreover, the possible mechanisms including anti-proliferation, anti-angiogenesis and apoptosis inducing will be further studied.
Methods
Materials
Monomethoxy poly(ethylene glycol)-poly(ε-caprolactone) polymers (MP, average Mw = 4000) were synthesized in our lab previously.32 DOTAP were purchased from Avanti Polar Lipids (Alabaster, AL). Branched polyethylenimine (PEI, average molecular weight 25 kDa), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased Sigma-Aldrich (St Louis, MO). All other chemicals were purchased from Sigma-Aldrich unless otherwise mentioned. OptiMem®, Dulbecco's Modified Eagle's Medium (DMEM) (ATCC® 30-2002™) and serums were purchased from Thermo Fisher Scientific. C26 Mus musculus colon carcinoma cell line (ATCC® CRL-2638™) and 293t human embryonic kidney cell line (ATCC® CRL-3216™) were purchased from the American Type Culture Collection (ATCC). BALB/c mice were obtained from Beijing HFK Bio-technology Co. Ltd. (Beijing, China) and maintained under specific pathogen-free conditions. All animal procedures were approved and controlled by the Institutional Animal Care and Treatment Committee of Sichuan University and carried out according to the Animal Care and Use Guidelines of Sichuan University.Plasmid construction
The plasmid pVAX1-mIL22BP expressing murine IL-22BP was constructed by cloning IL-22BP coding sequence into pVAX1 vector. Murine IL-22BP cDNA (Origene, USA) was amplified (forward: GGG AAA GCT TAT GAT GCC TAA GCA TTG CCT TCT AGG, reverse: GGG ATC TAG ATC ATG GAA TGT GCA CAC ATC TCT CC) and digested with Hind III and XbaI. pVAX1 plasmid was used as the empty vector. All plasmids were propagated in E. coli and purified by an EndoFree Plasmid Giga kit (Qiagen, Chatsworth, CA).Preparation and characterization of DMP micelles
DOTAP modified mPEG-PCL micelles (DMP) were prepared according our previous reports.30 Briefly, 45 mg of MPEG-PCL polymer and 5 mg DOTAP were co-dissolved in methylene dichloride (KeLong Chemicals, Chengdu, China) and solvent was removed under rotary evaporation for 45 min. The lipid film was re-hydrated with distilled water under 55 °C to self-assemble the micelles with a final concentration of 10 mg mL−1. The prepared micelles were stored at 4 °C for future use. The size and surface charge of prepared DMP micelles were determined by Malvern ZS90 (Malvern, Worcestershire, UK). Measurements were performed at 25 °C after equilibration. All results were the mean of three test runs. The morphology of DMP micelles were observed under a transmission electron microscope (TEM) (H-6009IV, Hitachi, Japan).Gene retarding assay
The DNA binding ability of DMP micelle was evaluated by agarose retarding assay. The DMP micelle/plasmid complex were electrophoresed on 1% (w/v) agarose gel for 30 min at 100 V. In each well, 1 mg of plasmid was mixed with different ratios of DMP micelles. Gel was then stained with ethidium bromide (0.5 mg mL−1) and illuminated by a UV illuminator (Bio-Rad ChemiDox XRS, USA).Cytotoxicity assays
293t cell line was used as healthy tissue cells to evaluate the cytotoxicity of DMP micelles. The study was established by MTT assay. Briefly, 293t cells were seeded into a 96-well plate at a density of 5 × 103 cells per well in 0.1 mL of complete medium (DMEM containing 10% FBS) and incubated for 24 h. The cells were then exposed to a series of DMP micelles or PEI25K with different concentrations for 24 h, and the viability of cells was measured using an MTT assay. Briefly, 20 μL of MTT solution was added to each well and incubated at 37 °C for 4 hours. The formazan was solubilized by adding 200 μL DMSO and shaken at room temperature for 30 minutes. The absorbance was read at 570 nm by the Spectramax M5 Microtiter Plate Luminometer (Molecular Devices, USA). Absorbance of untreated cells was considered as 100%.In vitro transfection
24 hours before transfection, 293t or C26 cells were seeded into a 24-well plate at a density of 1 × 104 cells per well in 0.5 mL of complete medium (DMEM containing 10% FBS). GFP encoding plasmid was used as a reporter gene. Particle equivalent to 1 μg of EGFP plasmid was added to each well in the presence of OptiMEM medium. The mass ratio of DMP/pGFP and PEI25K/GFP were 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1. The medium was then replaced with complete medium 4 hours post-transfection. 12 hours or 24 hours later, pictures of each well were taken under microscope and the transfection efficiency was determined by flow cytometry (NovoCyte Flow Cytometer, ACEA Biosciences, USA).
:1 and 2:1. The medium was then replaced with complete medium 4 hours post-transfection. 12 hours or 24 hours later, pictures of each well were taken under microscope and the transfection efficiency was determined by flow cytometry (NovoCyte Flow Cytometer, ACEA Biosciences, USA).
Real-time PCR
To determine the intracellular level of murine IL-22BP mRNA, total RNA was extracted from C26 cells or tumor samples using TRIzol™ Reagent (Thermo Fisher Scientific, USA) and individual cDNAs were synthesized with a SuperScript II reverse transcriptase assay (Sigma-Aldrich). Real-time quantitative PCR was performed with a SYBR GreenER quantitative PCR SuperMix Universal kit (Sigma-Aldrich). Reactions were run with a standard cycling program: 50 °C for 2 minutes, 95 °C for 10 minutes, 40 cycles of 95 °C for 15 seconds, and 60 °C for 1 minutes, on an AB7500 real-time PCR system (Applied Biosystems, Foster City, CA). The PCR primers to detect IL-22BP (forward: CCA GAA GG TCC GAT TTC AGT C, reverse: GCA GTC AAC TTT ATC TTC CCA TTG) and GAPDH (forward; 5′-ATG GGG AAG GTG AAG GTC G-3′, reverse; 5′-TAA AAG CAG CCC TGG TGA CC-3′) were synthesized and purified by TSINGKE Biological Technology (Chengdu, P. R. China).ELISA assay
The level of expressed IL-22BP in cell culture supernatant was determined through ELISA assay. C26 cells were seeded into a 24-well plate at a density of 1 × 104 cells per well in 0.5 mL of complete medium (DMEM containing 10% FBS). Particle equivalent to 1 μg of pVAX1-mIL22BP plasmid was added to each well in the presence of OptiMEM medium (DMP:plasmid = 10:1, w/w). The medium was then replaced with full medium 4 hours post transfection. 72 hours post transfection, cell culture supernatant from each well was collected and measured using a mouse Mouse Interleukin-22 receptor subunit alpha-2 (IL22RA2) ELISA kit (CUSABIO Life science, TX) according to manufacturer's instructions.
Anti-proliferation assay
C26 cells were seeded into a 96-well plate with a density of 1 × 104 cells per well. After transfection with DMP/mIL-22BP complex (0.5 μg DNA per well), cells were subjected to MTT cell proliferation assay 72 hours post-transfection. After incubation, 20 μL of MTT solution was added to each well and incubated at 37 °C for 4 hours. The formazan was solubilized by adding 200 μL DMSO and shaken at room temperature for 30 minutes. The absorbance was read at 570 nm by the Spectramax M5 Microtiter Plate Luminometer (Molecular Devices, USA). Absorbance of untreated cells was considered as 100%.For IL-22 blocking assay, C26 cells were seeded into a 96-well plate with a density of 1 × 103 cells per well and cultured with complete DMEM medium overnight. 2 hours before transfection, cells were pre-incubated with recombinant mouse IL-22 protein (R&D Systems, MN) at a final concentration of 100ng mL−1. Cells either transfected with DMP/mIL-22BP complex (0.5 μg DNA per well) or not were then subjected to MTT cell proliferation assay 72 hours post-transfection. After incubation, 20 μL of MTT solution was added to each well and incubated at 37 °C for 4 hours. The formazan was solubilized by adding 200 μL DMSO and shaken at room temperature for 30 minutes. The absorbance was read at 570 nm by the Spectramax M5 Microtiter Plate Luminometer (Molecular Devices, USA). Absorbance of IL-22 protein untreated cells was considered as 100%.
Clonogenic assay
DMP/mIL-22BP complex equivalent to 0.5 μg of pIL-22BP was administered to 1 × 103 C26 cells seeded in 6-well plate. 4 hours post-transfection, medium was refreshed with complete DMEM culture medium. The cells were continuing cultured for 2 weeks to form colonies. Colonies were washed with PBS for two times before stained with 10% crystal violet blue for 15 minutes. This assay was repeated for three times and the number of clones as well as inhibition rate in each well were then calculated.In vitro apoptosis assay
The cell apoptosis inducing ability of DMP/mIL-22BP complex was investigation by flow cytometry. C26 cells were pre-seeded into a 6-well plate with a density of 5 × 104 cells per well. After transfection with DMP/mIL-22BP complex (0.5 μg plasmid per well), DMP/pVAX1 complex (0.5 μg plasmid per well), normal saline (NS) and null DMP micelle (in equivalent amount with related complex) separately for 4 hours, the medium was replaced by complete medium. 72 hours later, cells were stained with propidium iodide and Annexin V-FITC (Sigma-Aldrich). The apoptotic cancer cells were measured by flow cytometry (NovoCyte Flow Cytometer, ACEA Biosciences, USA).In vivo tumor inhibition assay
The anti-cancer ability of DMP delivered IL-22BP gene was evaluated on C26 mouse colon carcinoma abdominal cavity metastatic model. Briefly, BALB/c mice of 6–8 weeks old were intraperitoneally injected with 1 × 105 C26 cells. On day 3, mice were randomized into 4 groups (5 mice per group) and numbered. DMP/DNA complexes equivalent to 5 μg of pVAX1-mIL22BP plasmid was prepared as aforementioned were injected intraperitoneally every day for 7 treatments. Mice receiving equivalent normal saline, DMP micelles or DMP/pVAX1 complex were regarded as control group. On day 20, all mice were sacrificed by cervical vertebra dislocation, and their tumors were immediately harvested, weighed, and analyzed. The volumes of ascites in each group were also measured and collected.Histological analysis
Tumor tissue harvested from in vivo inhibition studies and were fixed and embedded in paraffin. Wax-embedded tissue sections were dewaxed and rehydrated before immunohistochemistry staining. To analyze apoptotic cells within tumor tissues, sections were stained with DeadEnd™ Fluorometric TUNEL System kit (Promega) according to the manufacturer's manual. The fluorescent image from each group was acquired through a fluorescence microscope (Olympus, Japan). For IL-22BP, CD31, CD8 and CD4 staining, tumor sections were blocked and subsequently incubated with corresponding rabbit anti-mouse primary antibodies (Abcam, USA) at 4 °C overnight. Appropriate horseradish peroxidase-conjugated secondary antibody was then applied. The micro-vessel density was visualized and determined through a fluorescence microscope (Olympus, Japan).Statistical analysis
Data were expressed as the means with 95% confidence intervals. Statistical analysis was performed with two tailed t-test or one-way analysis of variance (ANOVA) using Prism 5.0c Software (GraphPad Software, La Jolla, CA). For all results, statistical significance was defined by a value of P < 0.05.Results
Preparation and characterization of DMP micelles
DMP cationic micelles were prepared based on MPEG-PCL and DOTAP as previously described.30 According to their structures, the amphiphilic MPEG-PCL diblock copolymer composes of a hydrophobic PCL segment and a hydrophilic PEG segment.Meanwhile, DOTAP is also amphiphilic with a hydrophobic carbochain and a hydrophilic cationic head. Thus, rehydration of the lipid film composed of DOTAP and MPEG-PCL rendering these two amphiphilic materials self-assemble into micelles together, forming core–shell structured particles as presented in Scheme 1. Within these micelles, DOTAP with cationic hydrophilic head provides positive charges for DNA binding capacity. The prepared DMP micelles were characterized in detail. As shown in Fig. 1a, the dynamic diameter of DMP was 46.4 ± 3.7 nm with a polydispersity index of 0.215. The measured zeta potential was 44.1 ± 1.5 mV (Fig. 1b). We then studied the morphology of DMP micelles. As shown in Fig. 1d, monodispersed and spherical shaped DMP micelles could be observed in the TEM picture. The calculated diameter of DMP particle on TEM image was approximate 50 nm, which was consistent with its particle size.
 | ||
| Scheme 1 The preparation process of cationic DMP micelle. | ||
 | ||
| Fig. 1 Characterization of prepared DMP micelle. (a) Size distribution; (b) zeta potential distribution; (c) gene retarding assay; (d) TEM image; (e) cytotoxicity evaluated by MTT assay. | ||
In order to evaluate the binding ability of DMP micelles to DNA plasmid, a gel retarding assay was performed. As shown in Fig. 1c, after electrophoresis, when the molar ratio of DMP: plasmid DNA was 10:1, almost no bright DNA band was observed, suggesting that the negatively charged plasmid DNA was completely bond by DMP micelles through electronic interaction. This prescription ratio was chosen for further application in our study. Besides, a MTT assay was performed to evaluate the safety of DMP micelles in vitro. As shown in Fig. 1e, when applied on 293t cells, DMP micelle were much less toxic with its IC50 exceeded the concentration of 1.2 mg mL−1, while that of PEI25K (‘‘gold standard’’ transfection agent33,34) was estimated to be smaller than 0.1 mg mL−1. This indicated that DMP micelles were much safer that conventional used PEI25K transfection agent which showed a tremendous toxicity.
In vitro transfection study
The gene delivery capacity of prepared DMP micelles were evaluated on both normal and cancer cells in vitro. As shown in Fig. 2b and c, both 293t (normal tissue derived) and C26 (cancer tissue derived) cells could be efficiently transfected by DMP/pGFP complex. The transfection efficiency on 293t cell and C26 cells was 40.3 ± 2.5% and 35.0 ± 7.1% compared to that of 43.2 ± 4.3% and 15.5 ± 1.4% for PEI25K, respectively. A direct observation of the transfection ability of DMP micelles on both cells were shown in Fig. 2a with the aid of GFP-based reporter plasmid. It could be also observed that PEI25K transfected cells demonstrated obviously stronger cytotoxicity than DMP groups. There results suggested that DMP micelle was superior in both transfection ability and safety to PEI25K, showing high potential in gene delivery. | ||
| Fig. 2 Gene delivery ability of prepared DMP micelle on C26 and 293t cells. (a) fluorescent images of transfected cells, pEGFP plasmid was used as a reporter gene (at 100× magnification); (b) transfection efficiency on 293t cell calculated by flow cytometry; (c) transfection efficiency on 293t cell calculated by flow cytometry; (d) intracellular mRNA level of IL-22BP in C26 cells; (e) IL-22BP protein levels in supernatant of C26 cell culture. | ||
Within our study, the neutralizing and inhibition effect on IL-22 is based on efficient expression of IL-22BP from transfected cancer cells. Thus, to clarify whether pVAX1-mIL22BP plasmid was successfully delivered into C26 cells by DMP micelles, we first evaluated the intracellular mRNA level of IL-22BP gene after transfection. As shown in Fig. 2d, comparing to untreated group, 72 hours post transfection, a strong IL-22BP mRNA level over 480 folds (P < 0.001) were detected in DMP/mIL-22BP complex treated group, showing high delivery and expression efficiency. Meanwhile, since IL-22BP protein is expressed in a secretory form, we then checked the IL-22BP protein level in the cell culture supernatant by IL-22RA2 ELISA assay. According to our results (Fig. 2e), comparing to untreated group, 72 hours post transfection, high levels of mouse IL-22BP protein with up to nearly 3000 folds (P < 0.001) could be detected in DMP/IL-22BP complex treated group, suggesting the delivered plasmids were efficiently expressed as expected in C26 cells.
Anti-cancer ability of DMP/mIL-22BP complex in vitro
Since IL-22BP is capable of neutralizing intracellular IL-22, we first evaluated whether DMP/mIL-22BP complex could inhibit cancer cell growth IL-22/IL-22R blocking. As shown in Fig. 3a, adding IL-22 cytokine into cell culture resulted in an average proliferation rate of 213% in untreated C26 cells. However, only 40.2% in average of proliferation rate was detected in DMP/mIL-22BP complex treated group. In combine with IL-22BP expression results above, it suggested that expressed IL-22BP efficiently blocked IL-22 signal as expected. | ||
| Fig. 3 DMP/IL-22BP complex efficiently inhibit the growth of C26 cancer cells in vitro. (a) expressing of IL-22BP blocked IL-22-based cell proliferation signal; (b) inhibition effect of DMP/IL-22BP complex detected by MTT assay; (c) and (d) DMP/IL-22BP complex efficiently induced apoptosis in C26 cells; (e) inhibition effect of DMP/IL-22BP complex evaluated by clonogenic assay, the numbers of clones in each well were counted (f) and translated into inhibition rate (g). | ||
Meanwhile, although evidences have shown that IL-22BP is capable of inhibiting IL-22/IL-22R1 complexing, it is still unclear that whether intracellularly expression of IL-22BP will directly interfere cell function. Thus, we first evaluated its affection on cancer cells in vitro. To test its anti-proliferation effect on C26 colon cancer cells, a MTT assay was conducted. As shown in Fig. 3b, after 72 hours, obvious proliferation inhibition was observed in DMP/mIL-22BP complex treated group, with an average inhibition rate of 75.71% comparing to control group (P < 0.001). It indicated that DMP/mIL-22BP complex equivalent to 0.5 μg of IL-22BP gene was able to kill more than 50% of C26 cell in vitro. This effect was not reached by other groups with average inhibition rates of 18.8% for DMP/pVAX1 group and 4% for DMP micelle group. The anti-proliferation capacity of DMP/mIL-22BP complex was also evaluated by clonogenic assay. As shown in Fig. 3e, 14 days after transfection, much fewer clones could be observed in DMP/mIL-22BP complex treated well than other wells. The average number of clones in DMP/mIL-22BP complex well was 72 while that of NS control, DMP/pVAX1 and DMP micelle wells were 331, 275 and 295 (Fig. 3f), respectively. In this experiment, single C26 cells were cultured and grown into small clones which could be stained by crystal violet blue. The fewer clones being visualized implies stronger anti-proliferation capacity, suggesting an average inhibition rate of 77.3% (P < 0.01 versus DMP group and P < 0.005 versus DMP/pVAX1 group, Fig. 3g). Our results suggested that when lacking of extracellular IL-22 signal, DMP delivered IL-22BP gene could directly inhibit cancer cell proliferation alone.
To clarify whether this anti-proliferation effect of DMP/mIL-22BP complex on C26 cells was conducted by apoptosis inducing, cells in different treatment group was analyzed by flow cytometry with PI/Annexin V staining. As shown in our results, DMP/mIL-22BP complex induced obvious apoptosis in C26 cells (Fig. 3d). After been exposed to IL-22BP complex for 72 h, a total of 31 ± 4.9% of C26 cells were detected in early and late apoptosis phase (P < 0.001), while other groups failed to exhibit equivalent capacity (shown in Fig. 3c). Our results suggested that DMP micelle could efficiently deliver IL-22BP coding plasmid into C26 cells in vitro, inhibiting cell proliferation through apoptosis induction. In summary, our results indicated that both IL-22 blocking as well as directly apoptosis inducing were involved in the anti-cancer mechanisms of DMP delivered IL-22BP.
CLPP/VSVMP mRNA complex inhibits C26 tumor growth in vivo
The anti-cancer activity of DMP/mIL-22BP complex was evaluated on C26 abdominal cavity metastases model by intraperitoneal administration. Fig. 4a shows representative images of abdominal cavity metastases of C26 colon carcinoma in each treatment group. It was obvious that the mice treated with DMP/mIL-22BP complex suffered mildest abdominal cavity metastases than other groups. As shown in Fig. 4b, compared with other group, IL-22BP treatment group was much lower in metastases tumor weight (P < 0.01), with an average weight of 1.7 ± 0.5 g than those of NS group (4.7 ± 0.5 g), DMP group (4.6 ± 1.3 g) and DMP/pVAX1 group (4.7 ± 0.8 g). Meanwhile, as shown in Fig. 4c, there was also an obviously decrease in the ascites volume of DMP/mIL-22BP complex treated mice. The volume of ascites in mice treated with IL-22BP complex was 0.5 ± 0.2 mL compared with 1.5 ± 0.5 mL in control group, 1.6 ± 0.5 mL in DMP group and 1.6 ± 0.3 mL in the mice treated with DMP/pVAX1 complex. It can also be observed that the mice without IL-22BP complex treatment suffered from obvious blood-like ascites, suggesting serious tumor infiltrating and inflammation. These results indicated that DMP/mIL-22BP complex efficiently suppressed tumor growth of abdominal cavity metastases in vivo. | ||
| Fig. 4 DMP/IL-22BP complex efficiently inhibits C26 colon cancer growth in vivo. (a) images of abdominal cavity metastases of C26 colon carcinoma in each treatment group, representative tumor nodules were marked by write arrows; (b) average tumor weights in each group; (c) average volume of ascites in each group; (d) mRNA level of IL-22BP in tumor tissues from each group; (e) expression of IL-22BP in tumors detected by immunohistochemistry analysis (at 100× magnification). | ||
The expression of IL-22BP in tumor tissues were confirmed by qPCR analysis and immunohistochemistry staining. According to our results (Fig. 4d), significant enhanced IL-22BP mRNA levels were detected in DMP/mIL-22BP complex treatment groups, with over 220 folds (P < 0.01) of increase comparing to control group, respectively. The high mRNA levels being detected indicated that IL-22BP plasmid was efficiently delivered into tumor cells by DMP micelle. Meanwhile, as shown in Fig. 4e, strong IL-22BP positive pots could be observed in tumor tissues from DMP/mIL-22BP treatment group, suggesting efficient secretly expression of IL-22BP protein after administration. These evidences indicated that IL-22BP gene was successfully delivered by DMP micelles into C26 colon cancer tissues and expressed as expected.
According to previous reports and our in vitro results, the anti-cancer ability of IL-22BP could be possibly attribute to IL-22 neutralizing and direct apoptosis inducing. Thus, we further evaluated these mechanisms in tumor samples by immunohistochemistry staining. First, as shown in Fig. 5, treatment with DMP/mIL-22BP complex induced a significantly increase in apoptosis within tumor tissues compared to other groups as determined by the TUNEL assay. This performance is consistence with our in vitro results. Then, the DMP/mIL-22BP complex treatment groups also demonstrated anti-angiogenesis effects in tumors compared to other groups as determined by CD31 staining mTOR. The micro-vessel density characterized by CD31 positive staining was significantly attenuated in the IL-22BP treatment group when compared with control groups. Therefore, our results suggested that DMP/IL-22BP complex could also inhibit tumor growth through anti-angiogenesis mechanism. Moreover, comparing to other groups, obvious CD8+ and CD4+ cells could be spotted in the tumor tissues of DMP/mIL-22BP group, showing strong lymphocyte infiltration in tumor microenvironment. Since lymphocyte infiltration is highly correlated with immune reaction, these phenomena suggested that boosting anti-cancer immune response were also involved in IL-22BP-based therapy.
 | ||
| Fig. 5 Immunohistochemistry analysis of in vivo anti-cancer mechanisms of DMP/IL-22BP complex on C26 abdominal cavity metastases mode. Studies on anti-angiogenesis (CD31), apoptosis (TUNEL) and lymphocyte filtration (CD4+ & CD8+) were performed (at 100× magnification). | ||
Discussion
In recent years, gene therapy has provided an alternative choice for cancer treatment. Seeking novel therapeutic targets as well as appropriate gene delivery vector remains to be important issue for cancer gene therapy. In this work, the murine IL-22BP gene was delivered by a newly developed non-viral gene vector DMP, establishing a novel cancer gene therapy strategy (Scheme 2). Our results showed that DMP micelle-based delivery and secretly expression of IL-22BP could efficiently inhibit tumor growth through multiple mechanisms, showing strong potential in cancer gene therapy.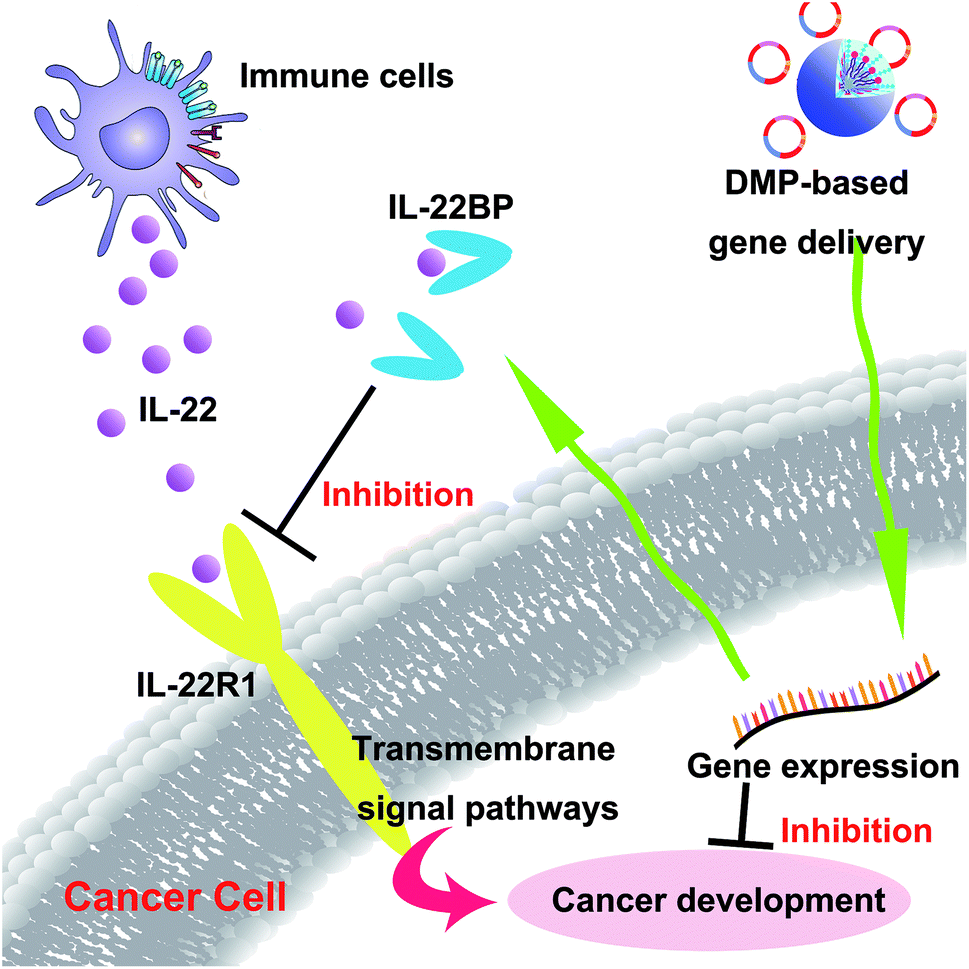 | ||
| Scheme 2 DMP micelle delivered IL-22BP gene efficiently inhibits tumor progression blocking IL-22 and apoptosis inducing. | ||
Owing to high specificity and safety, gene therapy has provided an alternative strategy for the treatment of cancer. In recent years, much progresses have been made using viral- or non-viral delivery systems, with some candidates being already approved for clinical use.29,35–38 In the area of plasmid-based gene therapy, several strategies have been attempted and evaluated. Among these, one of the most frequently applied strategy is delivering therapeutic exogenous or endogenous nucleic materials into cells to induce cell apoptosis.39–41 Several genes including VSVMP,42 p53 (ref. 43), survivinT34A30,44 and HSV1-tk45,46 have been tested and proved to be effective. This method provides “suicide” signals within cancer cells and achieves therapeutic effect through triggering programed cell death. Another conventional cancer gene therapy strategy is delivering antigen encoding plasmid into tumors. This “vaccine” method aims to stimulate anti-cancer immune response by expressing cancer specific markers such as peptide or polysaccharides which are then recognized and presented by APC cells.47,48 For example, Pirouz Daftarian et al. reported using dendrimer platform to deliver several plasmid DNAs encoding including TRP2, gp70 and OVA to develop DNA-based vaccines;49 Ang Li et al. used Mg/Al layered double hydroxides nanoparticles to deliver OVA encoding plasmid.50 Besides, a third gene therapy method involves stimulating anti-cancer immune response through direct enhancing the activities of immune cells, such as T cells and NK cells.51–53 To achieve this, cytokines or peptides required by the proliferation or maturation of immune cell were delivered to tumor microenvironment, resulting upregulated tumor infiltration and cytotoxic cytokine expression.54–56 Despite all these advantages and efficacies been demonstrated, several shortcomings still exist among them. For instance, the number of available suicide genes is severely limited with relative single mechanism. It is easy for cancer cells to active complementary pathway for replenishment, thus eradicate apoptosis signal.57 Then, the application of certain vaccine-based gene therapy usually limited to given tumor cells due to the specificity of antigens, which set obstacles to its use in other kinds of cancers, and immune response might be varied according to different vaccines. Meanwhile, immune stimulation effect mediated by vaccine is indirect. The therapeutic effect of gene vaccine relies much on antigen presenting process, while the latter is intensively correlated with the properties of antigens. Moreover, it should also be taken into consideration the potent of PD-1/PD-L1 blockage on immune system, especially T cell-based response.58 In previously reported colon cancer gene therapy studies, both “suicide” gene and immune activation strategies have been applied. Although potential therapeutic effects have been achieved, limitations were shown in the meantime. For example, most of the apoptosis inducing studies were restricted in the limited genes such as VSVMP. More suicide genes should be developed. Besides, immune activation-based colon cancer gene therapy strategy worked mainly in abdominal cavity metastases model, while little is reported about that in other tumor models. One possible reason is that the immune stimulation mechanisms rely much on ascites environment. In our study, we developed a novel therapeutic target based on IL-22 and its soluble receptor IL-22BP. As a member of cytokines, IL-22 is largely different with most of its counterparts since it acts directly on non-immune cells rather than immune cells.10,15 The important role of IL-22 in the progress of innate immune defenses, inflammation as well as tissue regeneration has been well characterized before. More importantly, evidences have been raised about the potential role of IL-22 in cancer. By binding to its transmembrane receptor IL-22R1, IL-22 transduce signals extracellular growth signals into cancer cells, resulting in enhanced tumor growth, suppressed apoptosis and metastasis. Thus, these properties of IL-22 suggest its potential as a therapeutic target. In our study, IL-22BP was delivered to neutralize IL-22 within microenvironment, so as to block IL-22/IL-22R1 signal axis. According to our results, DMP/IL-22BP DNA complex demonstrated strong anti-cancer effect on C26 colon cancer models. Meanwhile, succedent therapeutic effects including apoptosis, anti-angiogenesis and lymphocyte infiltration were also observed. More importantly, we further indicated that IL-22BP gene alone could inhibit cancer cell growth through inducing apoptosis in vitro, which has seldom been noticed by previous reports. This further suggested the flexible mechanism of IL-22BP gene. Comparing to other strategies, IL-22BP induced direct blockage on cancer growth signal without the help of immune cells. Meanwhile, the expression patent of IL-22 is mostly upregulated in inflammatory or cancer conditions, thus the impact of exogenous IL-22BP on health tissues would be much lower. These results suggested that this novel therapeutic strategy would be also potent in the treatment of other colon cancer models. After all, our results demonstrated IL-22 to be a novel target for cancer gene therapy with multiple mechanisms.
The efficiency and safety of gene delivery vectors also play crucial roles in gene therapy, while nanotechnology has contributed tremendously to this area.29,59–63 In this study, a newly developed DMP cationic micelle was used to deliver IL-22BP gene. As described previously, DMP micelle was formulated by self-assembling DOTAP with biodegradable polymer mPEG-PCL.32 The preparation process was simple with little organic solvent involved since both of the two components are amphiphilic. In our previous work, DMP micelle has been successfully applied in gene therapy study of colon cancer. In the present work, our results demonstrated that DMP micelles could efficiently deliver IL-22BP encoding plasmid into C26 colon cancer cells both in vitro and in vivo with high safety. Transfection with DMP/IL-22BP complex resulted in high IL-22BP mRNA level in C26 cells and tumor nodules, and obvious tumor growth inhibitions were achieved upon i.p administration. Moreover, no obvious pathology changes were detected in main organs(data not shown). These results again demonstrated the efficacy and safety of DMP cationic micelles. Comparing to DOTAP-based cationic liposome, the amphiphilic block polymer mPEG-PCL in DMP micelles provides better embedding effect for DOTAP, thus exhibiting proper positive charges. It is highly possible that this formulation structure contributes to its strong DNA binding efficacy as well as low cytotoxicity. In total, our results indicated that the newly developed cationic micelle DMP was capable of delivering IL-22BP gene into C26 colon cancer tissues, providing a potential choice for cancer gene therapy.
Conclusions
In this work, the murine IL-22BP encoding plasmid was successfully delivered by a newly developed cationic micelle DMP. The DMP/IL-22BP complex could efficiently inhibit the growth of C26 abdominal cavity metastases cancer model both in vitro and in vivo with high safety. Our results demonstrated the therapeutic potential of IL-22BP in cancer gene therapy. DMP micelle delivered IL-22BP gene offered an alternative strategy for cancer gene therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation (NSFC81502677), the National Key Research and Development Program of China (2016YFA0201402), the National Key Specialty Construction Project of Clinical Pharmacy (No 30305030698), the Science Foundation of Chengdu (2016-HM01-00438-SF).References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef PubMed.
- R. L. Siegel, K. D. Miller, S. A. Fedewa, D. J. Ahnen, R. G. Meester, A. Barzi and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 177–193 CrossRef PubMed.
- S. L. Ginn, I. E. Alexander, M. L. Edelstein, M. R. Abedi and J. Wixon, J. Gene Med., 2013, 15, 65–77 CrossRef CAS PubMed.
- L. Naldini, Nature, 2015, 526, 351 CrossRef CAS PubMed.
- E. Smalley, Nat. Biotechnol., 2017, 35, 998–999 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541 CrossRef CAS PubMed.
- C. Morrison, Nat. Biotechnol., 2015, 33, 217–218 CrossRef CAS PubMed.
- V. Kumar, S. Palazzolo, S. Bayda, G. Corona, G. Toffoli and F. Rizzolio, Theranostics, 2016, 6, 710 CrossRef CAS PubMed.
- S. V. Kotenko, L. S. Izotova, O. V. Mirochnitchenko, E. Esterova, H. Dickensheets, R. P. Donnelly and S. Pestka, J. Immunol., 2001, 166, 7096–7103 CrossRef CAS.
- J. C. Martin, G. Bériou, M. Heslan, C. Chauvin, L. Utriainen, A. Aumeunier, C. Scott, A. Mowat, V. Cerovic and S. Houston, Mucosal Immunol., 2014, 7, 101 CrossRef CAS PubMed.
- P. Pelczar, M. Witkowski, L. G. Perez, J. Kempski, A. G. Hammel, L. Brockmann, D. Kleinschmidt, S. Wende, C. Haueis and T. Bedke, Science, 2016, 354, 358–362 CrossRef CAS PubMed.
- X. Yang and S. G. Zheng, Autoimmun. Rev., 2014, 13, 615–620 CrossRef CAS PubMed.
- S. V. Kotenko, L. S. Izotova, O. V. Mirochnitchenko, E. Esterova, H. Dickensheets, R. P. Donnelly and S. Pestka, J. Biol. Chem., 2001, 276, 2725–2732 CrossRef CAS PubMed.
- Y. Peng and C. M. Croce, Signal Transduction Targeted Ther., 2016, 1, 15004 CrossRef PubMed.
- C. Lim and R. Savan, Cytokine Growth Factor Rev., 2014, 25, 257–271 CrossRef CAS PubMed.
- Y. Zheng, D. M. Danilenko, P. Valdez, I. Kasman, J. Eastham-Anderson, J. Wu and W. Ouyang, Nature, 2007, 445, 648 CrossRef CAS PubMed.
- L. Geboes, L. Dumoutier, H. Kelchtermans, E. Schurgers, T. Mitera, J. C. Renauld and P. Matthys, Arthritis Rheumatol., 2009, 60, 390–395 CrossRef CAS PubMed.
- G. F. Sonnenberg, M. G. Nair, T. J. Kirn, C. Zaph, L. A. Fouser and D. Artis, J. Exp. Med., 2010 DOI:10.1084/jem-20092054.
- L. Dumoutier, D. Lejeune, D. Colau and J.-C. Renauld, J. Immunol., 2001, 166, 7090–7095 CrossRef CAS.
- W. Xu, S. R. Presnell, J. Parrish-Novak, W. Kindsvogel, S. Jaspers, Z. Chen, S. R. Dillon, Z. Gao, T. Gilbert and K. Madden, Proc. Natl. Acad. Sci., 2001, 98, 9511–9516 CrossRef CAS PubMed.
- K. Sugimoto, A. Ogawa, E. Mizoguchi, Y. Shimomura, A. Andoh, A. K. Bhan, R. S. Blumberg, R. J. Xavier and A. Mizoguchi, J. Clin. Invest., 2008, 118, 534–544 CAS.
- M. Sertorio, X. Hou, R. F. Carmo, H. Dessein, S. Cabantous, M. Abdelwahed, A. Romano, F. Albuquerque, L. Vasconcelos and T. Carmo, Hepatology, 2015, 61, 1321–1331 CrossRef CAS PubMed.
- J. Dambacher, F. Beigel, K. Zitzmann, T. Olszak, T. Prüfer, C. Steib, M. Storr, B. Göke, H. Diepolder and M. Bilzer, J. Gastroenterol., 2006, 44, 43 Search PubMed.
- L. A. Zenewicz, G. D. Yancopoulos, D. M. Valenzuela, A. J. Murphy, S. Stevens and R. A. Flavell, Immunity, 2008, 29, 947–957 CrossRef CAS PubMed.
- H. Yu, D. Pardoll and R. Jove, Nat. Rev. Cancer, 2009, 9, 798 CrossRef CAS PubMed.
- H. Yu, M. Kortylewski and D. Pardoll, Nat. Rev. Immunol., 2007, 7, 41 CrossRef CAS PubMed.
- M. Karin and F. R. Greten, Nat. Rev. Immunol., 2005, 5, 749 CrossRef CAS PubMed.
- M. Gyrd-Hansen and P. Meier, Nat. Rev. Cancer, 2010, 10, 561 CrossRef CAS PubMed.
- S. Shi, K. Shi, L. Tan, Y. Qu, G. Shen, B. Chu, S. Zhang, X. Su, X. Li and Y. Wei, Biomaterials, 2014, 35, 4536–4547 CrossRef CAS PubMed.
- X. Duan, P. Wang, K. Men, X. Gao, M. Huang, M. Gou, L. Chen, Z. Qian and Y. Wei, Nanoscale, 2012, 4, 2400–2407 RSC.
- K. Men, W. Liu, L. Li, X. Duan, P. Wang, M. Gou, X. Wei, X. Gao, B. Wang and Y. Du, Nanoscale, 2012, 4, 6425–6433 RSC.
- M. Gou, X. Wei, K. Men, B. Wang, F. Luo, X. Zhao, Y. Wei and Z. Qian, Curr. Drug Targets, 2011, 12, 1131–1150 CrossRef CAS PubMed.
- J. Kim, J. Park, H. Kim, K. Singha and W. J. Kim, Biomaterials, 2013, 34, 7168–7180 CrossRef CAS PubMed.
- K. Zwiorek, J. Kloeckner, E. Wagner and C. Coester, J. Pharm. Pharm. Sci., 2004, 7, 22–28 CAS.
- P. J. Brooks, N. N. Yang and C. P. Austin, Hum. Gene Ther., 2016, 27, 7–13 CrossRef CAS PubMed.
- Q. Zheng, D. Lin, L. Lei, X. Li and S. Shi, J. Biomed. Nanotechnol., 2017, 13, 1565–1580 CrossRef PubMed.
- E. Ren, J. Wang and G. Liu, Chin. Chem. Lett., 2017, 28, 1799–1800 CrossRef CAS.
- Z.-Y. He, Z.-H. Jin, M. Zhan, Z. Qin, Z. Li and T. Xu, Chin. Chem. Lett., 2017, 28, 1851–1856 CrossRef CAS.
- S. D. Jo, S. H. Ku, Y.-Y. Won, S. H. Kim and I. C. Kwon, Theranostics, 2016, 6, 1362 CrossRef CAS PubMed.
- W.-Y. Kuo, L. Hwu, C.-Y. Wu, J.-S. Lee, C.-W. Chang and R.-S. Liu, Theranostics, 2017, 7, 647 CrossRef CAS PubMed.
- S. M. Freeman, in Cancer Gene Therapy, Springer, 2002, pp. 411–422 Search PubMed.
- M. Gou, K. Men, J. Zhang, Y. Li, J. Song, S. Luo, H. Shi, Y. Wen, G. Guo and M. Huang, ACS Nano, 2010, 4, 5573–5584 CrossRef CAS PubMed.
- F. F. Lang, J. M. Bruner, G. N. Fuller, K. Aldape, M. D. Prados, S. Chang, M. S. Berger, M. W. McDermott, S. M. Kunwar and L. R. Junck, J. Clin. Oncol., 2003, 21, 2508–2518 CrossRef CAS PubMed.
- B. Blum, O. Bar-Nur, T. Golan-Lev and N. Benvenisty, Nat. Biotechnol., 2009, 27, 281 CrossRef CAS PubMed.
- D. Klatzmann, C. A. Valery, G. Bensimon, B. Marro, O. Boyer, K. Mokhtari, B. Diquet, J.-L. Salzmann, J. Philippon and S. G. O. G. T. F. Glioblastoma, Hum. Gene Ther., 1998, 9, 2595–2604 CrossRef CAS PubMed.
- Y. Wang, H.-h. Su, Y. Yang, Y. Hu, L. Zhang, P. Blancafort and L. Huang, Mol. Ther., 2013, 21, 358–367 CrossRef CAS PubMed.
- D. Fioretti, S. Iurescia, V. M. Fazio and M. Rinaldi, J. BioMed. Biotechnol., 2010, 2010, 174378 Search PubMed , 16 pages.
- C. Bode, G. Zhao, F. Steinhagen, T. Kinjo and D. M. Klinman, Expert Rev. Vaccines, 2011, 10, 499–511 CrossRef CAS PubMed.
- P. Daftarian, A. E. Kaifer, W. Li, B. B. Blomberg, D. Frasca, F. Roth, R. Chowdhury, E. A. Berg, J. B. Fishman and H. A. Al Sayegh, Cancer Res., 2011, 71, 7452–7462 CrossRef CAS PubMed.
- A. Li, L. Qin, W. Wang, R. Zhu, Y. Yu, H. Liu and S. Wang, Biomaterials, 2011, 32, 469–477 CrossRef CAS PubMed.
- C. H. Lamers, R. A. Willemsen, B. A. Luider, R. Debets and R. L. Bolhuis, Cancer Gene Ther., 2002, 9, 613 CrossRef CAS PubMed.
- K. Suzuki, H. Nakazato, H. Matsui, M. Hasumi, Y. Shibata, K. Ito, Y. Fukabori, K. Kurokawa and H. Yamanaka, J. Leukocyte Biol., 2001, 69, 531–537 CAS.
- P.-U. Malmström, A. S. Loskog, C. A. Lindqvist, S. M. Mangsbo, M. Fransson, A. Wanders, T. Gårdmark and T. H. Tötterman, Clin. Cancer Res., 2010, 16, 3279–3287 CrossRef PubMed.
- M. Luo, X. Liang, S.-T. Luo, X.-W. Wei, T. Liu, J. Ren, C.-C. Ma, Y.-H. Yang, B.-L. Wang and L. Liu, J. Biomed. Nanotechnol., 2015, 11, 2011–2023 CrossRef CAS PubMed.
- D. H. Sterman, E. Alley, J. P. Stevenson, J. Friedberg, S. Metzger, A. Recio, E. K. Moon, A. R. Haas, A. Vachani and S. I. Katz, Clin. Cancer Res., 2016, 22, 3791–3800 CrossRef CAS PubMed.
- J.-Y. Ding, Z.-H. Wang, Z.-Z. Zhang, X.-R. Cui, Y.-Y. Hong and Q.-Q. Liu, Oncotarget, 2017, 8, 108108 Search PubMed.
- S. Grant, J. Clin. Invest., 2008, 118, 3003–3006 CrossRef CAS PubMed.
- S. L. Topalian, C. G. Drake and D. M. Pardoll, Curr. Opin. Immunol., 2012, 24, 207–212 CrossRef CAS PubMed.
- S. Shang, L. Monfregola and M. H. Caruthers, Signal Transduction Targeted Ther., 2016, 1, 16019 CrossRef PubMed.
- A. Khatri, A. Mishra and V. S. Chauhan, J. Biomed. Nanotechnol., 2017, 13, 35–53 CrossRef CAS PubMed.
- A. Srivastava, A. Babu, J. Filant, K. M. Moxley, R. Ruskin, D. Dhanasekaran, A. K. Sood, S. McMeekin and R. Ramesh, J. Biomed. Nanotechnol., 2016, 12, 1159–1173 CrossRef CAS PubMed.
- C. Oliveira, A. J. Ribeiro, F. Veiga and I. Silveira, J. Biomed. Nanotechnol., 2016, 12, 841–862 CrossRef CAS PubMed.
- S. Duan, M. Song, J. He, N. Zhou, S. Zhou, J. Zhao, Y. Fang, Y. Peng, X. Huang and G. Luo, J. Biomed. Nanotechnol., 2016, 12, 700–709 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |